
Abstract
Lipopolysaccharides (LPS), as lipoglycan microbe-associated molecular pattern molecules, trigger activation of signal transduction pathways involved in defence that generate an enhanced defensive capacity in plants. The transcriptional regulation of the genes for tryptophan synthase B, TSB1, and the cytochrome P450 monooxygenases CYP79B2 and CYP71B15, involved in the camalexin biosynthetic pathway, were investigated in response to LPS treatment. GUS-reporter assays for CYP71B15 and CYP79B2 gene promoter activation were performed on transgenic plants and showed positive histochemical staining in response to LPS treatment, indicating activation of the promoters. Quantitative PCR revealed that transcripts of TSB1, CYP79B2 and CYP71B15 exhibited differential, transient up-regulation. TSB1 transcript levels were up-regulated between 6 and 9 h after LPS-induction, while CYP71B15 and CYP79B2 both exhibited maxima at 12 h. To obtain information on the gene-to-metabolite network, the effect of the transcriptome changes on the metabolome was correlated to camalexin production. Increases in camalexin concentration were quantified by ultra pressure liquid chromatography-mass spectrometry and both absorbance spectra and elemental composition confirmed its identity. The concentrations increased from 0.03 to 3.7 μg g−1 fresh weight over a 24-h time period, thus indicating that the up-regulation of the biosynthetic pathway in response to LPS was accompanied by a time-dependent increase in camalexin concentration. Metabolomic analysis through principal component analysis-derived scores plots revealed clusters of sample replicates for 0, 6, 12, 18 and 24 h while loadings plots for LPS data identified camalexin as a biomarker that clearly demonstrated the variability between samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
On exposure to biotic stresses, plants trigger a cascade of metabolic reactions, some of which lead to the biosynthesis of secondary metabolites with roles in self-defence (Zhao and Last 1996; Hammerschmidt 1999; Bednarek et al. 2009). Phytoalexins are secondary metabolites synthesised de novo in response to microbial attack by activation of certain biosynthetic pathways. This is due to the activation of particular sets of genes encoding enzymes involved in the biosynthesis of these antimicrobial chemicals (Thomma et al. 1999). Cruciferae phytoalexins are all indole based with a carbon, nitrogen and sulphur-containing constituent on the 3′ position of the indole ring (Hammerschmidt 1999; Pedras et al. 2007). This similarity of all Cruciferae phytoalexins suggests that a common indolic alkaloid precursor is shared (Rauhut and Glawischnig 2009).
Camalexin (3′-thiazol-2′-yl-indole) is the primary phytoalexin of Arabidopsis thaliana and an integral part of the plant defences in A. thaliana (Glawischnig 2007). De novo synthesis of camalexin in response to pathogen attack is part of an elaborative network of defence mechanisms, which involves signalling pathways responsive to salicylic acid (SA), jasmonate (JA), ethylene (ET), and reactive oxygen species (ROS) (Glawischnig 2007). Camalexin accumulates during both compatible and incompatible interactions and the important role thereof in certain host: pathogen interactions have been described (Tsuji et al. 1992; Mert-Turk et al. 2003; Kliebenstein et al. 2005; Glazebrook 2005). The compound was found to exhibit both antibacterial as well as antifungal activity and was not present in the hosts until after infection or elicitation (Rogers et al. 1996).
Camalexin acts as an anti-bacterial agent by disrupting the cell membranes of pathogens, leading to a decrease in cell viability (Rogers et al. 1996). It is synthesised in close proximity to the site of pathogen infection, and the local concentrations strongly depend on the spatial distribution of the growing pathogens (Kliebenstein et al. 2005; Glawischnig 2007). These localized concentration levels in infected tissues can exceed the amounts required to inhibit bacterial growth in vitro (Rogers et al. 1996; Glawischnig 2007). However, it can be subject to degradation, biotransformation (Pedras and Khan 2000) and export by phytopathogens (Stefanato et al. 2009), as counter measures to reduce the effective concentration thereof.
There is genetic variation in camalexin biosynthesis among accessions of A. thaliana (Schuhegger et al. 2007), and many genes and enzymes involved in the process have been identified (Glazebrook and Ausubel 1994; Radwanski and Last 1995; Glawischnig et al. 2004, 2007). The biosynthesis of camalexin is associated with induction of the tryptophan biosynthetic genes such as ASA1 (anthranilate synthase alpha subunit) and TSA and TSB (alpha and beta subunits of tryptophan synthase) (Zhao and Last 1996; Schuhegger et al. 2007). Glawischnig et al. (2004) demonstrated that camalexin is derived from tryptophan and that the cytochrome P450 enzymes CYP79B2 and CYP79B3 are involved in converting tryptophan to indole-3-acetaldoxime (IAOx). IAOx is converted by CYP71A13 to indole-3-acetonitrile (IAN), and the last step in camalexin biosynthesis is catalysed by another cytochrome P450 enzyme, multifunctional CYP71B15, that acts upon either dihydrocamalexic acid (DHCA) or cysteine-indole-3-acetonitrile (cysIAN) (Böttcher et al. 2009).
LPS is a structurally conserved lipoglycan in Gram-negative bacteria which is exposed to the external environment and is thus involved with external interactions of the bacteria. LPS is one of the pathogen-associated molecular pattern molecules (PAMPs) capable of eliciting activation of elements (PAMP-triggered immunity or PTI) in the plant innate immune system (Newman et al. 2007; Sanabria et al. 2010). LPS can act as an elicitor to induce and potentiate basal defence responses, suppress the hypersensitive response (HR) in dicot plants and restrict pathogen growth in treated plants (reviewed in Newman et al. 2007). Furthermore, LPS from endophytic or pathogenic bacteria as well as rhizobacteria have been described as inducers of systemic acquired resistance (SAR) and induced systemic resistance (ISR), respectively (Coventry and Dubery 2001; Mishina and Zeier 2007; van Wees et al. 2008). In SAR and ISR, plant defences are preconditioned by prior infection or exposure to pathogen-derived molecules that result in resistance against subsequent challenges or infections.
Purified LPS has been used as a PAMP to investigate the induction of plant innate immune responses. This LPS was found to trigger a rapid influx of Ca2+ into the cytoplasm of tobacco cells, as well as the production of ROS and NO during an oxidative burst reaction and K+/H+ exchange and alkalization of the extracellular culture medium (Gerber et al. 2004; Zeidler et al. 2004). LPS also has specific effects on reversible protein phosphorylation events underlying the perception systems involved in its interaction of plant cells (Gerber and Dubery 2004; Gerber et al. 2004, 2006, 2008), e.g. the phosphorylation and activation of an extracellular signal-related MAP kinase (Piater et al. 2004).
Previously, gene expression studies in LPS-treated Nicotiana tabacum cells (Sanabria and Dubery 2006), A. thaliana plants and cultured cells (Zeidler et al. 2004; Livaja et al. 2008; Madala et al. 2011, 2012), revealed the induction of an array of defence- and biotic stress-associated genes. The specific LPS-induced alterations in plant defence- and resistance-related responses indicate the activation of an important signalling and response system in plant: pathogen interactions. Here we report on the LPS-responsive biosynthesis of camalexin as part of such a response system that contributes to our understanding of the mechanistic basis of LPS-triggered PTI in A. thaliana.
Materials and methods
Plant material and growth conditions
Arabidopsis thaliana (Col-0) (Lehle Seeds, Round Rock, TX, USA) was used as the wild type in all experiments. Seeds of CYP79B2:GUS and CYP71B15:GUS lines, all in Col-0 background, were obtained from Prof. E. Glawischnig, Munich Technical University, Germany. Plants were grown in germination mix soil (Culterra, Muldersdrift, South Africa) in a plant room (23°C at 50% humidity and 60 μmol m−2 s−1 fluorescent illumination) on a 8 h light/16 h dark cycle. Six-week-old plants were used for experiments.
Plant elicitation
LPS from Burkholderia cepacia (Coventry and Dubery 2001) was dissolved at 1 mg mL−1 in a 1 mM CaCl2 and 2.5 mM MgCl2 solution. For time studies, the stock solution of LPS was further diluted with dH2O to the desired concentration (100 μg mL−1) and plants were treated by spraying the LPS solution on the leaves. At the desired time points (0, 6, 12, 18 and 24 h), the leaves were removed from the plants for extraction procedures. As a positive control plants were sprayed with 5 mM silver nitrate, prepared in 0.1% “insure” buffer wetter and spreader (Efekto, Johannesburg, South Africa).
Cis-element analysis
The promoter sequences of ASA (At1g25220), TSB (At5g54810), CYP79B2 (At4g39950), CYP71B15 (At3g26830), CYP79B3 (At2g22330), CYP71A13 (At2g30770) and CYP83B1 (At4g31500), were retrieved from TAIR (http://www.arabidopsis.org). The locus numbers were then used in TAIR to obtain the promoter sequence of 1,000 bp (1 kb) up-stream from the 5′-UTR including the sequence up-stream from the start codon. The length of promoter was chosen due to most A. thaliana promoters having cis-elements within the first 1,000 bps (Maleck et al. 2000). Cis-element analyses were performed using PlantPAN (http://plantpan.mbc.nctu.edu.tw/gene_group/index.php) (Chang et al. 2008).
β-Glucuronidase reporter gene assays
Leaves of the CYP71B15 and CYP79B2 GUS reporter gene plants were pressure infiltrated by means of a blunt-ended syringe with 100 μg mL−1 LPS or 5 mM AgNO3. Leaves were left overnight (24 h), and transferred to 1.5 mL microfuge tubes. 1 mL X-Gluc staining solution (5 mg 5-bromo-4-chloro-3-indolyl glucuronide, 10 μL dimethyl formamide (Sigma) 7.5 mL 50 mM NaH2PO4, pH 7.0) was added to the tubes to completely submerge the leaves (Jefferson 1987). The samples were incubated at 37°C overnight and the staining solution removed. In order to effect the removal of chlorophyll, 1.5 mL 75% ethanol was added to the leaves at 70°C until the leaves turned clear. Leaves were photographed for visualization of stained regions.
Gene expression analysis
RNA was isolated from A. thaliana leaf tissue for the time periods of 0, 6, 12, 18 and 24 h after LPS elicitation by the Trizol method of isolation (Chomczynski and Sacchi 1987). Aliquots were separated on 1% TAE buffered agarose gels to visualise the integrity of the RNA.
For semi-quantitative RT-PCR, mRNA was subjected to reverse transcription (Improm II kit, Promega). Expression analyses of the TSB1, At5g54810 (forward primer: 5′-GGAGTCCACTCTGGAACAGC-3′, Tm 57.6°C; reverse primer: 5′-CAAGCAACCAGAACATCAGG-3′, Tm 54°C), CYP79B2, At4g39950 (forward primer: 5′-ACGATCATTTAACCGCTTGG-3′, Tm 53.2°C; reverse primer: 5′-CAAACATTGCTTCCATGTGC-3′, Tm 53.1°C), CYP71B15, At3g26830 (forward primer: 5′-GGCTGAAGCGGTCATAAGAG-3′, Tm 55.6°C; reverse primer: 5′-TCCAGGCTTAAGATGCTCGT-3′, Tm 56°C) and 18S rRNA (forward primer: 5′-CGGCTACCACATCCAAGGAA-3′, Tm 57.1°C, reverse primer: 5′-GCTGGAATTACCGCGGCT-3′, Tm 58.5°C) genes were obtained by PCR using the GoTaq Flexi DNA Polymerase kit (Promega). Primers were designed using “Primer3″ (http://frodo.wi.mit.edu), and were synthesised by Integrated DNA Technologies (Leuven, Belgium). The PCR program was set up with an initial denaturation step of 95°C for 2 min, followed by 35 cycles of denaturation (95°C, 1 min), annealing (53–59°C adjusted for primer Tm, 1 min) and extension (72°C, 1 min). A final extension step of 72°C for 5 min was performed followed by a hold at 4°C before use. Aliquots of the 6, 9, 12 and 18 h products were analysed on 1.5% TBE buffered agarose gels to visualise the obtained cDNA amplicons. The amplicons were sequenced for confirmation.
For quantitative real-time PCR, qRT-PCR was performed in a Rotor-Gene (Corbett Research, Sydney, Australia) instrument with the primers as previously mentioned and the Quantace Sensimix kit (BioLine, London, UK) using the SYBR green dye. Experimental design and RT-qPCR of the reference and target genes were performed according to the MIQE (minimum information for publication of quantitative real-time experiments) guidelines (Bustin et al. 2009). The RT-qPCR program was set up with an initial enzyme activation step of 95°C for 10 min, followed by 40 cycles of denaturation (95°C, 15 s), annealing (53–59°C adjusted for each primer set Tm, 30 s) and extension (72°C, 6 s). A melt analysis was performed by increasing the temperature from 72°C to 99°C. Relative quantification of the amplicons was performed by comparison to both a reference gene (18S rRNA) and a calibrator ratio. A normalized calibrator ratio was obtained by dividing the normalized fold induction for each time point by the 0-h normalized fold induction (i.e. 0 h calibrator ratio gives a value of 1) for each target gene. Statistical analysis of the data (Yuan et al. 2006) was by means of analysis of variance (ANOVA).
Camalexin extraction, quantification and analysis
Following induction, leaves were removed, weighed (approximately 1–5 g), covered with 1:2 (m/v) 80% methanol and homogenized using an UltraTurrax homogenizer. The homogenates were centrifuged for 5 min at 17,136g, and the supernatants transferred to clean microcentrifuge tubes. The pellets were re-suspended in methanol and the extraction and centrifugation steps repeated. The supernatants were combined, and the methanol evaporated at 45°C in a Speed-Vac centrifuge under reduced pressure (Jouan, Nantes, France). The residue was re-suspended in UPLC grade methanol (Sigma) to 400 μL, filtered through 0.22 μm filters (Millipore) and analysed using UPLC–MS.
For UPLC–MS identification and quantification of camalexin, an authentic sample of camalexin was obtained from Prof. P. Simoneau (Universite D’Angers, France). A standard stock solution (100 μg mL−1) of camalexin was prepared in UPLC grade methanol (Sigma) and dilutions were made to obtain a standard series ranging between 0.1 and 15 μg mL−1.
Separation of crude extracts was carried out on an Acquity UPLC column (BEH C18 2.1 × 50 mm, 1.7 μm; Waters) (Beets and Dubery 2012). The volume injected was 10 μL and gradient elution was performed at a flow rate of 0.4 mL min−1 with the following solvent system: (A) 0.1% formic acid–water, (B) acetonitrile; 10% B for 0.1 min, 10–98% B in 6.9 min and holding at 98% B for 1 min, followed by a decrease to 10% B in 2 min. Under these conditions, the camalexin peak eluted at a retention time (R t) of 3.47 min.
The UPLC Synapt High Definition Ion Mobility MS system (Synapt-TOF–MS, Waters) allows for the determination of the accurate mass, fragmentation pattern, elemental composition (i-fit) as well as shape and isomer analysis of analytes. Experimental conditions for mass spectrometric detection were as follows: ESI (electrospray ionization) conditions: positive mode, capillary voltage 1 kV, sampling cone 20 V, MCP (micro channel plate detector) detector voltage 1,600 V, source temperature 120°C, desolvation temperature 350°C, cone gas flow 50 L h−1, desolvation gas flow 450 L h−1. Detection was performed in positive ion V mode in the m/z range of 100–1,000, with a scan time of 0.1 s in centroid mode. For the lockmass, a solution of leucine-enkephalin (556.277 Da, mass window 0.5 Da) was infused through the lockmass probe.
Mass accuracy of all the m/z values in all the acquired spectra was automatically corrected during acquisition based on calibration curves, lockmass and dynamic range extended. The mass accuracy window was set at 0.5 Da. The molecular formula assignments were obtained with the MassLynx software. For the molecular formula assignments, the search was restricted to C, H, O, N and S, and the best fit was obtained on both mass accuracy and isotope intensity pattern (i-fit). For the m/z of 201.0484, only one possible molecular formula which could fit with naturally occurring metabolites was obtained.
Metabolomic and multivariate data analysis
The UPLC-QTOF-MS data of LPS-induced A. thaliana samples were analysed to identify discriminant variables. MS acquisition software included MassLynx, MarkerLynx and MetaboLynx (Waters). For data processing, MarkerLynx parameters were R t 2–10 min, mass range 100–500 Da, mass tolerance 0.02 Da, R t window 0.2 min, mass window of 0.050 Da, and isotopic peaks were excluded from the analysis. A list of the intensities of the peaks detected was generated, using R t and m/z data pairs as the identifiers of each peak. The resulting two-dimensional matrix of measured mass values and their intensities for each sample were further exported to SIMCA-P software 12.0 (Umetrics). Multivariate statistical analysis were performed which included principal component analysis (PCA) Scores and Loadings plots.
Results
In silico promoter analysis of genes involved in camalexin biosynthesis
In silico analyses of the promoter regions of genes which enzyme products feed into the camalexin pathway (ASA and TSB1), act within the pathway (CYP79B2, CYP79B3, CYP71A13 and CYP71B15) and drain from the pathway (CYP83B1) (Fig. S1) were performed in order to identify possible cis-elements responsible for induction by LPS. A summary of the cis-element analysis in 1,000 bp upstream promoter regions of the genes (Fig. S2) is shown in Table 1. Cis-elements were selected based on occurrence (GT1, GATA, CORE, ARR1AT) and relevance to defence (W, MYB, RAV1). An over representation of defence-related W-, MYBcore- and RAV1AAT- motif elements was observed in some of the promoters. The promoter region of TSB1 contains only one W-box, but CYP71B15 and CYP79B2 each contained five, while ASA and CYP831B1 contain six and ten, respectively. Interestingly, no JA response elements (JERE) (AGACCGCC) or ET response elements (ERE) (AGCCGCC) was located in the promoter regions of the genes linked to the core pathway of camalexin biosynthesis.
Histochemical analysis
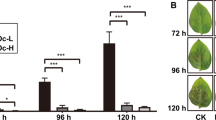
Histochemical staining of LPS-treated CYP71B15 and CYP79B2 GUS reporter gene plants indicated positive staining at 12 h, increasing in intensity up to 24 h.
Gene expression analysis
Aliquots of the amplicons obtained from a two-step semi-quantitative RT-PCR with RNA isolated at 6, 9, 12 and 18 h were separated on agarose gels to visualise the amplified cDNA. Single bands for the TSB1 (217 bp), CYP79B2 (192 bp), CYP71B15 (163 bp) and 18SrRNA (185 bp) amplicons were obtained. Compared with the 18S rRNA, that exhibited constant expression, TSB1 and CYP79B2 transcripts showed up-regulation at 9 and 18 h, respectively, while those of CYP71B15 showed a slight increase at 12 and 18 h after LPS treatment (results not shown).
In independent RT-qPCR experiments, cDNAs prepared from 0, 6, 9, 12 and 18 h RNA pools were analysed for quantitative expression analysis of the TSB1, CYP79B2 and CYP71B15 genes. 18S rRNA was found to be a stable reference gene, showing no change during elicitation of the leaf tissue with LPS. qPCR data were analysed by comparing the expression of the target genes against the expression of the 18S rRNA reference gene (Fig. 2a–c). In addition, a normalized calibrator ratio for each gene at various time points was calculated, which indicated the same trends. The summarized results are presented in Table S1.
TSB1 was differentially expressed in response to LPS in relation to a normalized control sample (0 h), and was induced over a time interval of 6–12 h with an initial increase in fold induction of 3.7 at 6 h and maintained up to 4.1 at 12 h. The induction decreases between 12 and 18 h to 1.3-fold. CYP79B2 was induced over a time interval of 6–18 h with a maximum normalized fold induction of 3.7 at 12 h followed by a decrease to 1.3 at 18 h. CYP71B15 was induced over a time-interval of 6–12 h with a maximum normalized fold induction of 4.4 at 12 h, followed by a decrease to 1.3 at 18 h when normalized to the reference gene. ANOVA analysis indicated that the transcript levels at all the time points from 6 h onwards are significantly different to the control (P < 0.05 or P < 0.01; Yuan et al. 2006). ANOVA analysis of each individual time point is indicated in Fig. 2a–c.
Camalexin identification and quantification
Camalexin present in standard solutions and in crude extracts of elicited leaf tissues was analysed as described under “Materials and methods”. The experimental conditions were optimised using a camalexin standard that eluted at R t 3.46 min. A standard curve was constructed (best fit trend line, y = 2567.9 + 438.81, R 2 = 0.9877), using the average peak area from three sets of a camalexin concentration series. Positive identification of camalexin was by means of photo diode array (PDA) UV spectroscopy. Elemental composition analysis using the MassLynx software was performed on the MS spectrum peak at 201.0484 Da. An i-fit (Norm) of 0 indicated the best fit for the reported mass at a tolerance of 0.9 mDa. The formula C11H9N2S is the protonated form of camalexin, thus verifying the compound’s identity in the plant extract.
Extracts from 0-, 6-, 12-, 18- and 24-h treated leaf samples were analysed for camalexin as marker (Fig. 3) and quantified. LPS-induced plants produced camalexin levels up to 4 μg g−1 FW over a period of 24 h, with the greatest increase occurring between 12 and 18 h (Fig. 4).
Metabolomics and multivariate data analysis
Following PCA, the first two principal components (PC1 and PC2), explaining 67.4% of the variance, were used to derive a scores plot (Fig. 5). The samples were found to be differentially clustered into five groups corresponding to the different time intervals (0–24 h) with no significant intra-group variation. There is a general trend of clusters corresponding to the different time points to increase along the PC1 (principal component 1) axis as the induction time increases, concurrent with an initial increase (6, 12, 18 h) followed by a decrease (24 h) along the PC2 axis with increasing induction time. The PCA Loadings plot (Fig. 6) for the extracts from LPS-treated leaves indicates camalexin (based on R t and m/z values) as a biomarker that makes a large contribution to the total information of the metabolome in the extracts.
Discussion
LPS contributes to the microbe-derived molecules to which plant cells are exposed upon bacterial infection (Boller and Felix 2009). LPS is known to trigger both SA and JA signalling events [important for the enhancement of induced disease resistance (van Wees et al. 2000)] as indicated by the activation of gene expression of the PR-1 (pathogenesis-related 1) (Zeidler et al. 2010) and PDF1.2 (plant defensin) (Dubery et al. unpublished) marker genes. Compared with the biochemical steps required for the synthesis of camalexin (Fig. S1), much less is known about the signalling components upstream of the camalexin biosynthesis pathway (Glawischnig 2007; Xu et al. 2008). Camalexin production cannot be triggered by exogenous application of SA, JA or ET and appears to be controlled by a pathway that exhibits little cross-talk with SA-, ET- and JA-dependent signalling events (Thomma et al. 1999). Targets of this signalling cascade that involves ROS, acting upstream of SA, are both the camalexin- as well as tryptophan biosynthetic genes (Glawischnig 2007).
Leaves from CYP79B2:GUS and CYP71B15:GUS transgenic A. thaliana plants were induced with LPS for 12 and 24 h (Fig. 1). The blue stain that developed utilizing the GUS reporter enzyme is indicative of LPS-responsive signal transduction events resulting in defence-associated transcription factors binding to the promoter sites and activation of the reporter gene.

Histochemical staining of LPS-treated CYP71B15 and CYP79B2 GUS reporter gene plants. LPS-induced leaves (at 12 and 24 h) show positive histochemical staining whilst the water-treated controls only showed slight staining at wounding sites

Expression kinetics of TSB1 (a), CYP79B2 (b), and CYP71B15 (c) in response to LPS elicitation. RNA extracted from LPS-treated leaves at 0, 6, 9, 12 and 18 h was reverse transcribed and used in RT-qPCR for expression analysis. The 18S rRNA gene was used for reference and normalization. Error bars represent standard deviation of three biological and technical repeats. Anova statistical analysis is indicated as *(P < 0.05) or **(P < 0.01)

UPLC–MS chromatograms of LPS-treated leaf extracts. Total ion chromatograms of extracts obtained from 0, 6, 12, 18 and 24 h LPS-treated tissues (a–e). The camalexin peak eluted at R t = 3.47 min. The mass of 201.0484 corresponds to C11H9N2S, the protonated form of camalexin

Increase in camalexin concentration in response to LPS. Camalexin concentrations in LPS-treated sample extracts at various time points from 0 to 24 h. Values indicated are the averages of three biological replicates. Error bars indicate standard deviation
Upstream regulatory sequences nested in inducible promoters form a complex regulation network to respond to a variety of agents, including ET, JA, SA and various bacterial and fungal elicitors. Singh (1998) proposed that the specificity of the plant response to different pathogens can be ascribed to combinatorial interactions among the cis-elements and their corresponding transcription factors (TFs). A positive correlation exists between genes that respond to diverse stimuli (stress responses and signalling) and the amount of cis-elements in their promoters (Walther et al. 2007). Based on the variety and quantity of putative regulatory element groups in the promoters, the genes evaluated in this study (ASA, TSB1, CYP79B2, CYP71A13, CYP71B15 and CYP83B1) can potentially respond to a large and diverse repertoire of TFs (Fig. S2, Table 1).
Cis-acting elements found to occur in a high frequency in the gene promoters investigated in this study includes the GATA-box, associated with high level expression and suggesting strong promoters or inducible promoters. The ARR1AT-binding element is a response regulator, binding to transcriptional activators (Sakai et al. 2000). The core consensus binding sequence (ATTA) binds zinc finger-homeodomain TFs, some of which are known to be regulated by pathogen stimulation (Park et al. 2007). The GT1consensus cis-element occurs in many light-regulated genes and also plays a role in SA- and pathogen-induced gene expression. Binding of GT-1-like factors to the PR-1a promoter influences the level of SA-inducible gene expression (Zhou 1999).
The TFs which play an important role in defence signalling include WRKY, ERF, bZIP, MYB and RAV1 (Rushton and Somssich 1998; Singh et al. 2002) and the interactions of these TFs to their cognate cis-elements are key steps in the process of defence signalling. The absence of JERE and ERE cis-elements in the promoters analysed, is in line with the observation that external applications of JA or ET do not trigger camalexin synthesis (Thomma et al. 1999). MYB proteins, binding to the MYBconsensus motifs are key factors in regulatory networks controlling development, metabolism and responses to biotic and abiotic stresses (Dubos et al. 2010). The RAV1AAT/RAV1 binding motif (CAACA) binds TFs known to play roles in biotic and abiotic stresses (Sohn et al. 2006). W-box elements (TTGACC/T) form part of the binding regions of the WRKY TFs known to play a major role in regulating the plant defence transcriptome (Eulgem et al. 2000). Furthermore, the W-box was reported to be overrepresented in the promoters of up-regulatable defence genes such as PR-1 (Maleck et al. 2000). The fact that the promoter regions of CYP71B15 and CYP79B2 each contained five W boxes compared with the one of TSB1 suggests that over-representation of the W-box elements could be a component in determining the full expression of the genes in response to bacterial attack, where LPS is perceived as a MAMP, possibly via a ROS and/or SA-signalling pathway. Based on the results in Table 1, TSB1, CYP79B2 and CYP71B15 were chosen for further study.
Tryptophan synthase converts indole and serine to tryptophan (Radwanski and Last 1995), which feeds not only the camalexin biosynthetic pathway, but also acts as a precursor for indole-3-acetic acid and defence compounds such as indole alkaloids and indole glucosinolates (Fig. S1; Zhao and Last 1996). Regulatory networks exist to increase Trp synthesis beyond constitutive levels under conditions of increased demand for Trp-derived defence metabolites such as camalexin and the indole glucosinolates (Bender and Celenza 2009). The pattern of activation of the TSB1 gene in response to LPS is presumably to support the flow of carbon into camalexin and indole glucosinolate synthesis without significantly perturbing other branches of Trp-metabolites (Bender and Celenza 2009).
Trp-derived secondary metabolites are inter-related through a common precursor, indole-3-acetaldoxime (IAOx), and CYP79B2/B3 is a monooxygenase that converts tryptophan to IAOx via a trp-N-hydroxylation step (Hull et al. 2000; Mikkelsen et al. 2000). IAOx represents a major branch point (Fig. S1) between the synthesis of the camalexin and indole glucosinolates (Glawischnig et al. 2004; Bednarek et al. 2009).
CYP71B15, corresponding to PAD3 (phytoalexin deficient 3), is monooxygenase/decarboxylase that catalyses the last step of the pathway, converting DHCA (Schuhegger et al. 2006) or Cys(IAN) to camalexin (Fig. S1; Böttcher et al. 2009). Its induction profile can be explained by the fact that the gene product fulfils a more dedicated function in the camalexin pathway.
Overall, the induction kinetics of CYP79B2 and CYP71B15 (which each contains five W-boxes in their promoters) were more similar to each other compared with that of TSB1 (which contains only one W-box). This could reflect a regulatory mechanism coordinating Trp biosynthesis with a Trp-derived secondary metabolic pathway, i.e. the distinct coordinated but independent regulation of a secondary pathway (CYP79B2 and CYP71B15), linked to a primary one (TSB1) (Bender and Celenza 2009).
For detection and identification of metabolites occurring in low concentrations, UPLC–MS is the most effective method (Grata et al. 2008; Böttcher et al. 2009; Beets and Dubery 2012) and was used to quantify camalexin in A. thaliana following LPS elicitation. The levels of camalexin produced by A. thaliana in response to LPS-treatment were within the range exhibited by other inducers and other cruciferous phytoalexins (Zhou et al. 1998; Mert-Turk et al. 2003; Glawischnig et al. 2004; Schuhegger et al. 2006). Col-0 has been reported to be a low, intermediate or high camalexin-producing ecotype of A. thaliana, depending on the infection, elicitor or treatment (Mert-Turk et al. 2003; Denby et al. 2004; Schuhegger et al. 2007). The capacity to synthesise camalexin appears to be dependent on the effect of the inducing agents on regulatory components involving elicitor recognition and signal transduction (Thomma et al. 1999; Schuhegger et al. 2007). Moreover, the differences in responses can be explained by the involvement of different signalling pathways for different pathogens and abiotic elicitors (Schuhegger et al. 2007).
Present models of plant responses towards pathogens indicate that A. thaliana undergoes major metabolic re-organization away from the production of constitutive glucosinolates and phenylpropanoids towards production of camalexin (Kliebenstein et al. 2005). To investigate the relevance of the transcriptional up-regulation of TSB1, CYP79B2 and CYP71B15 to the biosynthesis of camalexin, metabolomic analysis known as ‘metabolite target analysis’ was used. Here the analysis is restricted to metabolite(s) of a particular enzyme system that would be directly affected by abiotic or biotic perturbation (Goodacre et al. 2004).
Metabolomic analysis generates high-dimensional and complex data sets that are difficult to analyse and interpret by visual inspection or traditional univariate statistics (Fiehn et al. 2000). The various factors responsible for overall differences seen between samples are often intertwined amongst all the non-changed effects, complicating the analysis of variations caused by transient effects. Unsupervised multivariate PCA was used as a means of identifying patterns in the data and expressing the data from the different time points in such a way as to highlight their similarities and differences.
The PCA scores plot exhibits interpretable projections of samples in a reduced dimensionality. It is limited only to the most significant components and gives a visual image of the differences of all the samples (Fukusaki and Kobayashi 2005). Separation between the various time points as shown in Fig. 5 is indicative of on-going, time-dependent dynamic changes in response to LPS. The PCA Loadings (scatter) plot permits the evaluation of the contribution that each ion mass (metabolite) makes to the total information of the analysed metabolome, indicating the differences among samples in each metabolite level. Highlighted biomarkers, responsible for the group separation seen in the scores plot, can be identified in the Loading plots as for camalexin (Fig. 6). Camalexin was thus principally responsible for the clustering of the extracts obtained at different time points.

PCA scores plot of UPLC–MS analysed extracts from LPS-treated leaf extracts. The plot highlights the differences between the analytes present in extracts obtained at progressive time intervals with the clusters representing the time points in the series. R t and m/z data pairs are the identifiers of each analyte peak. For data processing, the parameters used were a R t interval of 2–10 and a 100–500 Da mass interval. The scores plot represents the analysis from three representative biological repeats, each with three technical repeats

Loadings plot of UPLC–MS analysed samples from LPS-treated leaf extracts. Putative biomarkers responsible for the group separation appear in the plot as outliers, with the camalexin outlier at 24 h indicated. Camalexin as a marker generates variability between 0, 6, 12, 18 and 24 h extracts due to time-dependent increase in its concentration. For data processing, the parameters used were a R t interval of 2–10 and a 100–500 Da mass interval. The loadings plot represents the analysis from three representative biological repeats, each with three technical repeats
Conclusion
The results contribute novel information about plant immune responses towards LPS as a MAMP and triggering agent of defence responses and add new insights into the biochemical action mechanism of LPS. The integration of bio-informatic, transcriptomic and metabolomic data provides novel information on gene-to-metabolite networks active upon LPS-triggered PTI. Collectively, the results indicate that perception of LPS activate plant signalling pathways, targeting the genes encoding enzymes in the biosynthetic pathway of tryptophan to camalexin, thereby contributing towards an enhanced chemical defence capacity in the plant. The resulting transient up-regulation of the TSB1, CYP79B2 and CYP71B15 transcripts was accompanied by time-dependent increases in camalexin concentration as bio-analytically determined by UPLC–MS. Multivariate data analysis indicates that camalexin is a major metabolite that contributes to the variability of the altered metabolome in response to LPS.
Abbreviations
- CYP:
-
Cytochrome P450 monooxygenase
- GUS:
-
Beta-glucuronidase
- LPS:
-
Lipopolysaccharide
- PAMP:
-
Pathogen-associated molecular pattern
- PCA:
-
Principal components analysis
- TSB:
-
Tryptophan synthase subunit B
- UPLC–MS:
-
Ultra performance liquid chromatography–mass spectrometry
References
Bednarek P, Piślewska-Bednarek M, Svatoš A, Schneider B, Doubský J, Mansurova M, Humphry M, Consonni C, Panstruga R, Sanchez-Vallet A, Molina A, Schulze-Lefert P (2009) A glucosinolate metabolism pathway in living plant cells mediates broad-spectrum antifungal defense. Science 323:101–106
Beets C, Dubery IA (2012) Quantification of camalexin, a phytoalexin from Arabidopsis thaliana: a comparison of five analytical methods. Anal Biochem 419:260–265
Bender J, Celenza JL (2009) Indolic glucosinolates at the crossroads of tryptophan metabolism. Phytochem Rev 8:25–37
Boller T, Felix G (2009) A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol 60:379–406
Böttcher C, Westphal L, Schmotz C, Prade E, Scheel D, Glawischnig E (2009) The multifunctional enzyme CYP71B15 (PHYTOALEXIN DEFICIENT3) converts cysteine-indole-3-acetonitrile to camalexin in the indole-3-acetonitrile metabolic network of Arabidopsis thaliana. Plant Cell 21:1830–1845
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Chang WC, Lee TY, Huang HY, Pan RL (2008) PlantPAN: plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genomics 9:561
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Coventry HS, Dubery IA (2001) Lipopolysaccharides from Burkholderia cepacia contribute to an enhanced defensive capacity and the induction of pathogenesis-related proteins in Nicotiana tabacum. Physiol Mol Plant Pathol 58:149–158
Denby KJ, Kumar P, Kliebenstein DJ (2004) Identification of Botrytis cinerea susceptibility loci in Arabidopsis thaliana. Plant J 38:473–486
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci 15:573–581
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey RN, Willmitzer L (2000) Metabolite profiling for plant functional genomics. Nature Biotech 18:1157–1161
Fukusaki E, Kobayashi A (2005) Plant metabolomics: potential for practical operation. J Biosci Bioeng 100:347–354
Gerber IB, Dubery IA (2004) Protein phosphorylation in Nicotiana tabacum cells in response to perception of lipopolysaccharides from Burkholderia cepacia. Phytochemistry 65:2957–2966
Gerber IB, Zeidler D, Durner J, Dubery IA (2004) Early perception responses of Nicotiana tabacum cells in response to lipopolysaccharides from Burkholderia cepacia. Planta 218:647–657
Gerber IB, Laukens K, Witters E, Dubery IA (2006) Identification of lipopolysaccharide-responsive phosphoproteins in Nicotiana tabacum cells. Plant Physiol Biochem 44:369–379
Gerber IB, Laukens K, De Vijlder T, Witters E, Dubery IA (2008) Proteomic identification of cellular targets of lipopolysaccharide-induced signalling in Nicotiana tabacum BY-2 cells. Biochim Biophys Acta Prot Proteom 1784:1750–1762
Glawischnig E (2007) Camalexin. Phytochemistry 68:401–406
Glawischnig E, Hansen BG, Olsen CE, Halkier BA (2004) Camalexin is synthesized from indole-3-acetaldoxime, a key branching point between primary and secondary metabolism in Arabidopsis. Proc Natl Acad Sci USA 101:8245–8250
Glazebrook J (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43:205–227
Glazebrook J, Ausubel FM (1994) Isolation of phytoalexin-deficient mutants of Arabidopsis thaliana and characterization of their interactions with bacterial pathogens. Proc Natl Acad Sci USA 91:8955–8959
Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotech 22:245–252
Grata E, Boccard J, Guillarme D, Glauser G, Carrupt PA, Farmer EE, Wolfender JL, Rudaz S (2008) UPLC-TOF-MS for plant metabolomics: a sequential approach for wound marker analysis in Arabidopsis thaliana. J Chromatogr B 871:261–327
Hammerschmidt R (1999) Phytoalexins: What have we learned after 60 years? Annu Rev Phytopathol 37:285–306
Hull AK, Vij R, Celenza JL (2000) Arabidopsis cytochrome P450 s that catalyze the first step of tryptophan-dependent indole-3-acetic acid biosynthesis. Proc Natl Acad Sci USA 97:2379–2384
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Kliebenstein DJ, Rowe HC, Denby KJ (2005) Secondary metabolites influence Arabidopsis/Botrytis interactions: variation in host production and pathogen sensitivity. Plant J 44:25–36
Livaja M, Zeidler D, von Rad U, Durner J (2008) Transcriptional responses of Arabidopsis thaliana to the bacteria-derived PAMPs harpin and lipopolysaccharide. Immunobiology 213:161–171
Madala NE, Leone MR, Molinaro A, Dubery IA (2011) Deciphering the structural and biological properties of the lipid A sub-component of lipopolysaccharides from Burkholderia cepacia strain ASP B 2D, in Arabidopsis thaliana. GlycoBiology 21:184–194
Madala NE, Molinaro A, Dubery IA (2012) Distinct carbohydrate and lipid-based molecular patterns within lipopolysaccharides from Burkholderia cepacia contribute to defense-associated differential gene expression in Arabidopsis thaliana. Innate Immun. doi: 10.1177/1753425910392609 (published online 6 July 2011)
Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, Dangl JL, Dietrich RA (2000) The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat Genet 26:403–410
Mert-Turk F, Bennett MH, Mansfield JW, Holub EB (2003) Camalexin accumulation in Arabidopsis thaliana following abiotic elicitation or inoculation with virulent or avirulent Hyaloperonospora parasitica. Physiol Mol Plant Pathol 62:137–145
Mikkelsen MD, Hansen CH, Wittstock U, Halkier BA (2000) Cytochrome P450 CYP79B2 from Arabidopsis catalyzes the conversion of tryptophan to indole-3-acetaldoxime, a precursor of indole glucosinolates and indole-3-acetic acid. J Biol Chem 275:33712–33717
Mishina TE, Zeier J (2007) Pathogen-associated molecular pattern recognition rather than development of tissue necrosis contributes to bacterial induction of systemic acquired resistance in Arabidopsis. Plant J 50:500–513
Newman MA, Dow JM, Molinaro A, Parrilli M (2007) Priming, induction and modulation of plant defence responses by bacterial lipopolysaccharides. J Endotoxin Res 13:69–84
Park HC, Kim ML, Lee SM, Bahk JD, Yun DJ, Lim CO, Hong JC, Lee SY, Cho MJ, Chung WS (2007) Pathogen-induced binding of the soybean zinc finger homeodomain proteins GmZF-HD1 and GmZF-HD2 to two repeats of ATTA homeodomain binding site in the calmodulin isoform 4 (GmCaM4) promoter. Nucleic Acids Res 35:3612–3623
Pedras MSC, Khan AQ (2000) Biotransformation of the phytoalexin camalexin by the phytopathogen Rhizoctonia solani. Phytochemistry 53:59–69
Pedras MSC, Zheng QA, Sarma-Mamillapalle VK (2007) The phytoalexins from Brassicaceae: structure, biological activity, synthesis and biosynthesis. Nat Prod Commun 2:319–330
Piater LA, Nürnberger T, Dubery IA (2004) Identification of a lipopolysaccharide responsive erk-like map kinase in tobacco leaf tissue. Mol Plant Pathol 5:331–341
Radwanski ER, Last RL (1995) Tryptophan biosynthesis and metabolism: biochemical and molecular genetics. Plant Cell 7:921–934
Rauhut T, Glawischnig E (2009) Evolution of camalexin and structurally related indolic compounds. Phytochemistry 70:1638–1644
Rogers EE, Glazebrook J, Ausubel FM (1996) Mode of action of the Arabidopsis thaliana phytoalexin camalexin and its role in Arabidopsis-pathogen interactions. Mol Plant Micr Interact 9:748–757
Rushton PJ, Somssich IE (1998) Transcriptional control of plant genes responsive to pathogens. Curr Opin Plant Biol 1:311–315
Sakai H, Aoyama T, Oka A (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J 24:703–711
Sanabria NM, Dubery IA (2006) Differential display profiling of the Nicotiana response to LPS reveals elements of plant basal resistance. Biochem Biophys Res Commun 344:1001–1007
Sanabria NM, Huang J-C, Dubery IA (2010) Self/nonself perception in plants in innate immunity and defense. Self/Nonself Imm Regul 1:40–54
Schuhegger R, Nafisi M, Mansourova M, Petersen BL, Olsen CE, Svatos A, Halkier BA, Glawischnig E (2006) CYP71B15 (PAD3) catalyzes the final step in camalexin biosynthesis. Plant Physiol 141:1248–1254
Schuhegger R, Rauhut T, Glawischnig E (2007) Regulatory variability of camalexin biosynthesis. J Plant Physiol 164:636–644
Singh KB (1998) Transcriptional regulation in plants: the importance of combinatorial control. Plant Physiol 118:1111–1120
Singh K, Foley RC, Onate-Sanchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5:430–436
Sohn KH, Lee SC, Jung HW, Hong JK, Hwang BK (2006) Expression and functional roles of the pepper pathogen-induced transcription factor RAV1 in bacterial disease resistance, and drought and salt stress tolerance. Plant Mol Biol 61:897–915
Stefanato FL, Abou-Mansour E, Buchala A, Kretschmer M, Mosbach A, Hahn M, Bochet CG, Metraux J-P, Schoonbeek HJ (2009) The ABC-transporter BcatrB from Botrytis cinerea exports camalexin and is a virulence factor on Arabidopsis thaliana. Plant J 58:499–510
Thomma BP, Nelissen I, Eggermont K, Broekaert WF (1999) Deficiency in phytoalexin production causes enhanced susceptibility of Arabidopsis thaliana to the fungus Alternaria brassicicola. Plant J 19:163–171
Tsuji J, Jackson EP, Gage DA, Hammerschmidt R, Somerville SC (1992) Phytoalexin accumulation in Arabidopsis thaliana during the hypersensitive reaction to Pseudomonas syringae pv syringae. Plant Physiol 98:1304–1309
Van Wees SCM, de Swart EAM, van Pelt JA, van Loon LC, Pieterse CM (2000) Enhancement of induced disease resistance by simultaneous activation of salicylate- and jasmonate-dependent defense pathways in Arabidopsis thaliana. Proc Natl Acad Sci USA 97:8711–8716
Van Wees SCM, Van der Ent S, Pieterse CMJ (2008) Plant immune responses triggered by beneficial microbes. Curr Opin Plant Biol 11:443–448
Walther D, Brunnemann R, Selbig J (2007) The regulatory code for transcriptional response diversity and its relation to genome structural properties in Arabidopsis thaliana. PLoS Genetics 3:e11
Xu J, Li Y, Wang Y, Liu H, Lei L, Yang H, Liu G, Ren D (2008) Activation of MAPK kinase 9 induces ethylene and camalexin biosynthesis and enhances sensitivity to salt stress in Arabidopsis. J Biol Chem 283:26996–27006
Yuan J, Reed A, Chen F, Stewart CN (2006) Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85
Zeidler D, Zähringer U, Gerber IB, Dubery IA, Hartung T, Bors W, Hutzler P, Durner J (2004) Innate immunity in Arabidopsis thaliana: lipopolysaccharides activate nitric oxide synthase (nos) and induce defense genes. Proc Natl Acad Sci USA 101:15811–15816
Zeidler D, Dubery IA, Schmitt-Kopplin P, von Rad U, Durner J (2010) Lipopolysaccharide mobility in leaf tissue of Arabidopsis thaliana. Mol Plant Pathol 11:747–755
Zhao J, Last RL (1996) Coordinate regulation of the tryptophan biosynthetic pathway and indolic phytoalexin accumulation in Arabidopsis. Plant Cell 8:2235–2244
Zhou DX (1999) Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci 4:210–214
Zhou N, Tootle TL, Tsui F, Klessig DF, Glazebrook J (1998) PAD4 functions upstream from salicylic acid to control defense responses in Arabidopsis. Plant Cell 10:1021–1030
Acknowledgments
This work was funded by the South African National Research Foundation. Mr M. George and Dr P. Steenkamp are thanked for assistance with UPLC–MS analysis. Dr N. Sanabria is thanked for help with qPCR analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material: The online version of this article contains supplementary material.
Table S1 Summary of expression levels of TSB1, CYP79B2 and CYP71B15 in response to LPS elicitation. Statistical analysis of RT-qPCR results was performed by ANOVA.
Fig. S1 Camalexin and indole glucosinolates biosynthetic pathways. (Derived from: Mikkelsen et al. 2000; Hull et al. 2000; Glawischnig et al. 2004; Schuhegger et al. 2007; Rauhut and Glawischnig 2009). Grey circles indicate steps investigated in this study. TSA/B = Tryptophan synthase alpha/beta subunit; CYP79B2/B3, CYP83B1, CYP71A13, CYP71B15 = Cytochrome P450 monooxygenases; SGT = UDPG-thiohydroximate glycosyltransferase; PAPS-ST = 3′-Phosphoadenosine 5′-phosphosulfate:desulfoglucosinolate sulfotransferase. Anthranilate synthase (AS) feeds indole into the pathway. CYP83B1 leads to indolyl glucosinolates.
Fig. S2 Schematic representation of selected cis-elements in upstream promoter regions (-1,000 bp). Cis-elements were selected based on occurrence and relevance to defence. a TSB1 (At5g54810), CYP79B2 (At4g39950) and CYP71B15 (At3g26830) investigated in this study. b ASA (At1g25220), CYP79B3 (At2g22330), CYP71A13 (At2g30770) and CYP83B1 (At4g31500), included for comparison purposes.
Rights and permissions
About this article
Cite this article
Beets, C.A., Huang, JC., Madala, N.E. et al. Activation of camalexin biosynthesis in Arabidopsis thaliana in response to perception of bacterial lipopolysaccharides: a gene-to-metabolite study. Planta 236, 261–272 (2012). https://doi.org/10.1007/s00425-012-1606-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-012-1606-1