
Abstract
Main conclusion
Successful application of the photosynthesis-to-fuels approach requires a high product-to-biomass carbon-partitioning ratio. The work points to the limiting amounts of heterologous terpene synthase in cyanobacteria as a potential barrier in the yield of terpene hydrocarbons via photosynthesis.
Cyanobacteria like Synechocystis sp. can be exploited as platforms in a photosynthesis-to-fuels process for the generation of terpene hydrocarbons. Successful application of this concept requires maximizing photosynthesis and attaining a high endogenous carbon partitioning toward the desirable product. The work addressed the question of the regulation of β-phellandrene synthase transgene expression in relation to product yield from the terpenoid biosynthetic pathway of cyanobacteria. The choice of strong alternative transcriptional and translational cis-regulatory elements and the choice of the Synechocystis genomic DNA loci for transgene insertion were investigated. Specifically, the β-phellandrene synthase transgene was expressed under the control of the endogenous psbA2 promoter, or under the control of the Ptrc promoter from Escherichia coli with the translation initiation region of highly expressed gene 10 from bacteriophage T7. These heterologous elements allowed for constitutive transgene expression. In addition, the β-phellandrene synthase construct was directed to replace the Synechocystis cpc operon, encoding the peripheral phycocyanin rods of the phycobilisome antenna. Results showed that a 4-fold increase in the cellular content of the β-phellandrene synthase was accompanied by a 22-fold increase in β-phellandrene yield, suggesting limitations in rate and yield by the amount of the transgenic enzyme. The work points to the limiting amount of transgenic terpene synthases as a potential barrier in the heterologous generation of terpene products via the process of photosynthesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyanobacteria have the potential to be industrially robust photosynthetic microorganisms, competent to drive oxygenic photosynthesis and the associated metabolism toward the synthesis of high impact products. Novel biosynthetic pathways leading to molecules with industrial and transportation applications have been introduced in cyanobacteria, mainly through the heterologous expression of exogenous genes. Cyanobacteria are especially promising host microorganisms for the production of small molecules that can be exuded or secreted (Melis 2012; Wijffels et al. 2013). These include ethanol (Deng and Coleman 1999; Dexter and Fu 2009; Hellingwerf and Teixeira de Mattos 2009; Gao et al. 2012), isoprene (Lindberg et al. 2010), lactic acid (Angermayr et al. 2012), sucrose (Ducat et al. 2012), ethylene (Takahama et al. 2003), acetone (Zhou et al. 2012), iso-butyraldehyde and isobutanol (Atsumi et al. 2009), 1-butanol (Lan and Liao 2012), 3-hydroxybutyrate (Wang et al. 2013), 2,3-butanediol (Oliver et al. 2013), 1,2-propanediol (Li and Liao 2013), fatty acids (Liu et al. 2011), and fatty alcohols (Tan et al. 2011). Product levels in the extracellular medium were reported in the mg L−1 range, up to g L−1 for ethanol, 2,3-butanediol and isobutanol (Gao et al. 2012; Oliver et al. 2013; Varman et al. 2013).
Terpene hydrocarbons are derived as secondary metabolites by many land plants (Gershenzon and Dudareva 2007). Their industrial applications range from feedstock for the synthetic chemistry industry, perfumes, pharmaceuticals, pesticides and hydrocarbon biofuels (Bohlmann and Keeling 2008; Peralta-Yahya et al. 2011). Heterologous production of the hemiterpene isoprene (C5H8) in Synechocystis (Lindberg et al. 2010) was the first metabolic engineering of cyanobacteria conferring CO2-based production of a terpene. Significant in this development was the quantitative and spontaneous separation of isoprene from the biomass and the liquid culture. Synechocystis was also employed to generate the monoterpene β-phellandrene (PHL) (Bentley et al. 2013) and the sesquiterpene β-caryophyllene (Reinsvold et al. 2011), providing proof-of-concept that cyanobacteria can be metabolically engineered to produce secondary monoterpenes and sesquiterpenes. However, only μg quantities of the desired product were reported to accumulate [e.g. 50, 25 and 0.5 μg g−1 of dry cell weight (dcw) per day for isoprene, β-phellandrene, and β-caryophyllene, respectively] (Lindberg et al. 2010; Bentley et al. 2013; Reinsvold et al. 2011). The limited yields highlighted the problem of insufficient carbon partitioning to heterologous product formation relative to cell growth and biomass accumulation (Melis 2013).
There are two naturally occurring metabolic pathways leading to the formation of isopentenyl-diphosphate and dimethylallyl-diphosphate, which are precursors to all terpenoids: the mevalonic acid pathway, which operates in the cytosol of eukaryotes and archaea, and the methyl-erythritol-4-phosphate (MEP) pathway, which is of prokaryotic origin and active in plant plastids (Lichtenthaler 2000). Cyanobacteria express the MEP pathway to synthesize a wide variety of terpenoid-like molecules essential for cell function, such as carotenoids, phytol, quinones, tocopherols, sterols, and other prenyl molecules. However, they do not naturally synthesize secondary terpene hydrocarbons, such as monoterpenes, since they are not endowed with monoterpene synthases (Van Wagoner et al. 2007). A number of terrestrial plant species naturally produce in their leaves the monoterpene β-phellandrene, as a constituent of their essential oils. β-Phellandrene is synthesized from the metabolite geranyl-diphosphate by a nucleus-encoded and plastid localized β-phellandrene synthase enzyme. Recently, the β-phellandrene synthase gene from lavender (Demissie et al. 2011) was codon optimized and introduced via homologous recombination in the psbA2 site of Synechocystis, under the control of the psbA2 promoter, conferring β-phellandrene synthesis and accumulation to these cyanobacteria (Bentley et al. 2013). This approach utilized the expression system and transgene insertion site that was developed earlier for the isoprene synthase expression and isoprene production in this laboratory (Lindberg et al. 2010). As a monoterpene molecule, β-phellandrene diffused from the cell interior and accumulated as a non-miscible product on the surface of a sealed culture (Bentley et al. 2013). Spontaneous β-phellandrene separation from the biomass and from the aqueous culture alleviates undesirable product inhibition or toxic effects on cellular metabolism. Moreover, as a culture surface floater molecule, β-phellandrene could easily be harvested from the extracellular liquid phase, a parameter that weighs heavily on the economics of a microbial production system.
It is evident from the above-mentioned preliminary work that cyanobacteria can serve as platforms for the synthesis of fuel and chemicals in a process driven by sunlight with CO2 and H2O as the primary feedstock. However, many of the genetic elements that control chromosomal transgene expression, including transcription, translation, and recombinant protein accumulation in cyanobacteria are not fully understood, as they behave differently than the extrachromosomal gene expression from autonomously replicating plasmids in model heterotrophic microorganisms, e.g. Escherichia coli. The present study is, therefore, part of an effort in this laboratory to address chromosomal transgene-expression and carbon-partitioning issues in specific biosynthetic pathways in photosynthetic systems (Melis 2013). The focal point of the work is regulation of heterologous β-phellandrene synthase expression, as determined by the selection of promoters, codon-use optimization, and genomic DNA insertion sites in Synechocystis to increase the level of the catalytic enzyme(s) beyond a point where it is limiting the overall rate and yield of product formation. The function of strong alternative promoters, conferring constitutive expression of the transgene(s), was investigated. Specifically, rate and yield of β-phellandrene were examined under conditions when the β-phellandrene synthase gene was expressed under the control of the endogenous psbA2 promoter or under the control of the heterologous Ptrc promoter from E. coli. The ribosome-binding site of Ptrc 5′UTR was further replaced by the translation initiation region of highly expressed gene 10 from bacteriophage T7. In addition, the β-phellandrene synthase construct was directed to replace the highly expressed Synechocystis cpc operon, encoding the peripheral phycocyanin rods of the phycobilisome antenna. In this case, the β-phellandrene synthase construct was expressed under the control of the cpc promoter. The resulting cpc-deleted transformants lacked phycocyanin from their light-harvesting pigments. Minimizing, or truncating, the light-harvesting antenna of the photosynthetic apparatus is an experimental approach by which to maximize photosynthetic productivities in high-density microalgal cultures under bright sunlight condition, including attenuation of the steep gradients of light intensity within the culture (Melis 2009). The outcome of this work was a demonstration of methods by which to increase the amount of the transgenic β-phellandrene synthase accumulating in the cells and, thereby, to improve the rate and yield of β-phellandrene production in cyanobacteria.
Materials and methods
Synechocystis strains, β-phellandrene synthase constructs, and culturing conditions
Synechocystis sp. PCC 6803 (Synechocystis) was used as the recipient strain and referred to as the wild type in this study (Williams 1988). The β-phellandrene synthase (PHLS)-encoding gene from Lavandula angustifolia (lavender) was codon optimized (Bentley et al. 2013) and cloned via NdeI and BamHI restriction in an operon configuration together with the gene conferring resistance to chloramphenicol in plasmid pBA2A2 (Lindberg et al. 2010), which contains 500 bp of the upstream and downstream sequences of the endogenous psbA2 gene. These flanking regions served in the psbA2 replacement by the exogenous transgene in the Synechocystis genome via double homologous recombination (Fig. 1a, b). After Synechocystis transformation, the resulting ∆psbA2-PHLS strain was endowed with the property of PHLS expression, regulated by the endogenous psbA2 promoter, and β-phellandrene production (Bentley et al. 2013).

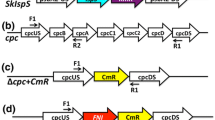
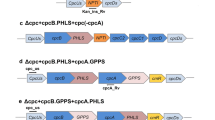
Graphic overview of recombinant constructs used in the present study. a Wild-type genomic DNA locus for the psbA2 gene. b Construct for the expression of PHLS under the control of the endogenous psbA2 promoter following homologous recombination and replacement of psbA2 gene. c Expression of PHLS under the control of the psbA2 promoter, where the cis-element controlling light-dependent RNA stability (AT-box) has been replaced by a GC sequence. Bold letters in the nucleotide sequence identify the AT (or GC)-box, the ribosome-binding site and the start codon for translation. d Expression of PHLS under the control of the Ptrc promoter in the psbA2 site. e Expression of PHLS under the control of the Ptrc promoter fused to the 5′UTR of bacteriophage T7 gene 10. Bold letters in the reported sequence mark the Ptrc promoter sequence (underlined are the −35 and −10 consensus elements), the EP upstream box, the ribosome-binding site, the ATG start codon for translation and the DB (downstream) box. f Wild-type genomic DNA locus for the cpc operon. g Expression of PHLS under the control of the endogenous cpc promoter following homologous recombination and replacement of the cpc operon. For all constructs, arrows show the location of primers used to test inserts and to genetically characterize the homoplasmy status of transformants (see Figs. 3, 4). Transformants were selected on the basis of resistance to chloramphenicol (cmR). The cmR gene was expressed downstream of the PHLS gene in each of the above operon configurations
The aforementioned plasmid was then amplified with primers pSynPHLS Fw _noATbox TTggcgcgccTAAGGAATTATAACCATATGTG and pSynPHLS Rv _noATbox TTggcgcgccTCGATGTTCAGATTGGAACTGAC, facing in opposite directions and substituting the AT-box (CAAATACA in Fig. 1b), which is responsible for light regulation in the psbA2 promoter (Horie et al. 2007), with the GGCGCGCC sequence (Fig. 1c) corresponding to AscI restriction site. The resulting PCR product was sequence verified, digested with AscI, ligated upon itself, and used to transform Synechocystis, obtaining the ∆psbA2(noAT box)-PHLS strain.
The psbA2 promoter (86 nucleotides upstream from the translation start codon) was replaced by the E. coli Ptrc-lacO1 (Amann et al. 1988), keeping the remaining upstream psbA2 region for homologous recombination in the Synechocystis genome. Ptrc-lacO1 is a hybrid promoter made with consensus sequences from E. coli Ptrp and Plac plus the LacI-binding operator O1. Unlike E. coli, the Ptrc-lacO1 promoter is poorly repressed in Synechocystis leading to constitutive expression (Guerrero et al. 2012). Nucleotide structure of the Ptrc-lacO1 promoter is shown in Fig. 1d with underlined −35 and −10 consensus elements. The psbA2-Ptrc-lacO1 fused sequence was synthesized by DNA2.0 (https://www.dna20.com) and used to replace the psbA2 sequence in the pBA2A2 plasmid (Fig. 1b) via XhoI and NdeI restriction. The resulting construct (Fig. 1d) was transformed into Synechocystis wild type obtaining ∆psbA2-trc-PHLS transformants.
The ribosome-binding site (RBS) of Ptrc-lacO1 was subsequently replaced by the 5′UTR and RBS of the highly expressed gene 10 of bacteriophage T7 (Sprengart et al. 1996) (Fig. 1e). The PHLS encoding sequence was additionally fused to the downstream box (DB): GCTAGC (Fig. 1e) from gene 10 of bacteriophage T7, adding two amino acids (Ala, Ser) at the 5′ end. The GCTAGC DB was shown to increase translation initiation in tobacco chloroplasts (Kuroda and Maliga 2001a). Synechocystis transformants for the latter construct are hereto referred as ∆psbA2-trc-T7gene10-PHLS.
Expression of the PHLS gene was further assayed under the control of the Synechocystis endogenous cpc promoter, upon replacement of the entire cpc operon (Fig. 1f), which encodes for components of the peripheral phycocyanin antenna rods. The upstream and downstream sequences of psbA2 in pBA2A2 plasmid (Fig. 1b) were replaced by 500 pb of the upstream and downstream sequences of the cpc operon, via either XhoI and NdeI or BamHI and SacI restriction, respectively (Fig. 1g). Synechocystis transformants for the latter construct are referred to as ∆cpc-PHLS. The DNA recombinant constructs in this work have been deposited and can be made available through Addgene (https://www.addgene.org/Anastasios_Melis) with the following Addgene accession numbers 52307: psbA2-PHLS (b); 52308: psbA2(noATbox)-PHLS (c); 52309: psbA2-Ptrc-PHLS (d); 52310: psbA2-Ptrc-T7-g10-PHLS (e); and 52311 cpc-PHLS (g).
Synechocystis transformations were performed according to established procedures (Eaton-Rye 2011). Wild type and transformants were maintained on 1 % agar BG11 media supplemented with 10 mM TES-NaOH (pH 8.2) and 0.3 % sodium thiosulphate. Liquid cultures in BG11 were buffered with 25 mM phosphate (pH 7.5) and incubated under continuous low-stream bubbling with air at 28 °C. Chloramphenicol was added to a final concentration of 30 μg mL−1 to select and maintain transformants. Cpc mutants reached homoplasmy on 1 % agar BG11 solid media supplemented with 5 mM glucose plus 60 μg mL−1 of chloramphenicol and upon incubation under weak blue light at 10 μmol photons m−2 s−1, designed to sensitize preferentially chlorophyll a rather than phycocyanin molecules.
PCR analysis of Synechocystis transformants for genomic DNA insertion and homoplasmy
A culture aliquot of 20 μL was mixed with an equal volume of 100 % ethanol by brief vortexing. Subsequently, 200 μL of a 10 % (w/v) suspension in water of Chelex®100 Resin (BioRad) was added to the sample prior to mixing and heating at 98 °C for 10 min to lyse the cells. Following centrifugation at 16,000g for 10 min to pellet debris, 5 μL of the supernatant was used as a genomic DNA template in a 25 μL PCR mix.
To test for DNA copy homoplasmy, primers in the PCR were chosen to be either external to the recombinant cassette and localized in the flanking genomic DNA regions (Fig. 1a) or placed within the deleted region on the genomic DNA to test for the presence of wild-type copies of the DNA (Fig. 1f). With primers external to the recombinant cassette, wild-type and transgenic DNA would yield different molecular size PCR products, affording insight as to the ratio of transgenic vs wild-type DNA in the cells (Fig. 3a). With primers placed within the deleted region on the genomic DNA, PCRs would test exclusively for the presence of wild-type copies of the Synechocystis DNA (Fig. 4a, b). This is considered to be a most sensitive test for transgenic DNA copy homoplasmy. In parallel, PCRs with a primer designed within the transgenic cassette would test for insert integration and would give a PCR DNA product only in the transformants but not in the wild type (Figs. 3b–d, 4c). A complete listing of primers with corresponding DNA sequences is given in Table 1.
SDS-PAGE and Western blot analysis
Cells were harvested by centrifugation and resuspended in (50 mM Tris–HCl pH 8, 50 mM NaCl, 10 mM CaCl2, 10 mM MgCl2) plus protease inhibitors (1 mM PMSF, 2 mM aminocaproic acid, 1 mM benzamidine). The cell suspension was treated first with lysozyme (Thermo Scientific) then with bovine pancreas DNAseI (Sigma) for 30 min each at room temperature. Cell disruption was achieved by French press (passing twice at 1,200 psi). The soluble fraction, containing PHLS (Bentley et al. 2013), was separated from membranes and cell debris by centrifugation at 20,000g for 10 min. Protein extracts were then solubilized in Laemmli loading buffer (1 % running buffer, 2 % SDS, 5 % β-mercaptoethanol, 10 % glycerol) and separated on SDS-PAGE (Bio-Rad, USA) prior to Coomassie staining. For Western blot analysis, proteins were transferred from the polyacrylamide gel to a nitrocellulose membrane and probed with PHLS-specific polyclonal antibodies (Bentley et al. 2013).
Analysis of photosynthetic pigments and measurement of photoautotrophic growth
Cells in the exponential growth phase were disrupted upon passing twice at 1,200 psi through a French press. Absorbance spectra of the crude homogenate in the range of 350–750 nm were then recorded, revealing the in vivo absorbance contributions of chlorophyll a, carotenoids and phycobilins without the expected scattering of light caused by intact cells. Chlorophyll a and carotenoid analysis was based on extraction in 90 % methanol (Meeks and Castenholz 1971). Photoautotrophic growth of wild type and transformants was measured from the optical density of the cultures at 730 nm.
Quantification of β-phellandrene production by Synechocystis transformants
Liquid cultures of Synechocystis in BG11 were grown photoautotrophically at incident light of 50 μmol photons m−2 s−1 under continuous low-stream air bubbling until reaching an optical density (OD) at 730 nm of 0.5. Cultures were subsequently grown in the 1 L aqueous/gaseous two-phase reactors developed in this lab (Bentley and Melis 2012), in which the 700 mL liquid culture is supplemented with 100 % CO2 gas. Delivery of the 100 % CO2 gas was by slow bubbling through the bottom of the liquid culture so as to fill the ~500 mL gaseous headspace. Cultures were sealed for 48 h and incubated under slow continuous mechanical mixing, at either 50 or 170 μmol photons m−2 s−1 of incident intensity, as indicated. At the end of the 48 h cultivation, β-phellandrene was removed from the culture as a floater molecule. This was achieved upon addition of a known amount of hexane as an over-layer on top of the liquid culture, while gently stirring for 2 h (Bentley et al. 2013). The addition of hexane helped in the collection of floating β-phellandrene molecules by increasing the volume of the top hydrophobic layer. β-Phellandrene was quantified from the absorption spectrum of the hexane extract in the UV region of the spectrum. An extinction coefficient of 15,698 L mol−1 cm−1 (15.7 mM cm−1) at 232.4 nm was calculated in the present study by titrating dilutions of a β-phellandrene standard (Chemos GmbH) from their absorbance spectra (Fig. 2a). The slope of the straight line in the plot of the absorbance at 232.4 nm as a function of β-phellandrene concentration (Fig. 2b) is consistent with some measurements in the literature (Gross and Schnepp 1978), but is rather different from the result of others (MacBeth et al. 1938; Booker et al. 1940). β-Phellandrene production was normalized to that of biomass that accumulated in the same period of time and expressed as g PHL per g dcw. For the gravimetric dcw measurement, 5 mL of culture aliquots was filtered through 0.22 μm Millipore filters and the immobilized cells were dried in a convection oven at 80 °C overnight prior to weighing with a Sartorius CP124S analytical balance. Qualitatively, β-phellandrene production was confirmed in the hexane extract by gas chromatography–mass spectrometry (GC–MS) analysis, according to established approaches (Demissie et al. 2011; Bentley et al. 2013).

Absorbance spectroscopy of β-phellandrene standard solutions (Chemos GmbH, #9964, d = 0.85 g mL−1, MW = 136.23 g mol−1). a Absorbance spectra of serial dilutions in hexane. b Maximum absorbance at 232.4 nm as a function of β-phellandrene concentration (mol L−1). The β-phellandrene extinction coefficient in hexane was estimated to be 15.698 L mol−1 cm−1, assuming that contaminants present in the standard are not significantly contributing to the absorbance at 232.4 nm
Results
Heterologous transformation of the PHLS gene in Synechocystis under the control of different promoters
Application of the endogenous psbA2 promoter in the presence or absence of the AT light-sensitivity box
The psbA gene, encoding the 32 kD D1 reaction center protein of photosystem II, occurs as a single copy in the plastid genome of plants and algae (Nixon and Jansson 1996). Synechocystis and other cyanobacteria, however, contain three versions of the psbA gene (psbA1, psbA2, and psbA3). The psbA2 and psbA3 genes translate into identical D1 proteins, i.e., encode for proteins with a redundant function, whereas psbA1 is not expressed (Jansson et al. 1998). Homologous recombination and replacement of the psbA2 gene in the Synechocystis genome by recombinant constructs allowed the expression of terpene synthase transgenes under the control of the light-inducible psbA2 promoter and its 5′UTR, without compromising photoautotrophy of the cells thanks to the redundancy afforded by the remaining psbA3 gene (Lindberg et al. 2010; Bentley et al. 2013).
The psbA gene displays a light-dependent up-regulation of expression, required to ensure a sufficient supply of new 32 kD D1 proteins, as this is needed for the replacement of photodamaged D1. This is especially the case under high irradiance, as the rate constant for photodamage is a linear function of light intensity (Melis 1999). Accordingly, transcription/translation of the psbA2 gene is most active under moderate and high irradiance, when approximately 95 % of psbA transcripts are produced by psbA2 and the remainder by psbA3 (Mohamed et al. 1993). Light sensitivity of expression of the psbA gene is afforded by the function of an “AT-rich light-sensitivity box” occurring in the 5′UTR of the gene. In this respect, cis-encoded antisense RNAs have been identified to target the 5′UTR of the psbA2 and psbA3 gene transcripts, thereby stabilizing transcript levels in the light by acting to prevent the function of an RNAse E/G-type endoribonuclease-mediated cleavage of the AU-rich sequence (AU-box) just upstream of the ribosome-binding site (Sakurai et al. 2012; Horie et al. 2007). Removal of the AT light-sensitivity box, or substitution with a GC-rich sequence, has been shown to increase psbA2 transcript stability and accumulation following a light-to-dark shift (Horie et al. 2007). However, the consequence of removal of the AT light-sensitivity box in terms of transgene expression and product yield under continuous illumination was not tested. In this study, the transgene encoding β-phellandrene synthase (PHLS) was integrated in the genomic psbA2 site (Fig. 1a) under the control of either the endogenous psbA2 promoter (∆psbA2-PHLS, Fig. 1b) or a modified promoter, where the AT-rich box was replaced by a GC-rich sequence [∆psbA2(noATbox)-PHLS, Fig. 1c].
Genomic DNA PCR analysis was performed to test for insert integration and DNA copy homoplasmy in these transformants. For this purpose, selected forward (us) and reverse (ds) primers were designed from the genomic DNA of Synechocystis wild type and PHLS transformants (Table 1). Results from this analysis are shown in Fig. 3. In lane 1, wild-type DNA was used as template. In lane 2, DNA from a ∆psbA2-PHLS transformant was used as template. In lanes 3–5, DNA template from three independent ∆psbA2(noATbox)-PHLS transformant lines was used. Figure 3a shows PCR products using primers A2_us and A2_ds (Fig. 1). Note the presence of a single 1,700 bp product in the wild type (lane 1) and the presence of a single ~3,100 bp product in the transformants (lanes 2–5). Absence of the 1,700 bp product from the transformants signified complete segregation and transgenic DNA copy homoplasmy in these transformant lines. Figure 3b shows PCR products using primers A2_us and PHLS_Rv (Fig. 1). Note the absence of a PCR product from the wild type (lane 1), and the presence of a single ~1,290 bp product in these transformant (lanes 2–5), confirming the presence of the insertion cassette in the correct genomic DNA region of Synechocystis.

Genomic DNA PCR analysis with selected forward (us) and reverse (ds) primers positioned on the genomic DNA of Synechocystis wild type and PHLS transformants. Lane 1 wild-type DNA was used as template. Lane 2 DNA from the ∆psbA2-PHLS transformant. Lanes 3–5 DNA from three independent ∆psbA2(noATbox)-PHLS transformant lines. Lanes 6–8 DNA from three independent ∆psbA2-trc-PHLS transformant lines. Lanes 9–11 DNA from three independent ∆psbA2-trc-T7gene10 transformant lines. a PCRs using primers A2_us and A2_ds. Note the presence of a single 1,700 bp product in the wild type (lane 1) and the presence of a single 3,100 bp product in the transformants (lanes 2–11). Absence of the 1,700 bp product from the transformants signified transgenic DNA copy homoplasmy in these lines. b PCRs using primers A2_us and PHLS_Rv. Note the absence of a PCR product in lane 1, and the presence of a 1,290 bp product from each of the transgenic lines. c PCR using primers A2_us and trc_Rv. Note the absence of a PCR product in lanes 1–5 and the presence of a single 445 bp product in lanes 6–11. d PCRs using primers A2_us and T7_Rv. Note the absence of a PCR product in lanes 1–8 and the presence of a single 512 bp product in lanes 9–11. The location of the primers used is shown in Fig. 1 (for primer sequences please see Table 1)
Application of the heterologous Ptrc promoter and a modified Ptrc promoter with the 5′UTR and RBS of gene 10 from bacteriophage T7
The hybrid promoter Ptrc contains the consensus −35 sequence (TGACA) from Ptrp and the consensus −10 sequence (TATAA) from Plac (both from E. coli), which are separated by a 17-bp spacer, and the LacI-binding operator O1 (Amann et al. 1988; Huang et al. 2010) (Fig. 1d). Strong Ptrc expression is induced by isopropyl β-d-1-thiogalactopyranoside (IPTG) in E. coli, but expression from this promoter is constitutive in Synechocystis (Guerrero et al. 2012), suggesting distinct differences at the molecular induction level between the two prokaryotic cell types. In the present work, 86 nucleotides of the psbA2 promoter and 5′UTR, upstream of the start codon for translation, were substituted by the Ptrc-lacO1 sequence. No repressor of the Ptrc-lacO1 is synthesized by Synechocystis and lacO1 just acts as a spacer between the transcription and the translation initiation sites. The Ptrc-lacO1 sequence was fused to the upstream psbA2 region as part of the cassette for homologous recombination leading to the replacement of endogenous psbA2 gene (∆psbA2-trc-PHLS, Fig. 1d). Further, the RBS of the Ptrc was substituted by the 5′UTR from gene 10 of bacteriophage T7, containing the EP upstream box “TTAACTTTA”, which is complementary to 16 s rRNA, and the RBS sequence “AAGGAGA” (Sprengart et al. 1996) (∆psbA2-trc-T7gene10-PHLS, Fig. 1e). Gene 10 encodes for the highly expressed coat protein of the T7 virus. In addition, the 10–15 codons downstream of the translation start (named downstream box) were shown to affect accumulation of foreign proteins in E. coli and tobacco chloroplasts (Sprengart et al. 1996; Kuroda and Maliga 2001a, b). The GCUAGC DB from T7 gene 10 leads to 5 nucleotides of complementarity to chloroplast and Synechocystis 16 s rRNA (anti-DS, nt 1416–1430). It was shown to enhance protein expression in tobacco chloroplasts (Kuroda and Maliga 2001a) and we assumed that the same would be true for Synechocystis. This DB box sequence (adding an Alanine and a Serine at the N-terminus of the protein) was used in the present paper to express the PHLS gene under the control of the Ptrc promoter and T7 gene-10 5′UTR (∆psbA2-trc-T7gene10-PHLS, Fig. 1e). Transgene integration in the psbA2 genomic locus and DNA copy homoplasmy were verified by genomic DNA PCRs (Fig. 3). DNA template from three independent ∆psbA2-trc-PHLS and ∆psbA2-trc-T7gene10-PHLS transformant lines was used in lanes 6–8 and 9–11, respectively. Figure 3c shows PCRs using primers A2_us and trc_Rv (Fig. 1d). Note the absence of a PCR product in lanes 1–5 and the presence of a single ~445 bp product in lanes 6–11. Figure 3d shows PCRs using primers A2_us and T7_Rv (Fig. 1e). Note the absence of a PCR product in lanes 1–8 and the presence of a single ~512 bp product in lanes 9–11 (for primer sequences please refer to Table 1). These results show cassette insertion in the targeted locus of the Synechocystis DNA. Together with the results of Fig. 3a, they further suggest transgenic DNA copy homoplasmy in these transformants and the absence of wild-type copies of the Synechocystis DNA.
Application of the endogenous cpc operon promoter in the PHLS transgene expression
The phycobilisome light-harvesting antenna of Synechocystis and other cyanobacteria is a peripheral bilin-protein supercomplex, attached to the stromal side of the thylakoid membrane facing photosystem II (Glazer and Melis 1987). The phycobilisome core is composed of allophycocyanin (APC) trimers, comprising core cylinders that rest on the surface of the thylakoids. From the core cylinders, six peripheral rods radiate away from the thylakoid membrane, each composed of hexamers of phycocyanin (PC) (Glazer and Melis 1987; Grossman et al. 1993; MacColl 1998; Adir 2005). The phycocyanin constituent α- and β-subunits and their linker polypeptides are encoded by the cpc operon (Fig. 1f, locus 724094–727466 of the Synechocystis genome, http://genome.microbedb.jp/cyanobase). Phycocyanin serves as a peripheral light-harvesting antenna in the phycobilisome of Synechocystis and other cyanobacteria. It is one of the most abundant soluble proteins in cyanobacteria, suggesting strong expression elements controlling the cpc operon transcription and translation initiation processes. Several transcription regulatory elements of the cpc operon have been identified and described (Nakajima et al. 2001; Imashimizu et al. 2003; Münch et al. 2005). In the present study, the PHLS transgene was cloned, together with the gene encoding resistance to chloramphenicol, in-between 500 bp upstream and downstream sequences of the cpc operon, with the 500 bp serving for homologous recombination and replacement of the cpc operon in Synechocystis with the PHLS transgenic construct (Δcpc-PHLS, Fig. 1g). In this design, the PHLS gene would be expressed under the control of the cpc promoter, taking advantage of the high transcriptional activity of this genomic environment. Deletion of the endogenous cpc operon genes would lead to a phycobilisome phenotype lacking phycocyanin and possessing a truncated light-harvesting antenna size. Integration of the PHLS transgene in the cpc genomic locus, deletion of the native cpc genes encoded by this operon, and achievement of homoplasmy were tested by genomic DNA PCRs. Figure 4, lanes 1–3, shows PCR products with DNA extracted from three independent Δcpc-PHLS transformant lines. Figure 4, lane 4, shows PCR products with DNA from the wild type. Figure 4a shows PCRs with primers cpc_us and cpcA_Rv designed from the endogenous cpc operon (Fig. 1f). Only the wild type generated a product at 1,289 bp. Figure 4b shows PCRs with primers cpcC1_Fw and cpc_ds also designed from the endogenous cpc operon (Fig. 1f). Here again, only the wild type generated a product at 1,270 bp. Lack of PCR products from the PHLS transformants (Fig. 4a, b, lanes 1–3) suggests the absence of wild-type DNA copies in these transformants. Figure 4c shows PCRs with primers cpc_us and PHLS_Rv designed from within the PHLS transgene (Fig. 1g). In this case, all three lines of the transformants, and only the transformants, generated a PCR product but not the wild type, showing successful integration of the Δcpc-PHLS cassette in the Synechocystis genome. These results (Fig. 4) further suggest segregation of the transgenic DNA and DNA copy homoplasmy in all three lines of the transformants.

Application of the endogenous cpc operon promoter in the PHLS transgene expression and genomic DNA PCR analysis with selected forward (us) and reverse (ds) primers positioned on the genomic DNA of Synechocystis wild type and PHLS transformants. Lanes 1–3 DNA was extracted from three independent ∆cpc-PHLS transformant lines and used as template for the PCRs. Lane 4 PCR using extracted DNA from wild type as template. a PCRs using primers cpc_us and cpcA_Rv (Fig. 1f). b PCRs using primers cpcC1_Fw and cpc_ds (Fig. 1f). c PCRs using primers cpc_us and PHLS_Rv (Fig. 1g). The location of the primers used is shown in Fig. 1 (for primer sequences please see Table 1). Note the absence of 1,289 and 1,270 bp PCR products from the transgenic lines (lanes 1–3), when primers from within the cpc operon were used (a, b, respectively). These results suggest lack of wild-type DNA copies, and transgenic DNA copy homoplasmy in the transformants. Also note the presence of 1,414 bp PCR products from the transgenic lines (lanes 1–3), when primers from within the PHLS transgenic region were used (c)
Accumulation of recombinant PHLS protein in Synechocystis, as a function of different genomic insertion sites, promoters, and translation initiation regions
Expression of the PHLS transgene was assessed by SDS-PAGE and Western blot analysis. All transformants, but not the wild type, showed clear evidence for the presence of the PHLS protein in Synechocystis-soluble protein extracts (Fig. 5). Importantly, the PHLS transgenic protein could be detected from both the Coomassie stain of SDS-PAGE and the corresponding Western blot analysis. Figure 5a shows the soluble protein extract SDS-PAGE analysis of Synechocystis wild type and PHLS transformants. A protein band migrating to 64 kD is attributed to the transgenic β-phellandrene synthase and is clearly present in all transformants (Fig. 5a, lanes 2–8), but absent from the wild-type extract (Fig. 5a, lane 1). Other notable changes in the protein composition of the samples are the absence of the phycocyanin linker polypeptides CpcC2 and CpcC1, as well as the absence of the α-phycocyanin and β-phycocyanin (CpcB-A) subunits from the Δcpc-PHLS transformants (Fig. 5a, lanes 7, 8). Western blot analysis with specific polyclonal antibodies against the PHLS recombinant protein (Fig. 5b) confirmed increase in the steady-state amounts of the PHLS protein from a relative level of 100 % in the ∆psbA2-PHLS transformant (Fig. 5b, lane 2) up to about 400 % in the ∆psbA2-trc-T7gene10-PHLS (Fig. 5b, lanes 5, 6) and Δcpc-PHLS transformants (Fig. 5b, lanes 7, 8).

Protein analysis of Synechocystis wild type and PHLS transformants. a Coomassie-stained SDS-PAGE of soluble protein extracts from Synechocystis wild type and PHLS transformants. Lane 1 wild type. Lane 2 ∆psbA2-PHLS. Lane 3 ∆psbA2(noATbox)-PHLS. Lane 4 ∆psbA2-trc-PHLS. Lanes 5–6 ∆psbA2-trc-T7gene10-PHLS. Lanes 7–8 Δcpc-PHLS. Molecular weight markers are reported on the leftmost lane. Phycobilisome subunits deleted in the Δcpc-PHLS strain are indicated, as well as the protein band of β-phellandrene synthase, visible in the Coomassie stain as a 64 kD protein (PHLS). b Western blot analysis with specific polyclonal antibodies raised against the PHLS recombinant protein. Lane loadings as in a above. The relative amount of expressed PHLS protein was normalized to that of lane 2 ∆psbA2-PHLS, and is reported in percentage units, listed underneath b
Photosynthetic pigments from Synechocystis wild type and PHLS transformants
There was a visual difference in the coloration of the Δcpc-PHLS transformant cultures, as compared to that of the wild type and the other transformants. Figure 6a shows, from left to right and top to bottom, photographs of wild-type, ∆psbA2-PHLS, ∆psbA2(noATbox)-PHLS, ∆psbA2-trc-PHLS, ∆psbA2-trc-T7gene10-PHLS, and Δcpc-PHLS transformants. Noted is the yellow-greenish coloration of the Δcpc-PHLS culture, attributed to the absence of phycocyanin, as opposed to the blue-green coloration of the other strains.

Analysis of photosynthetic pigments. a Photograph of Synechocystis wild type and PHLS transformant cultures, taken at an optical density (absorbance at 730 nm) of 2. From left to right and top to bottom, wild type, ∆psbA2-PHLS, ∆psbA2(noATbox)-PHLS, ∆psbA2-trc-PHLS, ∆psbA2-trc-T7gene10-PHLS, Δcpc-PHLS. Note the yellow-greenish coloration of the Δcpc-PHLS strain, attributed to the absence of phycocyanin, as opposed to the blue-green coloration of the other strains. b Absorbance spectra of total cell extracts, after cell disruption by French press, of wild type, ∆psbA2-trc-PHLS and ∆cpc-PHLS transformants. Spectra were normalized to chlorophyll a absorbance peak at 678 nm. c Absorbance spectra of chlorophyll a and carotenoids extracted in 90 % methanol, same strains as in b. Spectra were normalized to the chlorophyll a absorbance peak at 665.6 nm. Standard deviations within 10 %
Absorbance spectra of wild type, ∆psbA2-trc-PHLS and Δcpc-PHLS transformants were measured. Presence of the phycobilisome in the wild type and ∆psbA2-trc-PHLS cells was evident from the pronounced 625 nm absorbance band, originating from phycocyanin in these samples (Fig. 6b). Conversely, the absence of phycocyanin in the Δcpc-PHLS transformants was evident from the lack of the aforementioned 625 nm absorbance band. Absorbance spectra of chlorophyll a and carotenoids extracted in 90 % methanol (Fig. 6c) showed no marked differences, except perhaps for the slightly elevated Soret absorbance by the PHLS transformants, relative to that of the wild type, suggesting a slightly greater carotenoid-to-Chl ratio in the former. Otherwise, it appears that transformation of Synechocystis with the various forms of the PHLS cassette had no adverse effect on the pigment organization of the photosynthetic apparatus.
Properties of photoautotrophic growth of wild type and PHLS transformants
Photoautotrophic growth rates were measured in wild type and transformants grown either at 50 μmol photons m−2 s−1 or 170 μmol photons m−2 s−1 of incident irradiance. When grown at 50 μmol photons m−2 s−1, the ∆psbA2-trc-PHLS strains grew as efficiently as the wild type (Fig. 7a, circles, squares). Under the same conditions, the Δcpc and Δcpc-PHLS transformants grew with only about a third of the rate of the wild type and ∆psbA2-trc-PHLS strains (Fig. 7a, diamonds, triangles). A much slower rate of growth for the Δcpc and Δcpc-PHLS transformants, as compared to the wild type, is attributed to the absence of the phycocyanin antenna and the light-limiting conditions that the Δcpc and Δcpc-PHLS transformants are apparently encountering. When grown at 170 μmol photons m−2 s−1, rate of growth accelerated for the wild type by about 35 % (Fig. 7b, circles), whereas rate of growth for the Δcpc and Δcpc-PHLS transformants accelerated by 280 % (Fig. 7b, diamonds, triangles). These results are consistent with the notion of disproportionate light-limitation in the Δcpc and Δcpc-PHLS strains, as compared to the wild type, a limitation that is gradually alleviated as the growth irradiance increases. Of interest is also the observation that rate of growth for the ∆psbA2-trc-PHLS at 170 μmol photons m−2 s−1 (Fig. 7b, squares) was only marginally increased (by about 10 %) from that at 50 μmol photons m−2 s−1 (Fig. 7a, squares). Expression of the PHLS transgene and β-phellandrene synthesis in the cells do not appear to exert any adverse effects on cell growth, as Δcpc-PHLS transformants behaved like the simple Δcpc knock-out (Fig. 7, diamonds, triangles).

Growth curves of Synechocystis wild type and PHLS transformants, as measured from the optical density OD730 of the cultures at 730 nm. a Growth curves at 50 μmol photons m−2 s−1 incident intensity of wild type, ∆psbA2-trc-PHLS and Δcpc-PHLS transformants, compared to a Δcpc knock-out where the cpc operon has been replaced by the gene conferring antibiotic resistance for selection without the addition of the PHLS transgene. b Growth curves at 170 μmol photons m−2 s−1 incident light for the same strains as in a. Averages and standard deviations were calculated from three independent biological replicates for each genotype. Cultures were inoculated to an OD at 730 nm of about 0.2, as initial cell concentration in the growth kinetic analysis. Best fit of the points from the cell-density measurements were straight lines, reflecting a deviation from exponential growth due to increased cell density and shading, gradually limiting the effective light intensity through the culture
β-Phellandrene production in PHLS transformants
β-Phellandrene production by Synechocystis transformants was assayed spectrophotometrically from the absorbance spectra of hexane extracts, as described in the “Materials and methods”. β-Phellandrene was chemically identified as a transformant-specific product by GC–MS analysis. Conveniently, the product was collected as a non-miscible molecule floating on top of the aqueous medium of transformant cultures. The floating β-phellandrene product was diluted with hexane and siphoned off the culture. Figure 8 shows typical absorbance spectra of hexane extracts from the surface of wild type and PHLS transformants. The absorbance spectra of the hexane extracts were normalized to the dcw accumulated in the cultures that generated them. Amplitude of the absorbance spectra at 232.4 nm and application of Beer’s law were employed for product quantification. It is evident from the results of Fig. 8 that ∆psbA2-trc-T7gene10-PHLS and Δcpc-PHLS transformants generated β-phellandrene with greater efficiency than the rest. A complete listing of the yields and efficiencies of β-phellandrene production is given in Table 2.

β-Phellandrene production assays by Synechocystis transformants. β-Phellandrene was collected as a non-miscible product floating on top of the aqueous phase of transformant cultures. The product was diluted with hexane and siphoned off the culture. Three independent transformants for each genotype and at least two different biological replicates per transformant were analyzed. Absorption spectra of the hexane extracts were normalized on per g dcw. Black solid line hexane extract from the surface of wild-type cultures. Blue solid line extract from ∆psbA2-PHLS transformants. Blue dotted line ∆psbA2(noATbox)-PHLS. Red solid line ∆psbA2-trc-PHLS. Red dotted line ∆psbA2-trc-T7gene10-PHLS. Green solid line Δcpc-PHLS. Calculated β-phellandrene yields are reported in Table 2
β-Phellandrene was identified in the hexane extracts by GC–MS analysis, conducted according to Demissie et al. (2011). A β-phellandrene standard (Chemos GmbH) was used as reference in these measurements. In the GC analysis, the β-phellandrene standard showed a retention time of 9.10 min (Fig. 9a) and pronounced 77, 93, 136 signature MS lines (Fig. 9b). All hexane extracts from the surface of transformant Synechocystis cultures showed a peak with a retention time of 9.38 min (Fig. 9c). MS analysis of the GC peak with a retention time of 9.38 min showed pronounced 77, 93, 136 signature MS lines (Fig. 9d). This clearly showed the presence of β-phellandrene as a floater molecule in the transformant cultures. GC analysis of hexane extracts from the surface of a wild-type culture showed none of the above (Fig. 9e).

β-Phellandrene was identified in hexane extracts by GC-MS analysis, according to Demissie et al. (2011). A β-phellandrene standard in hexane (Chemos GmbH) was used as reference. a The β-phellandrene standard in hexane showed a GC retention time of 9.10 min, and pronounced 77, 93, 136 signature MS lines (b). c β-Phellandrene in the hexane extract from Synechocystis cultures showed a retention time of 9.38 min and pronounced 77, 93, 136 signature MS lines (d). e GC analysis of hexane extracts from the surface of a wild-type culture
Table 2 provides a summary of β-phellandrene production as a function of the transgene configuration in the constructs, and also as a function of cultivation conditions employed in this work. For growth under limiting irradiance at 50 μmol photons m−2 s−1, the ∆psbA2-PHLS transformant showed a β-phellandrene-to-biomass carbon-partitioning ratio of about 11 μg β-phellandrene per g dcw, whereas the ∆psbA2-trc-T7gene10-PHLS and Δcpc-PHLS transformants consistently showed the best β-phellandrene-to-biomass carbon-partitioning ratio in the range of 250–260 μg β-phellandrene per g dcw. This carbon partitioning to β-phellandrene is about 20-fold greater than what could be achieved with the original ∆psbA2-PHLS construct (Table 2, column 2). The ∆psbA2-trc-T7gene10-PHLS and Δcpc-PHLS transformants also showed greater absolute amounts of β-phellandrene harvested per volume of culture than what could be harvested from the other transformants (Table 2, column 3).
For growth under a greater irradiance at 170 μmol photons m−2 s−1, the ∆psbA2-PHLS transformant showed a β-phellandrene-to-biomass carbon-partitioning ratio of about 44 μg β-phellandrene per g dcw, consistent with earlier measurement from this lab (Bentley et al. 2013). This is a fourfold increase over the yield of this transformant measured at the lower intensity of 50 μmol photons m−2 s−1. The better β-phellandrene-to-biomass carbon-partitioning ratio measured at the higher growth irradiance is attributed to a greater expression of the PHLS gene, which is under the control of the light-sensitive psbA2 promoter. This result is consistent with the up-regulation of gene expression by the psbA2 promoter with increasing irradiance (Mohamed et al. 1993; Lindberg et al. 2010; Mulo et al. 2012). The results suggest that expression levels of the PHLS protein are the rate-limiting step in cells grown under 50 μmol photons m−2 s−1. In high-density mass cultures of cyanobacteria, even under direct and bright sunlight, steep gradients of light intensity would prevail through the culture (Melis et al. 1999), resulting in sub-illuminated zones where light intensity would be limiting (Melis 2009; Formighieri et al. 2012). Consequently, the choice of an alternative promoter to psbA2, which would be efficient in both low and high light, would be desirable.
At 170 μmol photons m−2 s−1, the ∆sbA2-trc-T7gene10-PHLS transformant showed a β-phellandrene-to-biomass carbon-partitioning ratio of about 249 μg β-phellandrene per g dcw. This yield is essentially the same as that achieved under the lower 50 μmol photons m−2 s−1 intensity. Use of heterologous regulatory elements allowed for a constitutive light-independent PHLS expression and β-phellandrene formation. Interestingly, yield of the Δcpc-PHLS transformant at 170 μmol photons m−2 s−1 was lowered to only about 79 μg β-phellandrene per g dcw. When compared to the yield at 50 μmol photons m−2 s−1 (259 μg β-phellandrene per g dcw, Table 2), this decline is attributed to the strong down-regulation in the expression of the cpc operon at higher light intensities, when the photosynthetic apparatus do not need to make large amounts of phycocyanin (Nakajima and Ueda 1999).
Discussion
Terpenoids constitute a wide class of compounds with several important industrial applications (Bohlmann and Keeling 2008; Peralta-Yahya et al. 2011). The possibility of genetic engineering of the cyanobacterium Synechocystis to produce terpene hydrocarbons from sunlight, CO2 and H2O is particularly attractive, as this microorganism has the ability to convert the primary products of photosynthesis into desirable fuel and chemicals for human consumption. Proof-of-concept with terpene hydrocarbons was demonstrated upon heterologous introduction in Synechocystis of genes conferring the ability to synthesize and release isoprene (Lindberg et al. 2010), the monoterpene β-phellandrene (Bentley et al. 2013), and the sesquiterpene β-caryophyllene (Reinsvold et al. 2011). Yields for the aforementioned terpenes, however, were low, in the range of 1–50 μg product per g dcw, an outcome that raised questions about the regulation of carbon partitioning in the cellular terpenoid metabolism (Melis 2013). As such, yield of terpenes was substantially lower from that reported for other small molecules heterologously produced in Synechocystis, e.g. ethanol, 2,3-butanediol, and isobutanol (Gao et al. 2012; Oliver et al. 2013; Varman et al. 2013). The low yield of terpene products could not be attributed to photosynthetic carbon allocation through the terpenoid biosynthetic pathway, which in Synechocystis was estimated to be 3–5 % of the total cellular carbon flux (Lindberg et al. 2010; Melis 2012, 2013; Wijffels et al. 2013). Earlier transgenic terpene synthase expression efforts in Synechocystis used the same strategy of expression, i.e., employed the endogenous psbA2 locus as the transgene insertion site and the same endogenous psbA2 5′UTR promoter (Lindberg et al. 2010; Reinsvold et al. 2011; Bentley et al. 2013). Although the 5′UTR of the psbA2 gene comprises a strong light-induced promoter, the level of transgene expression, as measured from the amount of transgenic protein accumulation, was not addressed.
Work reported in this paper showed that codon-use optimization and a strong promoter (e.g. psabA2) may not be sufficient for adequate transgene expression, so as to ensure that the product-generating enzymatic reaction is not rate limiting. In the present paper, a comparative quantitative study of recombinant β-phellandrene synthase expression and activity under a variety of promoters and insertion loci was undertaken. Transgenic protein accumulation was measured under the control of either the psbA2 or cpc endogenous promoters, and under the heterologous control of the Ptrc promoter from E. coli. Under these metabolic engineering conditions in Synechocystis, β-phellandrene hydrocarbons’ yield was measured as the product of cellular photoautotrophic metabolism. Results suggested that low levels of the recombinant PHLS protein were the rate- and yield-limiting step in the synthesis of β-phellandrene, when the psbA2 5′UTR promoter was used. Employment of the Ptrc promoter in the psbA2 locus, and also employment of the cpc promoter in the cpc locus, improved the level of the recombinant PHLS protein by 2.5- to 4-fold (Fig. 5, lane 2 vs 4–8) and also improved the rate and yield of β-phellandrene production by 10- to 20-fold (Table 2). These results strengthen the notion that PHLS expression and recombinant protein accumulation comprise the limiting step in β-phellandrene production achieved so far.
The work also compared the performance of the Ptrc promoter with different translation initiation sites. Combining the Ptrc promoter with the 5′UTR, RBS, and downstream box from the highly expressed gene 10 of bacteriophage T7 improved recombinant PHLS protein accumulation by 60 % (Fig. 5, lanes 5, 6) over that with the Ptrc promoter alone (Fig. 5, lane 4). This difference in the level of the recombinant PHLS protein accumulation was reflected in the rate and yield of β-phellandrene production, which was improved by 200–250 % (Table 2, ∆psbA2-trc-T7gene10-PHLS vs ∆psbA2-trc-PHLS). These results further corroborate the notion that PHLS expression and recombinant protein accumulation comprise the limiting step in β-phellandrene production.
The choice and design of a translation initiation region facilitating ribosome binding and translation initiation are equally important to the use of an efficient promoter for transcription in determining overall transgene expression. A 48 h yield of about 0.25 mg of β-PHL per g of dcw measured in this work is the highest value for a transgenic terpene achieved so far from photosynthesis-associated metabolism in Synechocystis. The construct using Ptrc and 5′UTR of bacteriophage T7 gene 10 to drive expression of PHLS, albeit in the psbA2 gene locus, proved to be superior in both recombinant protein accumulation and β-phellandrene production, when compared with the basic ∆psbA2-PHLS construct. Although widely used to insert transgenes in the Synechocystis genome, deletion of the psbA2 gene is not without consequence for cell fitness. Observed growth rate limitations at higher light intensities for the ∆psbA2 strain (Fig. 7b vs a) may be attributed to the lack of the PsbA2 protein. Replacement of the psbA2 gene with transgenes was not thought to compromise photoautotrophy (Lindberg et al. 2010; Reinsvold et al. 2011; Bentley et al. 2013). However, PsbA2 contributes in the up-regulated synthesis of the photosystem-II 32 kD D1 reaction center protein, needed for the more frequent turnover of photodamaged D1 proteins in high light (Melis 1999). psbA2 gene deletion could impair to some extent the photosystem-II repair from photodamage, especially so under high light intensities, where the psbA2 promoter is most active (Mohamed et al. 1993; Mulo et al. 2012), a phenotype that may not appear under low light conditions. Homologous recombination and insertion of a transgene in the psbA2 locus could therefore be a suboptimal choice.
A novelty in this work is the expression of the PHLS transgene by homologous recombination and replacement of the cpc operon, under the control of the cpc promoter. We reasoned that the cpc promoter and associated 5′UTR confer strong expression and are in a locus of high transcriptional activity, as expression of the encoded phycocyanin results in the accumulation of the most abundant protein in Synechocystis. Replacement of the cpc operon with the PHLS transgene resulted in the deletion of the PC peripheral rods from the phycobilisome. PC-deficient mutants have unchanged allophycocyanin antenna core cylinders (Nakajima and Ueda 1997) and uninterrupted non-photochemical energy quenching (Wilson et al. 2006; Tian et al. 2012), whose activity is associated with APC and the orange carotenoid protein contained therein (Bailey and Grossman 2008; Rakhimberdieva et al. 2010; Kirilovsky and Kerfeld 2012).
In the present work, and as a result of efforts to increase the level of recombinant β-phellandrene protein accumulation, we arrived at transformant strains in which the PHLS protein was visible and easily distinguishable in the Coomassie-stained protein profile of SDS-PAGE analysis (Fig. 5a, PHLS). Specifically, the intensity of the PHLS protein band equaled that of the phycobilisome linker polypeptides (CpcC1 and CpcC2, Fig. 5a). Such level of PHLS expression was required to sustain a β-phellandrene production yield of about 0.25 mg of β-PHL per g dcw in both the ∆cpc-PHLS and ∆psbA2-trc-T7gene10-PHLS strains. Nevertheless, a yield of 0.25 mg of β-PHL per g dcw, or 0.025 % β-PHL:biomass (w:w), accounts for only a small fraction of the carbon flux through the cell’s own terpenoid biosynthetic pathway, which handles 4–5 % of all photosynthetic carbon (Lindberg et al. 2010). It is suggested that even greater amounts of recombinant β-phellandrene synthase protein accumulation and/or other modifications in the availability of metabolite precursors are needed to further improve yields.
A requirement for high levels of β-phellandrene synthase accumulation may be explained upon consideration of the slow kcat catalytic rate constant of terpene synthases, measured to be in the range of kcat = 4–5 s−1 (Sasaki et al. 2005; Zurbriggen et al. 2012). In this respect, a kcat = 4–5 s−1 for terpene synthases is comparable to that of RuBisCO, which was reported to be in the range of kcat = 3 ± 0.5 s−1 (McNevin et al. 2006). Nature’s solution to the slow RuBisCO kcat was to enormously increase the relative amount of the RuBisCO protein in the chloroplast/cell, making RuBisCO one the most abundant proteins on Earth.
In summary, many of the genetic elements that control chromosomal transcription and translation in cyanobacteria are not fully characterized and appear to behave differently than extrachromosomal expression in model heterotrophic microorganisms, e.g. E. coli. The work emphasizes the requirement of a chromosomal expression system in photosynthetic microorganisms (needed for scale-up production), as opposed to extrachromosomal plasmid-based production in E. coli. The latter has been serving as the accepted model in synthetic biology, but it is inadequate and unacceptable for scale-up production, especially so with photosynthetic microorganisms. Accordingly, understanding the genetic elements that control transcription and translation in cyanobacteria and developing a chromosomal-based transgenic expression and production system were the aims of this work. Transformation of cyanobacteria (e.g. Synechocystis) with terpene synthases, and the associated metabolic engineering of photosynthesis, affords the prospect of generating compounds, such as the hemiterpene isoprene, monoterpenes (e.g. β-phellandrene), and sesquiterpenes (e.g. β-caryophyllene), which have obvious applications as fuel and feedstock for the synthetic chemistry, pharmaceutical, and cosmetics industries. Evident from this work is the requirement of transgene expression systems via which to further increase the recombinant terpene synthase protein accumulation in the chloroplast/cell, as a prerequisite to greater yields.
Abbreviations
- APC:
-
Allophycocyanin
- Chl:
-
Chlorophyll
- cmR:
-
Chloramphenicol resistance
- cpc:
-
Operon encoding the phycocyanin peripheral rods
- dcw:
-
Dry cell weight
- DB:
-
Downstream box
- GC–MS:
-
Gas chromatography–mass spectrometry
- OD:
-
Optical density
- PC:
-
Phycocyanin
- PHL:
-
β-Phellandrene
- PHLS:
-
β-Phellandrene synthase
- psbA2:
-
Gene encoding the D1/32 kD reaction center protein of PSII
- Ptrc:
-
Hybrid promoter made with consensus sequences from E. coli Ptrp and Plac
- RBS:
-
Ribosome-binding site
- T7g10:
-
Gene 10 from bacteriophage T7
References
Adir N (2005) Elucidation of the molecular structures of components of the phycobilisome: reconstructing a giant. Photosynth Res 85:15–32
Amann E, Ochs B, Abel KJ (1988) Tightly regulated tac promoter vectors useful for the expression of unfused and fused protein in Escherichia coli. Gene 69:301–315
Angermayr SA, Paszota M, Hellingwerf KJ (2012) Engineering a cyanobacterial cell factory for production of lactic acid. Appl Environ Microb 78:7098–7106
Atsumi S, Higashide W, Liao JC (2009) Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol 27:1177–1180
Bailey S, Grossman A (2008) Photoprotection in cyanobacteria: regulation of light harvesting. Photochem Photobiol 84:1410–1420
Bentley FK, Melis A (2012) Diffusion-based process for carbon dioxide uptake and isoprene emission in gaseous/aqueous two-phase photobioreactors by photosynthetic microorganisms. Biotechnol Bioeng 109:100–109
Bentley FK, García-Cerdán JG, Chen HC, Melis A (2013) Paradigm of monoterpene (β-phellandrene) hydrocarbons production via photosynthesis in cyanobacteria. BioEnergy Res 6:917–929
Bohlmann J, Keeling CI (2008) Terpenoid biomaterials. Plant J 54:656–669
Booker H, Evans LK, Gillam AE (1940) The effect of molecular environment on the absorption spectra of organic compounds in solution. Part I. Conjugated dienes. J Chem Soc 1453–1463. doi:10.1039/JR9400001453
Demissie ZA, Sarker LS, Mahmoud SS (2011) Cloning and functional characterization of β-phellandrene synthase from Lavandula angustifolia. Planta 233:685–696
Deng MD, Coleman JR (1999) Ethanol synthesis by genetic engineering in cyanobacteria. Appl Environ Microbiol 65:523–528
Dexter J, Fu P (2009) Metabolic engineering of cyanobacteria for ethanol production. Energy Environ Sci 2:857
Ducat DC, Avelar-Rivas JA, Way JC, Silver PA (2012) Rerouting carbon flux to enhance photosynthetic productivity. Appl Environ Microbiol 78:2660–2668
Eaton-Rye JJ (2011) Construction of gene interruptions and gene deletions in the cyanobacterium Synechocystis sp. strain PCC 6803. Methods Mol Biol 684:295–312
Formighieri C, Franck F, Bassi R (2012) Regulation of the pigment optical density of an algal cell: filling the gap between photosynthetic productivity in the laboratory and in mass culture. J Biotechnol 162:115–123
Gao Z, Zhao H, Li Z, Tan X, Lu X (2012) Photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energy Environ Sci 5:9857
Gershenzon J, Dudareva N (2007) The function of terpene natural products in the natural world. Nat Chem Biol 3:408–414
Glazer AN, Melis A (1987) Photochemical reaction centers: structure, organization and function. Annu Rev Plant Physiol 38:11–45
Gross KP, Schnepp O (1978) Absorption and circular dichroism spectra of the cis- and trans- butadiene chromophores α- and β-phellandrene. J Chem Phys 68:2647–2657
Grossman AR, Schaefer MR, Chiang GG, Collier JL (1993) The phycobilisome, a light-harvesting complex responsive to environmental conditions. Microbiol Rev 57:725–749
Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR (2012) Ethylene synthesis and regulated expression of recombinant protein in Synechocystis sp. PCC 6803. PLoS One 7:e50470
Hellingwerf KJ, Teixeira de Mattos MJ (2009) Alternative routes to biofuels: light-driven biofuel formation from CO2 and water based on the “photanol” approach. J Biotechnol 142:87–90
Horie Y, Ito Y, Ono M, Moriwaki N, Kato H, Hamakubo Y, Amano T, Wachi M, Shirai M, Asayama M (2007) Dark-induced mRNA instability involves RNase E/G-type endoribonuclease cleavage at the AU-box and SD sequences in cyanobacteria. Mol Genet Genomics 278:331–346
Huang HH, Camsund D, Lindblad P, Heidorn T (2010) Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res 38:2577–2593
Imashimizu M, Fujiwara S, Tanigawa R, Tanaka K, Hirokawa T, Nakajima Y, Higo J (2003) Thymine at -5 is crucial for cpc promoter activity of Synechocystis sp. strain PCC 6714. J Bacteriol 185:6477–6480
Jansson C, Salih G, Eriksson J, Wiklund R, Ghebramedhin H (1998) Use of Synechocystis 6803 to study expression of a psbA gene family. Methods Enzymol 297:166–182
Kirilovsky D, Kerfeld CA (2012) The orange carotenoid protein in photoprotection of photosystem II in cyanobacteria. Biochim Biophys Acta 1817:158–166
Kuroda H, Maliga P (2001a) Complementarity of the 16S rRNA penultimate stem with sequences downstream of the AUG destabilizes the plastid mRNAs. Nucleic Acids Res 29:970–975
Kuroda H, Maliga P (2001b) Sequences downstream of the translation initiation codon are important determinants of translation efficiency in chloroplasts. Plant Physiol 125:430–436
Lan EI, Liao JC (2012) ATP drives direct photosynthetic production of 1-butanol in cyanobacteria. Proc Natl Acad Sci USA 109:6018–6023
Li H, Liao JC (2013) Engineering a cyanobacterium as the catalyst for the photosynthetic conversion of CO2 to 1,2-propanediol. Microb Cell Fact 12:4
Lichtenthaler HK (2000) Sterols and isoprenoids. Biochem Soc Trans 28:785–789
Lindberg P, Park S, Melis A (2010) Engineering a platform for photosynthetic isoprene production in cyanobacteria, using Synechocystis as the model organism. Metab Eng 12:70–79
Liu X, Sheng J, Curtiss R (2011) Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci USA 108:6899–6904
MacBeth AK, Smith GE, West TF (1938) β-Phellandrene. J Chem Soc 119–123. doi:10.1039/JR9380000119
MacColl R (1998) Cyanobacterial phycobilisomes. J Struct Biol 124:311–334
McNevin D, von Caemmerer S, Graham Farquhar G (2006) Determining RuBisCO activation kinetics and other rate and equilibrium constants by simultaneous multiple non-linear regression of a kinetic model. J Exp Bot 57:3883–3900
Meeks JK, Castenholz RW (1971) Growth and photosynthesis in an extreme thermophile, Synechococcus lividus (Cyanophyta). Arch Microbiol 78:25–41
Melis A (1999) Photosystem-II damage and repair cycle in chloroplasts: what modulates the rate of photodamage in vivo?. Trends Plant Sci 4:130–135
Melis A (2009) Solar energy conversion efficiencies in photosynthesis: minimizing the chlorophyll antennae to maximize efficiency. Plant Sci 177:272–280
Melis A (2012) Photosynthesis-to-fuels: from sunlight to hydrogen, isoprene, and botryococcene production. Energy Environ Sci 5:5531–5539
Melis A (2013) Carbon partitioning in photosynthesis. Curr Opin Cheml Biol 17:453–456
Melis A, Neidhardt J, Benemann JR (1999) Dunaliella salina (Chlorophyta) with small chlorophyll antenna sizes exhibit higher photosynthetic productivities and photon use efficiencies than normally pigmented cells. J Appl Phycol 10:515–525
Mohamed A, Eriksson J, Osiewacz HD, Jansson C (1993) Differential expression of the psbA genes in the cyanobacterium Synechocystis 6803. Mol Gen Genet 238:161–168
Mulo P, Sakurai I, Aro EM (2012) Strategies for psbA gene expression in cyanobacteria, green algae and higher plants: from transcription to PSII repair. Biochim Biophys Acta 1817:247–257
Münch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, Jahn D (2005) Virtual footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics 21:4187–4189
Nakajima Y, Ueda R (1997) Improvement of photosynthesis in dense microalgal suspension by reduction of light harvesting pigments. J Appl Phycol 9:503–510
Nakajima Y, Ueda R (1999) Improvement of microalgal photosynthetic productivity by reducing the content of light harvesting pigment. J Appl Phycol 11:195–201
Nakajima Y, Fujiwara S, Sawai H, Imashimizu M, Tsuzuki M (2001) A phycocyanin-deficient mutant of Synechocystis PCC 6714 with a single-base substitution upstream of the cpc operon. Plant Cell Physiol 42:992–998
Nixon P, Jansson C (1996) Cyanobacterial transformation and gene regulation. In: Andersson B, Salter AH, Barber J (eds) Molecular genetics of photosynthesis. Oxford University Press, Oxford, UK, pp 197–224
Oliver JWK, Machado IMP, Yoneda H, Atsumi S (2013) Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. Proc Natl Acad Sci USA 110:1249–1254
Peralta-Yahya PP, Ouellet M, Chan R, Mukhopadhyay A, Keasling JD, Lee TS (2011) Identification and microbial production of a terpene-based advanced biofuel. Nat Commun 2:483
Rakhimberdieva MG, Elanskaya IV, Vermaas WF, Karapetyan NV (2010) Carotenoid-triggered energy dissipation in phycobilisomes of Synechocystis sp. PCC 6803 diverts excitation away from reaction centers of both photosystems. Biochim Biophys Acta 1797:241–249
Reinsvold RE, Jinkerson RE, Radakovits R, Posewitz MC, Basu C (2011) The production of the sesquiterpene β-caryophyllene in a transgenic strain of the cyanobacterium Synechocystis. J Plant Physiol 168:848–852
Sakurai I, Stazic D, Eisenhut M, Vuorio E, Steglich C, Hess WR, Aro EM (2012) Positive regulation of psbA gene expression by cis-encoded antisense RNAs in Synechocystis sp. PCC 6803. Plant Physiol 160:1000–1010
Sasaki K, Ohara K, Yazaki K (2005) Gene expression and characterization of isoprene synthase from Populus alba. FEBS Lett 579:2514–2518
Sprengart ML, Fuchs E, Porter AG (1996) The downstream box: an efficient and independent translation initiation signal in Escherichia coli. EMBO J 15:665–674
Takahama K, Matsuoka M, Nagahama K, Ogawa T (2003) Construction and analysis of a recombinant cyanobacterium expressing a chromosomally inserted gene for an ethylene-forming enzyme at the psbAI locus. J Biosci Bioeng 95:302–305
Tan X, Yao L, Gao Q, Wang W, Qi F, Lu X (2011) Photosynthesis driven conversion of carbon dioxide to fatty alcohols and hydrocarbons in cyanobacteria. Metab Eng 13:169–176
Tian L, van Stokkum IHM, Koehorst RBM, van Amerongen H (2012) Light harvesting and blue-green light induced non-photochemical quenching in two different c-phycocyanin mutants of Synechocystis PCC 6803. J Phys Chem 117:11000–11006
Van Wagoner RM, Drummond AK, Wright JLC (2007) Biogenetic diversity of cyanobacterial metabolites. Adv Appl Microbiol 61:89–217
Varman AM, Xiao Y, Pakrasi HB, Tang YJ (2013) Metabolic engineering of Synechocystis sp. strain PCC 6803 for isobutanol production. Appl Environ Microbiol 79:3908–3914
Wang B, Pugh S, Nielsen DR, Zhang W, Meldrum DR (2013) Engineering cyanobacteria for photosynthetic production of 3-hydroxybutyrate directly from CO2. Metab Eng 16:68–77
Wijffels RH, Kruse O, Hellingwerf KJ (2013) Potential of industrial biotechnology with cyanobacteria and eukaryotic microalgae. Curr Opin Biotechnol 24:405–413
Williams JGK (1988) Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. Methods Enzymol 167:766–778
Wilson A, Ajlani G, Verbavatz JM, Vass I, Kerfeld CA, Kirilovsky D (2006) A soluble carotenoid protein involved in phycobilisome-related energy dissipation in cyanobacteria. Plant Cell 18:992–1007
Zhou J, Zhang H, Zhang Y, Li Y, Ma Y (2012) Designing and creating a modularized synthetic pathway in cyanobacterium Synechocystis enables production of acetone from carbon dioxide. Metab Eng 14:394–400
Zurbriggen A, Kirst H, Melis A (2012) Isoprene production via the mevalonic acid pathway in Escherichia coli (Bacteria). BioEnergy Res 5:814–828
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Formighieri, C., Melis, A. Regulation of β-phellandrene synthase gene expression, recombinant protein accumulation, and monoterpene hydrocarbons production in Synechocystis transformants. Planta 240, 309–324 (2014). https://doi.org/10.1007/s00425-014-2080-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-014-2080-8