
Abstract
Ultraviolet-B (UV-B, 280–320 nm) radiation may have severe negative effects on plants including damage to their genetic information. UV protection and DNA-repair mechanisms have evolved to either avoid or repair such damage. Since autotrophic plants are dependent on sunlight for their energy supply, an increase in the amount of UV-B reaching the earth’s surface may affect the integrity of their genetic information if DNA damage is not repaired efficiently and rapidly. Here we show that overexpression of cyclobutane pyrimidine dimer (CPD) photolyase (EC 4.1.99.3) in Arabidopsis thaliana (L.), which catalyses the reversion of the major UV-B photoproduct in DNA (CPDs), strongly enhances the repair of CPDs and results in a moderate increase of biomass production under elevated UV-B.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ultraviolet (UV)-radiation is one of the most damaging agents for DNA and other biomolecules such as proteins and lipids (Prinsze et al. 1990; Smirnoff 1995). Ultraviolet B (UV-B) radiation reaching the earth’s surface is highly variable and influenced by many factors such as season and clouds. However, anthropogenic release of chlorine and fluorine-containing compounds (e.g. CFCs) probably caused the most significant depletion in stratospheric ozone and thus the increase in UV-B radiation in the past decades in particular in the Antartic (Anderson et al. 1991; McKenzie et al. 1999; Solomon et al. 2007).
Since photosynthetic plants use sunlight as energy source and thus cannot avoid UV-B exposure, sophisticated mechanisms for UV-protection and repair of damage have evolved (Jansen et al. 1998; Vonarx et al. 1998; Hollósy 2002; Emiliani et al. 2009). However, even subtle changes in the morphology and secondary metabolism under elevated UV-B could have important effects on competitive balance and susceptibility to pathogens in the long term (Orth et al. 1990; Panagopoulos et al. 1992; McCloud and Berenbaum 1994; Barnes et al. 1995; Ballaré et al. 1996).
Besides proteins, DNA in particular strongly absorbs UV-B, therefore it is the most important target for UV-B damage. Cyclobutane pyrimidine dimers (CPDs) and (6-4) photoproducts are the major UV-B photoproducts in DNA. They distort the DNA molecule so that transcription and replication are inhibited and induce mutations or even cell death (Thoma 1999; Friedberg et al. 2006). Accumulation of such photoproducts was documented for plant species endemic in Southern Argentina (Rousseaux et al. 1999), indicating that the mechanisms evolved for the protection and repair of DNA cannot completely compensate for increased UV-B levels. Not only can a loss of productivity be expected (Borneman and Teramura 1993; Jansen et al. 1998; Ballaré et al. 1999; Kumagai et al. 2001) but also an increase of recombination in the genome, which has been described by Ries et al. (2000). In plants, UV-B photoproducts can be removed from DNA by nucleotide excision repair (Gallego et al. 2000; Liu et al. 2000, 2001) or by photoreactivation (Ahmad et al. 1997; Landry et al. 1997; Nakajima et al. 1998). During photoreactivation, the DNA photolyases catalyse the repair of damaged DNA using the energy from photons in the blue/UV-A region (Sancar 2003). Two types of DNA photolyases were identified in plants specific for either CPDs or (6-4) photoproducts (Ahmad et al. 1997; Landry et al. 1997; Nakajima et al. 1998). It was shown that photoreactivation is the most efficient repair system for CPDs in plants (Pang and Hays 1991). Teranishi et al. (2004) and Iwamatsu et al. (2008) gave evidence that the UV-B sensitivity of different rice species and cultivars is dependent on their CPD photolyase genotypes. Consistent with these results, an increase of CPD photolyase activity in rice leads to a significant prevention of growth inhibition caused by UV-B irradiation (Hidema et al. 2007). The goal of this study was to investigate whether it is possible to increase the UV-tolerance of the dicot model plant Arabidopsis by upregulating a specific DNA repair mechanism. Considering that photoreactivation is very efficient in plants and mediated by a single enzyme, we have chosen overexpression of photolyase in transgenic plants in order to evaluate its potential to increase UV tolerance. Since CPDs are the major UV-B photoproducts (Friedberg et al. 2006) we took CPD photolyase for these studies, and our results show that the increased levels of CPD photolyase strongly promote the repair of CPD lesions in the DNA.
Materials and methods
Plant material, growth conditions, construction of transgenic lines and copy number analysis of T-DNA insertions
Arabidopsis thaliana L. wild type, the CPD photolyase mutant uvr2-1 (Landry et al. 1997; obtained from Arabidopsis Biological Resource Centre, Ohio State University, Columbus, OH, USA) and transgenic lines overexpressing Arabidopsis CPD photolyase (Ox lines), all in the Landsberg erecta (Ler) background, were used. For the construction of transgenic lines, the complete coding region of the At-PHR1 (Genbank Accession: AF053365) gene (Taylor et al. 1996; Ahmad et al. 1997) was amplified by PCR using as template pCR2.1 At-PHR1, described previously by Kleiner et al. (1999), and the following primers: forward (ACCCGGGAACA ATG GCG TCG ACA GTC TCA GTT) and reverse (AGAGCTC CTA AAC AAT AGT TAT CTT GGG). The resulting product was subcloned into vector pCR2.1-TOPO (Invitrogen, Carlsbad, CA, USA), sequenced and then inserted into the binary vector pPCV812 (Koncz et al. 1989) using the SmaI and SacI sites. In the resulting construct pPCV812 2x35S::At-PHR1 the At-PHR1 gene is under control of a tandem CaMV 35S promoter (Fang et al. 1989) (Fig. 1a). pPCV812 2x35S::At-PHR1 was transferred into the Agrobacterium strain GV3103 (Koncz and Schell 1986) and Arabidopsis plants were transformed using the floral dip method (Clough and Bent 1998). Seeds of the T0 plants were sown on agar plates containing MS salt mixture (Sigma, Munich, Germany) and 50 μg ml−1 hygromycin (Duchefa, Haarlem, The Netherlands). Plates were kept for 20 days in a growth chamber (14 h photoperiod; white light 150 μmol m−2 s−1) for selection. Green, rooted plants were transferred to soil for seed production after selfing. Seeds of the T1 generation were again selected with hygromycin and used to test for segregation. Seeds of homozygous T2 plants were used for further studies.

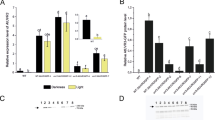
Copy number analysis of T-DNA insertion in transgenic 35S::PHR1 lines. a Scheme of the T-DNA construct used for Arabidopsis transformation. LB left border, RB right border, 2x 35S CaMV 35S tandem promoter, At-PHR1 coding region of At-PHR1 gene, T NOS nopaline synthase terminator, P NOS nopaline synthase promoter, HPT hygromycin phosphotransferase, P probe used for Southern blot. b Southern blot of wild type (WT) and photolyase overexpressor lines (Ox 2, 4, 5, 6). Total DNA was extracted from rosette leaves and digested with either NcoI or PstI. The resulting fragments were separated by agarose gel electrophoresis and transferred to a Hybond-N+ membrane. The membrane was probed with a DIG-labelled probe against the HPT gene located on the T-DNA close to the right border. Resulting bands are marked by asterisks
Plants used for the analysis of the effects of UV-stress on growth were grown in a growth chamber (14 h photoperiod, 300 μmol m−2 s−1, 60% humidity, 22°C during light phase and 18°C during dark phase) in 8 cm pots on a 2:1 mixture of commercially available garden soil and vermiculite. White light fluence rates were measured with a calibrated LI-COR LI-185B quantum sensor (LI-COR, Lincoln, Nebraska, USA). The plants used to analyse At-PHR1 expression and activity were cultured in a growth cabinet under short day conditions in 8 cm pots filled with 1:2 vermiculite and garden mould. Eight weeks after sowing, rosette leaves were harvested and used for experiments.
For the determination of copy number T-DNA insertions, the Ox lines were analysed by Southern blot (Southern 1975). A DNA-probe specific for the T-DNA was generated by PCR amplification of a 660 bp fragment of the HPT gene between the NcoI site and the stop codon (Fig. 1a). pPCV812 2x35S::At-PHR1 was used as template with primer forward (GCG ATC GCT GCG GAT CT) and reverse (CTT TGC CCT CGG ACG AGT GC). The PCR product was DIG-labelled using the DIG DNA labelling kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. For the extraction of total DNA leaves were frozen in liquid nitrogen and ground to a fine powder with a mixer mill (Retsch, Haan, Germany). The powder was suspended in CTAB buffer (20 mM Tris/HCl, pH 8.0; 20 mM EDTA; 1.4 M NaCl; 2% CTAB; 1% polyvinylpyrrolidone) and incubated for 30 min at 65°C. The suspension was extracted twice with chloroform, isoamyl alcohol (25:1, v/v) and the DNA was precipitated by adding 2-propanol. The pellets were washed with 70% ethanol, dried and dissolved in EB (10 mM Tris/HCl, pH 8.0). Subsequently, the samples were treated with DNase free RNase (Roth, Karlsruhe, Germany), and afterwards the enzyme and RNA fragments removed by ethanol precipitation. The resulting DNA pellets were dissolved in EB, and DNA was digested with PstI or NcoI. The digested DNA was separated on a 0.7% agarose gel. After electrophoresis the separated fragments were depurinated (0.125 M HCl, 15 min), denatured (0.5 M NaOH, 30 min), renatured (0.1 M Tris/HCl, pH 7.5; 1.5 M NaCl) and transferred to a Hybond N+ membrane (GE Healthcare, Freiburg, Germany) by capillary blotting with 20× SSC (1× SSC: 15 mM sodium citrate, 150 mM NaCl). The DNA was fixed to the membrane by incubating for 2 h at 80°C and the membrane was blocked with 35 ml Roti-Hybri-Quick (Roth) for 30 min at 62°C. The DIG-labelled probe was added to the blocking solution and hybridized overnight at 62°C. Afterwards the membrane was washed twice with 2× SSC, 0.1% SDS and 0.5× SSC, 0.1% SDS at 62°C. The bands were visualized using a α-DIG-alkaline phosphatase antibody and CDP-Star ready-to-use kit (both from Roche) according to the manufacturer’s instructions.
Analysis of At-PHR1 expression
To verify the functionality of the introduced At-PHR1 construct, its expression was checked on transcript and protein levels using real-time PCR and immunoblot analysis, respectively. RNA was extracted from 100 mg of leaf material cultured as described above using the RNeasy Plant Kit (Qiagen, Hilden, Germany) and further depleted from DNA using the Turbo DNA-free kit (Ambion, Austin, TX, USA). RNA was reverse-transcribed to cDNA with Superscript II (Invitrogen) using poly dT primers. Quantitative real-time PCR was performed using a Mastercycler ep Realplex (Eppendorf, Hamburg, Germany) PCR cycler and the Absolute qPCR SYBR Green mix (Abgene, Epson, UK) complemented with primers and cDNA. Both forward primer F1 (TGG GTC CCG TCG TTT ACT GG) and reverse primer R1 (GCG CGT TGG TTC TGT TTG CC) bind within the first exon of the At-PHR1 gene. The resulting PCR product has a size of 96 bp. Transcript levels of ubiquitin (UBQ) as internal standard were determined as well using the following oligonucleotides forward (CGG GAA AGA CGA TTA CTC TTG AGG) and reverse (GCA AGA GTT CTG CCA TCC TCC). The At-PHR1 signals were normalized to the UBQ signals. A second primer pair for At-PHR1 consisting of the F1 primer described above and a second reverse primer R2 (ATG GGT TTG GTG TGA GAT CG) that binds within the first intron of the At-PHR gene were used to test for contaminations with genomic DNA.
Polyclonal antiserum against the At-PHR1 protein was raised in rabbits (Eurogentec, Seraing, Belgium) using recombinant At-PHR1 protein expressed and purified as described by Kleiner et al. (1999). Since this antiserum showed some unspecific bands in immunoblot analysis of Landsberg erecta wild type, it was purified using At-PHR1 protein covalently bound to a cyanogen bromide activated matrix (Sigma). The purified antibody fraction was used for immunoblot analysis as follows: 80–120 mg of leaf material from plants grown as described above were ground in 200 μl ice cold extraction buffer [20 mM phosphate buffer, pH 7.4; 500 mM NaCl; 20% glycerol; complete protease inhibitor cocktail EDTA-free (Roche)]. Cell debris and insoluble compounds were removed by centrifugation (20,000g, 5 min, 4°C) and the protein concentration in the supernatant was determined with the Bio-Rad protein test (Bio-Rad, Hercules, CA, USA). 15 μg of soluble total proteins per lane was separated on a reducing 12% SDS polyacrylamide gel (Laemmli 1970) and transferred to a PVDF-membrane by semidry blotting (Bjerrum and Schäfer-Nilsen 1986). The membrane was blocked with 3% BSA in TBS-T (20 mM Tris/HCl, pH 7.5; 150 mM NaCl; 0.1% Tween 20) overnight at 4°C and washed three times with TBS-T. The primary α-CPD photolyase antibody (final dilution 1:1,000) was added, and the membrane was incubated while shaking for 1 h at room temperature. After three additional washings with TBS-T the secondary α-rabbit::HRP antibody (Sigma) diluted 1:10,000 in TBS-T was added and incubated as described for the primary antibody. Detection was achieved with the ECL plus kit together with Hyperfilm ECL (both from GE Healthcare). After probing with the α-CPD photolyase antibody the membrane was stripped by incubation at 60°C for 30 min in stripping buffer (65.5 mM Tris/HCl, pH 6.7; 100 mM 2-mercaptoethanol; 2% SDS) and the blot developed as described above using α-HSC70 antiserum (Agrisera, Vanns, Sweden) diluted 1:4,000 in TBS-T as primary antibody.
Localisation studies
To check whether the overexpressed At-PHR1 enzyme is located in the nucleus as expected the cellular localisation of an At-PHR1::GFP fusion protein expressed in protoplasts of an Arabidopsis cell culture was studied by Confocal Laser Scanning Microscopy. For the GFP construct the complete coding region of At-PHR1 was amplified by PCR using primers F (CCA TGG CGT CGA CAG TCT C) and R (CCA TGG AAA CAA TAG TTA TCT TGG G) and as template the pQE31 At-PHR1 vector (Kleiner et al. 1999). After subcloning into vector pGEM-T (Promega, Madison, WI, USA) the correctness of the PCR product was verified by sequencing. The coding sequence of At-PHR1 was introduced into the NcoI site of pAVA393 (von Arnim et al. 1998) 5′ and in frame with the coding region of the GFP4/GFP5 hybrid and 3′ of the tandem CaMV 35S promoter. Protoplasts of an Arabidopsis Landsberg erecta mesophyll cell culture were transformed with the resulting construct pAVA393 At-PHR1::GFP as described by (Rosenfeldt et al. 2008). The cellular localisation of the fusion protein was studied 16 h after transfection with a confocal microscope (Leica, Wetzlar, Germany) consisting of a DMRE stand equipped with an PL APO 63x/1.32-0.60 oil objective, a 65 nW Ar Laser and a TCS SP2 confocal scanner. The microscope was controlled by the Leica Confocal Software, which was used for analysis of the data as well.
Analysis of CPD formation and growth under UV-stress
To analyse the activity of the expressed At-PHR1 enzyme in vivo the accumulation of CPDs during UV-B irradiation was measured. Rosette leafs were placed on agar plates (0.5 × MS salts; 1% phytoagar) with the upper surface facing upwards. The leaves were irradiated with UV-B using Philips TL40/W12 fluorescent tubes (Philips, Eindhoven, The Netherlands) with a fluence rate of 9 μmol m−2 s−1. About 50% of the fluence rate emitted from these lamps is in the UV-B region (280–320 nm) and the remaining emission at longer wavebands in particular UV-A region. Samples consisting of four to five leaves were collected during UV-irradiation from time point 0 to time point 40 min in 10 min intervals, immediately frozen in liquid nitrogen and stored at −80°C till DNA extraction was performed essentially as described above for Southern analysis. After spectroscopic determination of DNA concentrations, the samples were diluted to 15 ng/μl. 50 μl of the DNA solution was pipetted into the wells of an Microlon 96 K ELISA-plate (Greiner, Frickenhausen, Germany) and the CPD content was determined as described in Klar et al. (2006). Each sample representing a single time point was analysed four times on the same plate. The values were set in relation to a reference well with a constant amount of CPDs and mean values were calculated for each time point (n = 5). These were plotted against time and the slope, representing the rate of CPD accumulation, and its standard error was calculated by a linear regression using Origin 7.5 (Origin Lab Corporation, Northampton, MA, USA). In order to allow easier comparison of the data, the accumulation rates were standardized against wild type.
Plants used for measuring rosette size, fresh and dry mass were grown individually in pots under long-day conditions in a growth chamber as described above. UV-B was supplied by 12 UV-emitting Philips TL40W/12 tubes. UV-irradiation was done exactly in the middle of the light phase. UV-B doses were measured with a calibrated MCS 55 UV-NIR spectrometer (Tec5, Oberursel, Germany). Data from measurements of rosette size and fresh mass were treated with ANOVA and Post hoc tests (Tukeys DSD test) using the program Statistica 6.0.
Results
Construction, selection and copy number analysis of photolyase-overexpressing Arabidopsis lines
The coding region of the CPD photolyase gene from Arabidopsis under the control of the CaMV 35S tandem promoter was placed together with a hygromycin resistance gene on the T-DNA of a binary vector and introduced into Arabidopsis plants (ecotype Landsberg erecta) via Agrobacterium-mediated transformation. Seeds of transformed plants were selected with hygromycin. The 15 independent hygromycin-resistant plants of the T1 generation were allowed to self fertilise. Offspring of the T1 generation was again selected with hygromycin and used for seed production and further analysis. Immunoblot analysis with antibodies raised against the Arabidopsis CPD photolyase, was used to screen transgenic lines that overexpress CPD photolyase. Four lines designated Ox2, Ox4, Ox5 and Ox6 shown in immunoblot analysis significantly enhanced CPD photolyase protein levels compared to wild type (see below Fig. 2b) and were used for further studies.

Expression of DNA photolyase. a At-PHR1 transcript levels in wild type (WT), photolyase overexpressor lines (Ox2, 4, 5 and 6), and the CPD photolyase mutant uvr2-1. Plants were grown under short day conditions for eight weeks. RNA was extracted from rosette leaves. Transcript levels were quantified by real time PCR and normalized to ubiquitin (UBQ) transcript levels. The normalized transcript level of the WT was set as 1. Bars represent standard errors. b Immunoblot analysis of At-PHR1 protein levels. Plants were grown as in a. Total soluble proteins were extracted from rosette leaves, separated by SDS-PAGE, transferred to a PVDF membrane and probed with a α-At-PHR1 antibody. Afterwards the membrane was stripped and reprobed with a α-HSC70 antibody to check for even loading and transfer to the membrane. The secondary antibody was labelled with horse radish peroxidase and ECL signals were detected by film exposure. Labelling of the lanes as in a
To analyse the number of insertions in transgenic 2x35S::At-PHR1 lines, Southern blot analysis was performed using total DNA from overexpressor lines and WT was digested with NcoI or PstI. The blot was probed with a DIG-labelled fragment of the HPT gene that is positioned close to the right border of the T-DNA (Fig. 1a). The resulting blot (Fig. 1b) showed no signal either for NcoI or for PstI cut WT DNA excluding unspecific binding of the probe. In the lanes containing NcoI cut DNA three bands can be detected for Ox2, one for Ox4, four for Ox5 and one for Ox6. In lanes containing PstI cut DNA, the number of detectable bands is the same as those in DNA cut with NcoI of the corresponding transgenic lines. For Ox6 an additional faint band can be found in the PstI-digested DNA. From these data, it can be concluded that Ox2, Ox4 and Ox5 carry three, one and four insertions of the T-DNA, respectively, while Ox6 carries at least one but more likely two insertions.
Photolyase expression in the overexpressor lines
To gain information about the expression of CPD photolyase in the transgenic Arabidopsis lines transcript and protein levels were measured by quantitative real-time PCR and semiquantitative immunoblot analysis, respectively. The signals obtained for the At-PHR1 transcripts were normalized against the UBQ signals and then set in relation to the normalized At-PHR1 transcript level of the wild type. Figure 2a shows a strong increase of At-PHR1 transcript levels for the Ox lines. The increase in At-PHR1 transcript levels is about 550-fold for Ox2 and at least 250-fold for the other lines in comparison with WT. This demonstrates that the expression of At-PHR1 can strongly be enhanced in Arabidopsis plants when driven by a strong and constitutive promoter such as the CaMV 35S promoter. To exclude that the strong signals obtained for At-PHR1 could be caused in part by contaminating genomic DNA, we performed a control PCR with the same upstream primer but used a downstream primer that bound within the first intron. No PCR products were detectable in these control PCR excluding any contamination of the samples by genomic DNA (data not shown).
We also analysed At-PHR1 protein levels by immunoblot analysis. As in the case of transcript measurements, the soluble proteins were extracted out of rosette leaves from plants that were grown for 8 weeks under short-day conditions. After ECL detection of the At-PHR1 signal the blot was stripped and developed against the housekeeping protein HSC70 to ensure equal loading and transfer of the samples. The blot against CPD photolyase (Fig. 2b) shows no signal for WT or the uvr2-1 mutant. The Ox lines 4 and 6 show a weak although clearly visible band while for Ox2 and Ox5 a relatively strong band is detectable. The stripped blot developed with a α-HSC70 antibody results in a clear band with a similar intensity for all the lanes confirming equal loading and transfer of the samples. The mobility of the detected proteins corresponds to their expected molecular masses.
Cellular localisation of At-PHR1
Currently it is not clear whether DNA photolyases exert their catalytic activity only in the nucleus or whether DNA in organelles is repaired as well. In order to investigate the subcellular localization of the At-PHR1 protein a GFP fusion of At-PHR1 (2x35S::At-PHR1::GFP) was transiently transfected into green protoplasts from an Arabidopsis mesophyll cell culture, and the localisation of the fusion protein was analysed by confocal laser scanning microscopy. As can be seen in Fig. 3a, the At-PHR1::GFP fusion protein is exclusively within the nucleus (channel 1, 500–522 nm, green signal). When GFP is expressed alone under the control of the same promoter the fluorescence can be found in the cytoplasm and in the nucleus but it is excluded from the nucleolus as expected (Fig. 3c). The confocal images shown in Fig. 3 are overlays between the GFP channel 1 and channel 2 (667–743 nm). Channel 2 is shown in red colour with emission from chlorophyll in the chloroplasts. The localization studies clearly demonstrate that At-PHR1 is a nuclear protein, as expected, and that the effects of CPD photolyase overexpression described below are exclusively caused by photorepair in the nucleus.

Cellular localization of At-PHR1. Confocal microscopic images of Arabidopsis protoplasts expressing At-PHR1::GFP (a, b) or GFP (c, d) in each case under the control of the CaMV 35S promoter. Images a and c show channel 1 (500–552 nm) in green and channel 2 (667–743 nm) in red as overlay. Images b and d are differential interference contrast pictures of the cells shown in a or c, respectively
Effects of CPD photolyase overexpression on CPD accumulation and growth under elevated UV-B
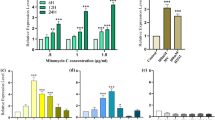
In order to test whether overexpression of CPD photolyase has any positive effects on Arabidopsis plants under enhanced UV-B, we analysed CPD accumulation as parameter for the protective activity of the At-PHR1 enzyme. Furthermore, rosette size, fresh mass and dry mass of the overexpressor lines were analysed. In order to measure the formation and accumulation of CPDs in planta under enhanced UV-B, individual rosette leaves were excised and removed from the plants and flattened on the surface of agar plates to ensure uniform exposure to the light source. As light source, UV-B emitting tubes were used that emit also UV-A (see “Materials and methods”). After irradiation CPDs were quantified in total DNA extracted from the leaves by ELISA using a monoclonal antibody against CPD (see “Materials and methods”). The accumulation of CPDs was plotted against time and the slope was calculated. The slope of the plot is inversely proportional to the activity of CPD photolyase assuming that the rate of CPD formation is the same in all analysed genotypes. The rate of CPD accumulation is elevated by about 45% in the uvr2-1 mutant compared to the wild type (Fig. 4). In the lines Ox4 and Ox5 the rate of CPD accumulation is reduced to 60% while in Ox2 and Ox6 the accumulation rate is about 40% of the wild type. These data show unambiguously that compared to the wild type the rate of CPD accumulation is increased in the uvr2-1 mutant but decreased in the CPD photolyase overexpressors.

Accumulation of CPDs during UV-B treatment of leaves. Plants were grown under short day conditions for 8 weeks. Individual rosette leaves were removed from the plant, flattened on the surface of half MS agar plates and irradiated with polychromatic UV-B/UV-A for varying time periods. Total DNA was extracted from the leaves and the CPD content was determined by ELISA using monoclonal antibodies against CPDs. The accumulation of CPDs was plotted against time and the rate of CPD formation calculated. The rate of CPD accumulation in wild type was set as 1 arbitrary unit (a.u.). The accumulation rates of other genotypes were normalized to wild type. WT wild type; uvr2-1 CPD photolyase mutant, Ox2, Ox4, Ox5, Ox6 photolyase overexpressor lines. Bars represent standard errors (n = 5)
To test whether the overexpression of CPD photolyase has any positive effect under enhanced UV-B on growth individual plants were grown in pots in a 14 h light/10 h dark cycle in a growth chamber. Plants were grown for the first 7 days without UV-B and then received a daily UV-B treatment emitted from Philips TL40/W12 lamps. UV-B treatment was done exactly in the middle of the light phase, allowing 7 h for photoreactivation before the dark phase. The position of the pots was rotated daily to eliminate any differences in photosynthetically active radiation as well as UV-B radiation applied to the plants. The rosette size was measured in 21-day-old plants. The influence of genotype and UV-B on plant growth was analysed by means of two-way ANOVA (Statistica 6.0). Every ANOVA yielding significant results was followed by Tukey HSD Post hoc test to evaluate significant differences between the means. Under conditions without UV-B, none of the transgenic lines showed statistically significant differences to wild type, indicating that overexpression of photolyase does not have any negative effect on growth (Fig. 5). UV-B doses of 1.8 and 3.6 kJ m−2 day−1, applied daily for 14 days, led to a significant reduction in rosette size for all genotypes (ANOVA, F 2, −165 = 163.47, P < 0.001). The photolyase mutant uvr2-1 suffered severely under these conditions (data not shown) and did not survive the higher UV-B dose (3.6 kJ m−2 day−1). The average rosette size of all CPD overexpressors was bigger than those of wild type under both UV-B treatments (Fig. 5b). However, statistical evaluation of the data showed that these differences were not significant (Genotype × UV-B interaction: ANOVA, F 8, 161 = 0.89, P > 0.05). Besides rosette size, we measured fresh and dry mass of wild type and photolyase overexpressing plants. For the wild type, daily UV-B doses of 1.8 and 3.6 kJ m−2 given over 16 days led to a reduction of the fresh mass to 26 and 3%, respectively. The overexpressors also showed a reduction of fresh mass under UV-B corresponding to 56, 49, 56 and 45% at 1,8 kJ m−2 day−1 and 7, 7, 10 and 8% at 3,6 kJ m−2 day−1 for the lines Ox2, Ox4, Ox5 and Ox6, respectively (Fig. 5a). As for rosette size, the UV-B effect was highly significant for all genotypes (ANOVA, F 2, −165 = 273.59, P < 0.001). The effect of UV-B on fresh mass differed significantly between genotypes (Genotype × UV-B interaction: ANOVA, F 8, 165 = 2.00, P = 0.049) with higher fresh mass of photolyase overexpressors compared to wild type under elevated UV-B. The dry mass strictly correlated with the fresh mass (dry mass about 10% of fresh mass) for both the wild type and the photolyase overexpressors (data not shown).

UV-B effects on plant growth. a Fresh mass of wild type (WT) and photolyase overexpressor lines (Ox2, 4, 5, 6). Plants were grown under long day conditions in a growth cabinet without UV-B for 7 days and then further cultivated for 16 days under the same conditions either without (-UV-B) or with a daily UV-B dose of 1.8 or 3.6 kJ m−2. The results present the average fresh mass of a plant. Error bars represent standard deviation of ten measured plants. b Rosette size of the various genotypes. Plants were grown under long day conditions in a growth cabinet without UV-B for 7 days and then further cultivated for 14 days under the same conditions either without (-UV-B) or with a daily UV-B dose of 1.8 or 3.6 kJ m−2. The results present the average rosette size of a plant. Error bars represent standard deviation of ten measured plants
Discussion
UV-B causes damage to nearly all biomolecules either direct via absorption or indirectly via the formation of reactive oxygen species (Apel and Hirt 2004; Friedberg et al. 2006). DNA is a very potent absorber of UV-B. In DNA, UV-B causes mainly two photoproducts, the cyclobutane pyrimidine dimers (CPDs) and the (6-4) photoproduct whereby CPDs are the major DNA photoproduct representing about 60–70% of the total damage. These DNA lesions disturb efficient and error-free transcription and replication, thus potentially leading to mutations and cytotoxic effects (Thoma 1999; Friedberg et al. 2006). In plants, UV-B induces several defence mechanisms such as the formation of UV-shielding pigments, thickening of cell walls, reduction of leaf and stem growth, leaf curling, and formation of UV-shielding structures such as trichomes. In general, UV-B treatment of plants causes loss of biomass production either indirectly through the induced changes in physiology towards UV-defence or directly through damage of proteins such as those of the photosynthetic apparatus or of nucleic acids (Jansen et al. 1998; Hollósy 2002;). Since UV-B can damage nearly every biomolecule, it is hard to predict which type of damage is most detrimental for the whole plant. However, studies on mutants that are hampered in repairing UV-lesions in DNA clearly show that this specific damage has quite a severe effect on the whole plant (Britt et al. 1993; Landry et al. 1997). Dark repair mechanisms such as nucleotide excision repair (Gallego et al. 2000; Liu et al. 2000, 2001) and photorepair mechanism for the removal of CPDs (Ahmad et al. 1997; Landry et al. 1997) and (6-4) photoproducts (Nakajima et al. 1998) exist in plants. It was shown that the dark repair mechanisms in plants are less effective for the repair of CPDs than the repair of this UV-lesion by DNA photolyase (Pang and Hays 1991). Studies on mutants defective in a defined DNA repair mechanism do not allow definitive conclusions about the effectiveness of the specific repair mechanism since disruption of one pathway could be compensated at least in part by another one. However, the approach used here of increasing one repair process for a specific photoproduct allows predictions of whether the UV-tolerance of plants can be enhanced or not. We have chosen the CPD photolyase for the following reasons: (i) DNA repair through photolyase is, in contrast to nucleotide excision repair, mediated by a single enzyme that does not require additional proteins or factors apart from the catalytic FAD and the 5,10-methenyl tetrahydrofolate antenna cofactor (Sancar 2003). Specifically, Arabidopsis CPD photolyase has been shown to fully complement photolyase-deficient E. coli cells (Ahmad et al. 1997). Thus, DNA photolyase does not depend on other plant-specific factors and allows a straightforward strategy to construct transgenic plants that express this enzyme under the control of a strong and constitutive promoter. (ii) The majority of UV-B photoproducts in DNA are CPDs, which are repaired specifically by this enzyme. (iii) Mutant studies have shown that accumulation of CPDs has an adverse effect on plant growth and health. (iv) CPD photolyase has already been shown to be a very effective enzyme for the removal of CPDs in the plant genome. (v) Repair by photolyase is a direct reversal of the damage and is therefore error-free. (vi) In contrast to excision repair photolyase uses light for catalysis and therefore operates without further energy costs.
The transgenic lines we have constructed and used here carry one (Ox4), two (Ox6), three (Ox2) and four (Ox5) copies of the T-DNA based on the Southern blot analysis shown in Fig. 1. The CaMV 35S promoter leads to a strong increase in At-PHR1 transcript levels in all transgenic lines that is about 250-fold (in Ox4 and Ox5), 300-fold (in Ox6) and 550-fold (in Ox2) higher than in wild type (Fig. 2a). However, we did not observe a clear correlation between the copy number and the expression levels of both mRNA and protein. For example line Ox5 with four T-DNA insertions has lower At-PHR1 transcript and protein levels than line Ox2 with three insertions. Likewise the protein levels of At-PHR1 analysed by immunoblot (Fig. 2b) do not perfectly reflect the transcript levels although some correlation does exist. For the wild type and the uvr2-1 mutant no signals were detected by immunoblot. This indicates that the amount of CPD photolyase in wild type is below the detection limit. The uvr2-1 mutant carries a 1 bp deletion in codon 28 resulting in a frameshift and premature termination (Ahmad et al. 1997). Thus, no full-length protein can be formed. This mutation also seems to have an adverse effect on the At-PHR1 transcript level since it is only about 1/3 of that from wild type (Fig. 2a). Closer examination of the overexpressor lines shows that Ox2 has the highest transcript and protein levels. The lower protein levels found for Ox4 and Ox6 correspond with lower transcript levels although Ox6 has more At-PHR1 transcripts than Ox4. The relatively high At-PHR1 protein level found in Ox5 compared to Ox4 and Ox6 is not consistent with its transcript level. We assume that the essentially non-existing correlation between copy number and expression levels could be caused by positioning effects on the chromosome or to homology-dependent gene silencing (Matzke and Matzke 1995). Since the amount of DNA photolyase protein in wild type plants was below the detection limit of immunoblot analysis, we cannot specify the fold-induction of photolyase protein levels in the overexpressor lines compared to wild type. Likewise, the induction rates given above for photolyase transcript levels most likely do not reflect the actual increase in photolyase protein levels due to possible instability of the protein or limited synthesis.
Our localisation studies demonstrate that the At-PHR1::GFP fusion protein is found exclusively in the nucleus (Fig. 3). Because GFP alone is evenly distributed in the cytosol and the nucleus but unlike At-PHR1::GFP excluded from the nucleolus, the observed localisation of the At-PHR1::GFP fusion protein is specifically mediated by At-PHR1. Since At-PHR1 does not contain a known or predicted nuclear localisation signal it remains unclear by which mechanism it enters the nucleus. If native At-PHR1 protein entered the nucleus by diffusion its affinity to DNA could retain and trap it there. Anacystis nidulans photolyase is active in human skin (Stege et al. 2000) suggesting that this enzyme can enter the nucleus although its prokaryotic origin most likely excludes the existence of a nuclear localisation signal. The At-PHR1::GFP fusion protein with a molecular mass of 84.1 kDa is almost certainly too large to diffuse passively through the nuclear pores. We therefore conclude that At-PHR1 is actively transported into the nucleus despite the lack of a consensus nuclear localisation signal. Our observation that At-PHR1 is not transported into chloroplasts is consistent with the data presented by Hada et al. (2000) who could not detect any photorepair in isolated chloroplasts. This is in contrast to a report by Draper and Hays (2000) where they describe a light and photolyase-dependent repair of DNA in chloroplasts and mitochondria suggesting access of this enzyme to the organelles. Considering our localisation data for At-PHR1 it is very unlikely that this photolyase is able to repair directly CPD lesions in organelles. We assume as a more likely effect of CPD photolyase efficient repair of the nuclear DNA and as a consequence of this sufficient expression of nuclear genes encoding proteins that are targeted to the organelles.
Under UV stress the photolyase-overexpressors showed a reduced accumulation of CPDs (Fig. 4). Irradiation conditions were used in these experiments that allowed simultaneous formation and repair of CPDs (UV-B given together with broad band UV-A). Thus, the measured CPD levels in the genomic DNA reflect the equilibrium between both processes and not the rate of repair in the various lines. The rate of repair could only be determined if the CPD substrate would be present in large excess, a scenario that we considered as very unlikely under natural conditions. The CPD photolyase overexpressor lines have better growth as can be seen by the higher fresh masses of the photolyase overexpressing lines in comparison to wild type plants (Fig. 5a). Thus, we conclude that increased photolyase levels can to some extent reduce the harmful effects of UV-B. Since the transgenic plants did not show any significant differences in growth from wild type when kept in the absence of UV-B, the increased photolyase levels do not seem to have any side effects on plant growth and development. The photolyase overexpressors showed strong reduction of growth under UV-B even though they accumulate much less CPDs than the wild type (Fig. 4). This is not unexpected since UV-B damages not only DNA, and CPDs are not the only UV-B photoproducts in DNA. (6-4) photoproducts probably have similar deleterious effects on replication and transcription as CPDs (Thoma 1999). Therefore, our approach to overexpress CPD photolyase, which can only enhance the repair of one specific UV-B photoproduct, cannot prevent all negative effects of UV-B on plant growth. Tanaka et al. (2002) described an Arabidopsis mutant, uvi1 that shows enhanced repair of CPDs and (6-4) photoproducts. Although the UV-B treatments and growth conditions these authors used are difficult to compare with our experimental setup, it seems that this mutant shows less reduction in growth than the CPD photolyase overexpressors analysed in our study. The data presented by Hidema et al. (2007) who overexpressed CPD photolyase in rice and our data presented here for Arabidopsis are consistent since in both cases the negative effects of UV-B on plant growth were diminished by elevated levels of CPD photolyase. Therefore, both studies together clearly demonstrate that even in distantly related species of higher plants that are model organisms for dicots (Arabidopsis) or monocots (rice) the level of endogenous photolyase is not sufficient to cope up with the harmful effects of increased UV-B.
Mammals have neither CPD photolyase nor (6-4) photolyase and thus depend on nucleotide excision repair to remove these UV lesions (Sancar 2003). Transgenic mice that express CPD photolyase, (6-4) photolyase, or both have strongly reduced stress responses and mutation induction in the UV-exposed skin through the more efficient repair of pyrimidine dimers (Schul et al. 2002; Jans et al. 2005) showing that the repair of these UV-lesions can be enhanced even in organisms that otherwise do not possess DNA photolyase. The differences we observed in UV-B induced growth inhibition between wild type and CPD photolyase overexpressing plants were significant. We assume that the much faster repair of CPDs in the photolyase overexpressors reduces the probability of mutation formation similar to the effect observed in mice. This could have major effects on the integrity of the genome when plants are exposed to elevated UV-B over several generations, and this topic is currently being investigated in our laboratory.
Abbreviations
- CPD:
-
Cyclobutane pyrimidine dimer
- DIG:
-
Digoxigenin
- PHR:
-
CPD photolyase
- UV-A:
-
Ultraviolet A
- UV-B:
-
Ultraviolet B
References
Ahmad M, Jarillo JA, Klimczak LJ, Landry LG, Peng T, Last RL, Cashmore AR (1997) An enzyme similar to animal type II photolyases mediates photoreactivation in Arabidopsis. Plant Cell 9:199–207
Anderson JG, Toohey DW, Brune WH (1991) Free radicals within the antarctic vortex: the role of CFCs in antarctic ozone loss. Science 251:39–46
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Ballaré CL, Scopel AL, Stapleton AE, Yanovsky MJ (1996) Solar ultraviolet-B radiation affects seedling emergence, DNA integrity, plant morphology, growth rate, and attractiveness to herbivore insects in Datura ferox. Plant Physiol 112:161–170
Ballaré CL, Scopel AL, Mazza C (1999) Effects of solar UV-B radiation on terrestrial ecosystems: case studies from South America. In: Rozema J (ed) Stratospheric ozone depletion: the effects of enhanced UV-B radiation on terrestrial ecosystems. Bachhuys Publishers, Leiden, pp 293–311
Barnes PW, Flint SD, Caldwell MM (1995) Early-season effects of supplemented solar UV-B radiation on seedling emergence, canopy structure, simulated stand photosynthesis and competition for light. Glob Chang Biol 1:43–53
Bjerrum JB, Schäfer-Nilsen C (1986) Buffer systems and transfer parameters for semidry electroblotting with a horizontal apparatus In: Dunn MJ (ed) Electrophoresis’86. VCH Weinheim, pp 315–327
Borneman JF, Teramura AH (1993) Effects of ultraviolet-B radiation on terrestrial plants. In: Young AR, Bjorn LO, Moan J, Nultsch W (eds) Environmental UV photobiology. Plenum Press, New York, pp 427–471
Britt AB, Chen JJ, Wykoff D, Mitchell D (1993) A UV-sensitive mutant of Arabidopsis defective in the repair of pyrimidine-pyrimidinone(6-4) dimers. Science 261:1571–1574
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Draper CK, Hays JB (2000) Replication of chloroplast, mitochondrial and nuclear DNA during growth of unirradiated and UVB-irradiated Arabidopsis leaves. Plant J 23:255–265
Emiliani G, Fondi M, Fani R, Gribaldo S (2009) A horizontal gene transfer at the origin of phenylpropanoid metabolism: a key adaptation of plants to land. Biol Direct 4:7
Fang RX, Nagy F, Sivasubramaniam S, Chua NH (1989) Multiple cis regulatory elements for maximal expression of the cauliflower mosaic virus 35S promoter in transgenic plants. Plant Cell 1:141–150
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger E (2006) DNA repair and mutagenesis. ASM Press, Washington, DC
Gallego F, Fleck O, Li A, Wyrzykowska J, Tinland B (2000) AtRAD1, a plant homologue of human and yeast nucleotide excision repair endonucleases, is involved in dark repair of UV damages and recombination. Plant J 21:507–518
Hada M, Hino K, Buchholz G, Goss J, Wellmann E, Shin M (2000) Assay of DNA photolyase activity in spinach leaves in relation to cell compartmentation-evidence for lack of DNA photolyase in chloroplasts. Biosci Biotechnol Biochem 64:1288–1291
Hidema J, Taguchi T, Ono T, Teranishi M, Yamamoto K, Kumagai T (2007) Increase in CPD photolyase activity functions effectively to prevent growth inhibition caused by UVB radiation. Plant J 50:70–79
Hollósy F (2002) Effects of ultraviolet radiation on plant cells. Micron 33:179–197
Iwamatsu Y, Aoki C, Takahashi M, Teranishi M, Ding Y, Sun C, Kumagai T, Hidema J (2008) UVB sensitivity and cyclobutane pyrimidine dimer (CPD) photolyase genotypes in cultivated and wild rice species. Photochem Photobiol Sci 7:311–320
Jans J, Schul W, Sert YG, Rijksen Y, Rebel H, Eker AP, Nakajima S, van Steeg H, de Gruijl FR, Yasui A, Hoeijmakers JH, van der Horst GT (2005) Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol 15:105–115
Jansen MAK, Gaba V, Greenberg BM (1998) Higher plants and UV-B radiation: balancing damage, repair and acclimation. Trends Plant Sci 3:131–135
Klar T, Kaiser G, Hennecke U, Carell T, Batschauer A, Essen LO (2006) Natural and non-natural antenna chromophores in the DNA photolyase from Thermus thermophilus. Chembiochem 7:1798–1806
Kleiner O, Butenandt J, Carell T, Batschauer A (1999) Class II DNA photolyase from Arabidopsis thaliana contains FAD as a cofactor. Eur J Biochem 264:161–167
Koncz C, Schell J (1986) The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204:383–396
Koncz C, Martini N, Mayerhofer R, Koncz-Kalman Z, Korber H, Redei GP, Schell J (1989) High-frequency T-DNA-mediated gene tagging in plants. Proc Natl Acad Sci USA 86:8467–8471
Kumagai T, Hidema J, Kang HS, Sato T (2001) Effects of supplemental UV-B radiation on the growth and yield of two cultivars of Japanese lowland rice (Oryza sativa L.) under the field in a cool rice-growing region of Japan. Agric Ecosyst Environ 83:201–208
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Landry LG, Stapleton AE, Lim J, Hoffman P, Hays JB, Walbot V, Last RL (1997) An Arabidopsis photolyase mutant is hypersensitive to ultraviolet-B radiation. Proc Natl Acad Sci USA 94:328–332
Liu Z, Hossain GS, Islas-Osuna MA, Mitchell DL, Mount DW (2000) Repair of UV damage in plants by nucleotide excision repair: Arabidopsis UVH1 DNA repair gene is a homolog of Saccharomyces cerevisiae Rad1. Plant J 21:519–528
Liu Z, Hall JD, Mount DW (2001) Arabidopsis UVH3 gene is a homolog of the Saccharomyces cerevisiae RAD2 and human XPG DNA repair genes. Plant J 26:329–338
Matzke MA, Matzke A (1995) How and why do plants inactivate homologous (trans)genes? Plant Physiol 107:679–685
McCloud ES, Berenbaum MR (1994) Stratospheric ozone depletion and plant-insect interactions: effects of UV-B radiation on foliage quality of Citrus jambhiri for Trichoplusia ni. J Chem Ecol 20:525–539
McKenzie R, Connor B, Bodeker G (1999) Increased summertime UV radiation in New Zealand in response to ozone loss. Science 285:1709–1711
Nakajima S, Sugiyama M, Iwai S, Hitomi K, Otoshi E, Kim ST, Jiang CZ, Todo T, Britt AB, Yamamoto K (1998) Cloning and characterization of a gene (UVR3) required for photorepair of 6–4 photoproducts in Arabidopsis thaliana. Nucleic Acids Res 26:638–644
Orth AB, Teramura AH, Sisler HD (1990) Effects of ultraviolet-B radiation on fungal disease development in Cucumber sativus. Am J Bot 77:1188–1192
Panagopoulos I, Bornman JF, Björn LO (1992) Response of sugar beet plants to ultraviolet-B (280–320 nm) radiation and Cercospora leaf spot disease. Physiol Plant 84:140–145
Pang Q, Hays JB (1991) UV-B-inducible and temperature-sensitive photoreactivation of cyclobutane pyrimidine dimers in Arabidopsis thaliana. Plant Physiol 95:536–543
Prinsze C, Dubbelman TM, Van Steveninck J (1990) Protein damage, induced by small amounts of photodynamically generated singlet oxygen or hydroxyl radicals. Biochim Biophys Acta 1038:152–157
Ries G, Heller W, Puchta H, Sandermann H, Seidlitz HK, Hohn B (2000) Elevated UV-B radiation reduces genome stability in plants. Nature 406:98–101
Rosenfeldt G, Muñoz Viana R, von Arnim AG, Mootz HD, Batschauer A (2008) Chemically induced and light-independent cryptochrome photoreceptor activation. Mol Plant 1:4–14
Rousseaux MC, Ballare CL, Giordano CV, Scopel AL, Zima AM, Szwarcberg-Bracchitta M, Searles PS, Caldwell MM, Diaz SB (1999) Ozone depletion and UVB radiation: impact on plant DNA damage in southern South America. Proc Natl Acad Sci USA 96:15310–15315
Sancar A (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev 103:2203–2237
Schul W, Jans J, Rijksen YM, Klemann KH, Eker AP, de Wit J, Nikaido O, Nakajima S, Yasui A, Hoeijmakers JH, van der Horst GT (2002) Enhanced repair of cyclobutane pyrimidine dimers and improved UV resistance in photolyase transgenic mice. EMBO J 21:4719–4729
Smirnoff N (1995) Antioxidant systems and plant response to the environment. In: Smirnoff N (ed) Environment and plant metabolism: flexibility and acclimation. Bios Scientific Publishers, Oxford, pp 217–243
Solomon S, Portmann RW, Thompson DW (2007) Contrasts between Antartic and Artic ozone depletion. Proc Natl Acad Sci USA 104:445–449
Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503–517
Stege H, Roza L, Vink AA, Grewe M, Ruzicka T, Grether-Beck S, Krutmann J (2000) Enzyme plus light therapy to repair DNA damage in ultraviolet-B-irradiated human skin. Proc Natl Acad Sci USA 97:1790–1795
Tanaka A, Sakamoto A, Ishigaki Y, Nikaido O, Sun G, Hase Y, Shikazono N, Tano S, Watanabe H (2002) An ultraviolet-B-resistant mutant with enhanced DNA repair in Arabidopsis. Plant Physiol 129:64–71
Taylor R, Tobin AK, Bray CM (1996) Nucleotide sequence of an Arabidopsis thaliana cDNA with high homology to the class II CPD photolyase present in higher eukaryotes. Plant Physiol 112:862
Teranishi M, Iwamatsu Y, Hidema J, Kumagai T (2004) Ultraviolet-B sensitivities in Japanese lowland rice cultivars: cyclobutane pyrimidine dimer photolyase activity and gene mutation. Plant Cell Physiol 45:1848–1856
Thoma F (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J 18:6585–6598
von Arnim AG, Deng XW, Stacey MG (1998) Cloning vectors for the expression of green fluorescent protein fusion proteins in transgenic plants. Gene 221:35–43
Vonarx EJ, Mitchell HL, Karthikeyan R, Chatterjee I, Kunz BA (1998) DNA repair in higher plants. Mutat Res 400:187–200
Acknowledgments
This work was supported in part by the DFG (grant BA 985/7-4), the Optodynamic Centre, University of Marburg, and by a fellowship given to O.K. by the State of Hessen. The provision of uvr2-1 seeds by the Arabidopsis Biological Resource Centre (Ohio State University, Columbus, OH, USA) is acknowledged. We thank Dr. Martin Schädler (Philipps-University Marburg, Germany) for statistical analyses, Dr. Csaba Koncz (Max-Planck-Institut für Züchtungsforschung, Cologne, Germany) for the binary vector pPCV812, the Agrobacterium strain GV3101 and the green mesophyll cell culture, Dr. Albrecht von Arnim (University of Tennessee, Knoxville, USA) for the pAVA 393 vector, Oxana Panajotowa and Elvira Stumpf for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kaiser, G., Kleiner, O., Beisswenger, C. et al. Increased DNA repair in Arabidopsis plants overexpressing CPD photolyase. Planta 230, 505–515 (2009). https://doi.org/10.1007/s00425-009-0962-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-009-0962-y