
Abstract
Cyanidioschyzon merolae is considered as a suitable model system for studies of organelle differentiation, proliferation and partitioning. Here, we have identified and characterized vacuoles in this organism and examined the partitioning of vacuoles using fluorescence and electron microscopy. Vacuoles were stained with the fluorescent aminopeptidase substrate 7-amino-4-chloromethylcoumarin l-arginine amide, acidotrophic dyes quinacrine and LysoTracker, and 4′,6-diamidino-2-phenyl indole, which, at a high concentration, stains polyphosphate. Vacuoles have been shown to be approximately 500 nm in diameter with a mean of around five per interphase cell. The vacuolar H+-ATPase inhibitor concanamycin A blocked the accumulation of quinacrine in the vacuoles, suggesting the presence of the enzyme on these membranes. Electron microscopy revealed that the vacuoles were single membrane-bound organelles with an electron-dense substance, often containing a thick layer surrounding the membrane. Immunoelectron microscopy using an anti-vacuolar-H+-pyrophosphatase antibody revealed the presence of the enzyme on these membranes. In interphase cells, vacuoles were distributed in the cytoplasm, while in mitotic cells they were localized adjacent to the mitochondria. Filamentous structures were observed between vacuoles and mitochondria. Vacuoles were distributed almost evenly to daughter cells and redistributed in the cytoplasm after cytokinesis. The change in localization of vacuoles also happened in microtubule-disrupted cells. Since no actin protein or filaments have been detected in C. merolae, this result suggests an intrinsic mechanism for the movement of vacuoles that differs from commonly known mechanisms mediated by microtubules and actin filaments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primitive red alga Cyanidioschyzon merolae is a small organism (2 μm in diameter) that lives in sulfate-rich acidic hot springs (pH 2.5, 42°C). C. merolae is an excellent model system to study the fundamental features of eukaryotic cells, especially with regard to organelle dynamics. The C. merolae cell does not have a cell wall and contains only one nucleus, one mitochondrion, and one chloroplast, as well as only one microbody, one Golgi apparatus with two cisternae, and one layer of endoplasmic reticulum, division of which can be highly synchronized by light/dark cycles (Kuroiwa et al. 1994, 1998; Suzuki et al. 1994; Miyagishima et al. 1999). In contrast, a typical eukaryotic cell contains many double membrane-bound and single membrane-bound organelles, division of which occurs at random and cannot be synchronized. In addition, the shape of the organelles is very diverse and complicated (Kuroiwa 1998). C. merolae is also a eukaryote with one of the smallest (16.5 Mb) and simplest genomes (Matsuzaki et al. 2004). Since most of the genes are present in low copy numbers and lack introns, this would facilitate proteome and transcriptome analyses. Although these features make C. merolae an ideal organism for elucidating the function, biosynthesis, and multiplication of organelles in eukaryotic cells, organelles functionally equivalent to vacuoles have not been well characterized.
The plant cell vacuoles are single membrane-bound organelles, which share several properties with lysosomes of animal cells and fungal vacuoles (Marty 1999). Plant cell vacuoles were discovered earlier under microscopy and were originally defined as a cell space empty of cytoplasmic matter. Most cell space in developed organs of higher plants is occupied with vacuoles. In contrast, typical mammalian lysosomes are small organelles about ∼500 nm in diameter (Holtzman 1989). Today increasing biochemical and molecular evidence suggests that vacuoles and lysosomes belong to a common group. Both exhibit an acidic luminal pH, which is maintained by proton pumps such as vacuolar H+-ATPase (V-ATPase) (Holtzman 1989). They are rich in hydrolytic enzymes and are responsible for intracellular degradation (Matile 1969). Vacuoles also function as reservoirs for ions and metabolites and are involved in cellular responses to environmental and biotic factors that provoke stresses. Furthermore, several proteins required for vacuolar protein transport in Saccharomyces cerevisiae seem to be conserved in higher plants and mammals (Sanderfoot and Raikhel 1999; Bethke and Jones 2000; Winter and Hauser 2006) and some of these proteins have been demonstrated to be involved in vacuolar or lysosomal assembly in those organisms (Babst 2005; Rojo et al. 2003; Suzuki et al. 2003). The presence of vacuoles in C. merolae cells is implied by the fact that the genome contains genes encoding vacuolar H+-pyrophosphatase (V-PPase), a full set of V-ATPases, as well as proteases and phosphatases (Misumi et al. 2005). It is not extensively discussed, however, vacuoles and lysosomes seems to have an evolutionary relationship as they have several enzymes in common and require homologous proteins for their assembly.
Red algae are one of the main photosynthetic eukaryotic lineages, as recent studies indicate their basal phylogenetic position within the eukaryotes (Nozaki et al. 2003). Therefore, characterizing vacuoles of C. merolae is expected to provide unique tools to address problems regarding origin, evolution, and fundamental features such as biogenesis and propagation of vacuoles and lysosomes. In this study, we used fluorescence microscopy and electron microscopy to identify and characterize the vacuoles in C. merolae. In addition we studied changes in number and localization of vacuoles throughout the cell cycle and our data suggest the presence of certain mechanisms that ensure partitioning of vacuoles.
Materials and methods
Cell culture and drug treatment
Cyanidioschyzon merolae 10D-14 strain (Toda et al. 1998) were maintained in swirling conical flasks in 2×Allen’s media (Allen 1959) at pH 2.5 and 42°C under continuous light. To make phosphate-depleted medium, 4 mM KH2PO4 was removed from 2×Allen’s media and instead, 4 mM KOH was added and pH was adjusted to 2.5. For synchronization, cells were diluted to an OD444 of 1.0 and cultured under a 12-h light/12-h dark cycle with vigorous aeration (Suzuki et al. 1994). Cells synchronized in the second dark period were used. The commencement of lateral chloroplast elongation was considered as the start of mitosis (Suzuki et al. 1994). For concanamycin A treatment, 1 mM concanamycin A (Wako Pure Chemical Industries, Ltd, Osaka, Japan) dissolved in DMSO was added to the culture in a final concentration of 1 μM and incubated for 5 h. For oryzalin treatment, 40 μM oryzalin (Wako Pure Chemical Industries) was added 2 h before the start of mitosis and aeration was changed to gentle agitation.
Fluorescence microscopy
Images were viewed using an epifluorescence microscope (BX51; Olympus, Tokyo, Japan) with a 3CCD digital camera (C7780; Hamamatsu Photonics, Hamamatsu, Japan) under ultraviolet excitation [for 4′,6-diamidino-2-phenylindole (DAPI), 7-amino-4-chloromethylcoumarin l-arginine amide (CMAC-Arg) and 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate (TMA-DPH)], blue excitation (for quinacrine and Alexa 488) or green excitation (for chloroplast autofluorescence, LysoTracker Red DND-99, and FM4-64). To observe DNA, cells were fixed for 1 min with 1% (w/v) glutaraldehyde in TAN buffer (20 mM Tris–HCl, 0.5 mM EDTA, 1.2 mM spermidine) and stained with 1 μg/ml DAPI for 1 min. To observe the yellow fluorescence of DAPI, cells were fixed for 5 min with 1% (w/v) glutaraldehyde, rinsed in distilled water and then stained with 3 μg/ml DAPI for 30 min at 37°C. For CMAC-Arg staining, 10 μM digitonin was added to the culture and incubated for 15 min. Cells were then fixed for 5 min with 1% (w/v) glutaraldehyde, rinsed in 2×Allen’s media and incubated with 100 μM CMAC-Arg (Molecular Probes, Eugene, OR) for 15 min at room temperature. For staining with quinacrine and LysoTracker Red DND-99, 30 mM Tris–HCl (pH 8.8), 10 μM digitonin and 40 μg/ml quinacrine or 1 mM LysoTracker Red DND-99 (Molecular Probes) were added to the culture and incubated for 15 min at room temperature. Cells were collected by weak centrifugation, resuspended in fresh media and incubated for 1 h at 37°C. For staining with TMA-DPH and FM4-64, 1 μg/ml TMA-DPH (Molecular Probes) or 1 μg/ml FM4-64 (Molecular Probes) was added to the culture. Immunofluorescence microscopy was performed as described previously (Nishida et al. 2005). Cells were fixed in 90% (v/v) methanol at −20°C for 5 min. Mouse anti-α-tubulin antibody (Molecular Probes) at a dilution of 1:100 was used as the primary antibody and goat anti-mouse IgG conjugated with Alexa 488 (Molecular Probes) at a dilution of 1:1,000 was used as the secondary antibody.
Electron microscopy
Transmission electron microscopy was performed as described previously (Miyagishima et al. 1999). Briefly, cell pellets were rapidly frozen in liquid propane cooled with liquid nitrogen, transferred to dried acetone containing 1% osmium tetraoxide at −80°C then embedded in Spurr’s resin (Polyscience Inc, Warrington, PA, USA). Thin sections (90 nm thick) were stained with uranyl acetate and lead citrate, and examined with an electron microscope (JEM-1200EX; JEOL, Tokyo, Japan). For immunoelectron microscopy, cells were rapidly frozen in liquid propane and fixed with dried acetone at −80°C (Miyagishima et al. 1999). Rabbit antibody against peptide (DVGADLVGKVE) of mung bean vacuolar H+-pyrophosphatase (V-PPase) (Takasu et al. 1997) was used for labeling; underlined amino acids represent conserved areas in the C. merolae sequence (GenBank accession number AB231859). Thin sections (90 nm thick) were labeled with the antibody at a dilution of 1:80 and then with 15-nm gold-conjugate goat anti-rabbit antibody, and stained with uranyl acetate. Cells were also fixed chemically with 0.5% glutaraldehyde in 1×Allen’s media buffered in 10 mM sodium cacodylate (pH 7.2) for 2 h at room temperature and 12 h at 4°C, and rinsed in the same buffer. Fixed cells were dehydrated in a graded ethanol series followed by propylene oxide and embedded in LR White resin (London Resin Company, London, UK). Thin sections were labeled with the antibody against V-PPase diluted 1:20 then with 10-nm gold-conjugate goat anti-rabbit antibody, and stained with uranyl acetate. Densities of labeling by antibodies against V-PPase were calculated as described previously (Tanaka et al. 1996).
Results
Identification and characterization of vacuoles
While C. merolae cells contain one nucleus, one mitochondrion, and one chloroplast, the mitochondrion is closely adjacent to the chloroplast (Fig. 1a). To visualize vacuoles, a range of fluorescent probes was used (Table 1). CMAC-Arg, quinacrine and a high concentration of DAPI presumably stained identical round-shaped organelles, which were about 430–550 nm in diameter (Table 1, Fig. 1b–d). These organelles were distributed in the cytoplasm during interphase and close to mitochondria in mitotic cells. A high concentration of DAPI stains polyphosphate, which is known to accumulate in vacuoles in various organisms. While DAPI-DNA fluorescence is blue-white with an emission maximum of 456 nm, DAPI-polyphosphate fluorescence is bright yellow with a fluorescence maximum of 525 nm (Tijssen et al. 1982). The yellow fluorescence of DAPI disappeared in the cells cultured in phosphate (Pi)-depleted medium (Fig. 1e) and became rapidly visible after readdition of phosphate (Fig. 1f), indicating that the yellow fluorescence originated from polyphosphate likely contained in vacuoles. LysoTracker is thought to be incorporated into membranes of acidic organelles according to the manufacturer’s protocol (Molecular Probes). Simultaneous staining with DAPI revealed LysoTracker signal surrounding the DAPI signal (Fig. 1g), suggesting that the membrane and matrix of the round structures were stained with LysoTracker and DAPI. These results suggest that these round organelles are lytic and acidic compartments containing polyphosphate. In general, vacuolar H+-ATPase (V-ATPase) is abundant in vacuolar membranes and is responsible for the maintenance of the proton gradient across the membranes to keep the inside acidic. To study whether the acidity of the round organelles depends on V-ATPase, cells were treated with concanamycin A, a specific inhibitor of V-ATPase (Fig. 2a, b). In the presence of the drug, quinacrine did not accumulate in these organelles (Fig. 2b), suggesting that V-ATPase is responsible for their acidification. Taken together, the organelles were designated as vacuoles.

Visualization of vacuoles by fluorescence microscopy. a A phase contrast-fluorescence microscopy image showing interphase (left) and mitotic cells (right) after DAPI staining. The red fluorescence is autofluorescence of chloroplasts. b Cells stained with CMAC-Arg and c cells stained with quinacrine. d–f Cells stained with a high concentration of DAPI: cells grown in 2×Allen’s medium (d); in Pi-depleted medium for 3 weeks (e) or cultured in Pi-containing medium for 14 h after culture in Pi-depleted medium for 3 weeks (f). g A cell stained with LysoTracker Red DND-99 and a high concentration of DAPI. PC Phase contrast, nu nucleus, mt mitochondrion, cp chloroplast. Bars 2 μm (a–c, f), 1 μm (g)

The effect of concanamycin A treatment on quinacrine accumulation. Cells were treated with DMSO (control a) or concanamycin A (b) for 5 h and subsequently stained with quinacrine. The autofluorescence of chloroplasts was subtracted from the images using Photoshop 6.0 (Adobe systems, Mountain View, CA, USA). PC Phase contrast. Bar 2 μm
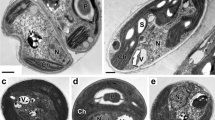
In order to reveal the detailed morphology of the vacuoles, cells were fixed using the rapid-freeze substitution method, embedded in Spurr’s resin, and observed by transmission electron microscopy. As previously reported, organelles such as nuclei, mitochondria, chloroplasts, and microbodies were easily identified (Fig. 3a, b, Kuroiwa et al. 1994, 1998; Miyagishima et al. 1999). Beside these known structures, we observed single membrane-bound organelles containing variable amounts of electron-dense entities (Fig. 3a–g), which we classified into two types. The first type had no visible specific structures around the membrane (Fig. 3c–e), whereas the second type had a layered structure surrounding the membrane (Fig. 3f–g). The first type and the second type of organelles were 325 ± 103 and 316 ± 81 nm in diameter, respectively. In mitotic cells, both types of organelles were observed close to the mitochondria (See below, Fig. 6a–i). We also embedded cells in LR White resin and performed immunoelectron microscopy using IgG against vacuolar H+-pyrophosphatase (V-PPase) (Fig. 3h–l, Table 2), which has been shown to be an intrinsic-vacuolar membrane-localized pump in plant cells (Maeshima 2000). Signals of PPase were detected around organelles, which were recognized as single membrane-bound organelles by their size and electron-dense inclusion. These results suggest that the observed single membrane-bound organelles are vacuoles.

Identification of vacuoles by electron microscopy. a, b Typical electron microscopic images of interphase cells. c–g Typical images of round single membrane-bound organelles. c, d Magnified images of (a) and (b). Organelles without a layer (c–e); and with a thick layer around the membrane (f, g). h–l Immunoelectron micrographs of cells stained with anti-V-PPase antibody. i Magnified image of (h). Cells were fixed with acetone (h–j) or glutaraldehyde (k, l). nu Nucleus, mt mitochondrion, cp chloroplast; mb microbody; st starch, arrowheads round single membrane-bound organelles. Bars 1 μm (b, h), 200 nm (g), 500 nm (l)
Proliferation and distribution of vacuoles
The number of organelles per cell should be controled to fulfill their functions. We also studied the proliferation and behavior of vacuoles during the cell cycle. To study the relationship between proliferation of vacuoles and cell cycle, after the synchronization of cell division, the number of vacuoles stained with quinacrine or DAPI was examined (Fig. 4). Synchronized cell division was observed from 35 to 37 h after the start of incubation. In C. merolae, the chloroplast, mitochondrion, microbody, and nucleus divide in this order before cytokinesis (Suzuki et al. 1994). Based on morphology, we sorted cells into six cell-cycle stages: interphase cells just before mitosis, cells with an elongated chloroplast, cells with a constricted chloroplast, cells after chloroplast division, cells at cytokinesis, and interphase cells after cytokinesis. Cells with an elongated chloroplast had a higher number of vacuoles (6.10 ± 1.73 and 6.05 ± 1.50 with quinacrine and DAPI, respectively) than interphase cells just before mitosis (4.77 ± 1.53 and 4.67 ± 1.29; P < 0.0001, Student’s t test). Alternatively, no significant difference was observed between the number of vacuoles in cells with a constricted chloroplast, in cells after chloroplast division, and in cells at cytokinesis. The number reduced by half in interphase cells just after mitosis.

Changes in the number of vacuoles per cell throughout the cell cycle. The number of vacuoles per cell visualized with quinacrine or DAPI was counted at different stages of the cell cycle. 1 Interphase cells just before mitosis; 2 cells with an elongated chloroplast; 3 cells with a constricted chloroplast; 4 cells after chloroplast division; 5 cells at cytokinesis; 6 interphase cells after cytokinesis. nu Nucleus, mt mitochondrion; cp chloroplast
As shown above, C. merolae vacuoles are distributed in the cytoplasm during interphase and localized close to the mitochondria in mitotic cells (Fig. 1b, c). This raised the possibility that certain mechanisms exist for proper partitioning of vacuoles. To ascertain details of the behavior of vacuoles, subcellular localization of vacuoles was studied using synchronized cells. In this experiment, we designated the nuclear side of the cell as upside, and the chloroplast side as downside. DAPI was used to observe the vacuoles. In the interphase, the vacuoles were distributed throughout the cytoplasm, mainly in the upper part of the cell (Fig. 5a). In cells with an elongated chloroplast (Fig. 5b, c), about half of the observed vacuoles were localized over the chloroplast, i.e. over the mitochondria. In cells in which the chloroplast had started to constrict and in cells with divided chloroplasts, about 80 and 90% of the observed vacuoles were located over the mitochondria, respectively (Fig. 5d, e). In cells at cytokinesis, the vacuoles were located close to the mitochondria and often situated near the division plane (Fig. 5f). In this stage, vacuoles were also observed in the upper part of the cytoplasm close to the division plane. To study how evenly the vacuoles were distributed to the daughter cells, we examined the difference in the number of vacuoles between the prospective daughter cells using cells in cytokinesis (Table 3). Thirty-four percent of cells in cytokinesis had the same number of vacuoles between the daughter cells and in 53% of cells; one daughter cell had one more vacuole than the other. This result shows that vacuoles are almost evenly distributed among daughter cells.

Changes in localization of vacuoles throughout the cell cycle visualized with DAPI. A cell in interphase (a); with an elongating chloroplast (b); with an elongated chloroplast (c); with a constricted chloroplast (d); with a divided chloroplasts (e); and at cytokinesis (f). PC phase contrast; CP autofluorescence of chloroplast. Bar 2 μm
The behavior of vacuoles was further examined by electron microscopy. Table 4 shows the percentage of each type of vacuole of the total number of observed vacuoles in each stage of the cell cycle stage. Vacuoles with or without a surrounding layer were both observed regardless of cell cycle. The percentage of vacuoles with a layer was slightly larger in cells with divided chloroplasts and cells in cytokinesis than in interphase cells and cells with an elongated chloroplast. In mitotic cells, both types of vacuoles were in close proximity to mitochondria, while in cells with an elongated chloroplast, the vacuoles were observed on the mitochondria and in other parts of the cytoplasm (Fig. 6a). In cells with constricting chloroplasts (Fig. 6b–d), cells with divided chloroplasts (Fig. 6g), and cells at cytokinesis (Fig. 6i), vacuoles were observed on top of mitochondria, while the microbodies were localized at the site of mitochondrial division (Miyagishima et al. 1999). In cells at cytokinesis, vacuoles were also observed in the cytoplasm close to the division plane. We found filamentous structures between vacuoles and mitochondria (Fig. 6d–h). In some peripheral regions of the mitochondria, a portion of the mitochondrial membrane was stretched and was in direct contact with the vacuoles (Figs. 6g, h, 7a–f).

Vertical sections of mitotic cells observed with electron microscopy. a A cell with an elongated chloroplast. b–d Cells with dividing chloroplasts. e, f Magnified images of (d). g A cell with divided chloroplasts. h Magnified image of (g). i A cell in cytokinesis. nu Nucleus, mt mitochondrion, cp chloroplast, mb microbody, st starch, arrowheads vacuoles, small arrows the filamentous structures, large arrow the stretched mitochondrial membrane. Bar 1 μm (i), 200 nm (f, h)

Cross sections of a single mitotic cell observed with electron microscopy. The vacuoles (arrowheads) were observed in the periphery of the mitochondrion, constricted by the mitochondrial dividing ring (double arrowheads) (a–f). b, d Magnified images of (a) and (c). mt Mitochondrion, cp chloroplast, arrows the contact sites of the mitochondrion and the vacuoles. Bars 1 μm (f), 200 nm (b, d)
Localization of the vacuoles in oryzalin-treated cells
Among components of cytoskeleton, only microtubules are observed in C. merolae (Matsuzaki et al. 2004; Nishida et al. 2005). To examine whether the change in localization of vacuoles during mitosis was dependent on mitotic spindles, we disrupted microtubules with oryzalin, an inhibitor of tubulin polymerization. When oryzalin was added to the synchronized culture 2 h before commencement of mitosis, cell-cycle progression was arrested after mitochondrial division, as described by Nishida et al. (2005), suggesting that the drug effectively disrupted microtubules. We also performed immunofluorescence microscopy using antibodies against α-tubulin. Microtubule filaments were observed in control cells (Fig. 8a), while no filaments could be observed in the oryzalin-treated cells (Fig. 8b). Vacuoles were also observed over the chloroplasts, i.e. over the mitochondria, in oryzalin-treated mitotic cells (Fig. 8c).

Localization of vacuoles in oryzalin-treated cells. Oryzalin was added 2 h before the start of mitosis. Immunofluorescence images of α-tubulin in control (a) and oryzalin-treated cells (b). c Oryzalin-treated mitotic cells stained with a high concentration of DAPI. PC Phase contrast, CP autofluorescence of the chloroplast. Bars 2 μm
Discussion
Identification and characterization of vacuoles
In this paper, we have identified and characterized vacuoles in C. merolae. They possess fundamental characteristics of typical vacuoles, in that they are single membrane-bound, lytic and acidic organelles. Vacuoles visualized with the blue fluorescence of CMAC were about 430 nm in diameter on average, whereas those visualized with yellow fluorescence of quinacrine and DAPI were about 550 and 530 nm, respectively (Table 1). This is not inconsistent, since a longer wavelength gives objects nonnegligibly larger appearance due to limits of resolution when the objects are as small as the light wavelength. Alternatively, fixation might cause the difference in image size (see Materials and methods). Each vacuole contained polyphosphate as shown by dual labeling with DAPI and LysoTracker (Fig. 1). We also showed drastic changes in the amount of polyphosphate in response to the outer phosphate concentration, implying that polyphosphate acts as a reservoir of phosphate. Several microorganisms such as S. cerevisiae (Vorisek et al. 1982), Neurospora crassa (Cramer et al. 1980), the green alga Chlorella pyrenoidosa (Peverly et al. 1978), and Dunaliella salina (Pick and Weiss 1991) accumulate polyphosphate in vacuoles. Incorporation of orthophosphate into polyphosphate was also reported in isolated lysosomes of human fibroblasts (Pisoni and Lindley 1992). Polyphosphate performs numerous and various biological functions. Among these, they function as a reservoir of energy and phosphate, a chelator of metals, and a regulator of cytoplasmic pH and osmolarity (Pick and Weiss 1991; Weiss et al. 1991; Kornberg 1995; Shirahama et al. 1996; Kulaev et al. 2004). In considering our results, accumulation of polyphosphate into vacuoles seems likely to occur in the early stages of eukaryotic evolution and have various biological functions in diverse organisms. By treatment with concanamycin A, a V-ATPase inhibitor, the vacuoles abolished their acidic state, suggesting that V-ATPase is responsible for acidity of the vacuoles, as is the case in many other organisms (Fig. 2). In S. cerevisiae, suppression of V-ATPase activity is known to have a pleiotropic effect on the vacuolar system (Nelson et al. 2000). Indeed, concanamycin A-treated cells stopped growing and died within 24 h (data not shown), suggesting that the active vacuolar system is vital for C. merolae.
When observed by electron microscopy, the average size of vacuoles was about 300 nm in diameter. The size could have been underestimated as not all vacuoles were sectioned in the median plane. The vacuoles had an electron-dense substance (Fig. 3). In many organisms, electron-dense contents are observed in vacuoles or lysosomes (Holtzmann 1989). In mammalian cells, lysosomes have a heterogeneous content, owing to differences in function and substrate (Holtzmann 1989). We also observed variations in amount and density of the electron-dense contents. Such variation might reflect different properties of the vacuoles. Alternatively, apparent differences in electron-dense substances might be partly due to instability of the contents during fixation. We classified the vacuoles into two types based on their outer structure (Fig. 3). One type of vacuole might turn into the other type of vacuole as vacuoles with a slight outer layer, which are supposed to be at an intermediate state, were also observed. Immunofluorescence microscopy revealed the localization of V-PPase on the vacuoles (Fig. 3, Table 2). Presence of V-PPase and V-ATPases in C. merolae vacuole strongly supports that the observed vacuoles are true vacuoles, although the term “vacuoles” is occasionally used for quite different organelles (Holtzman 1989).
Proliferation and distribution of the vacuoles
We investigated as to when vacuoles of C. merolae proliferate. Mitotic cells had a higher number of vacuoles than interphase cells, whereas no significant increase in number was detected in the progression of mitosis (Fig. 4). This indicated that proliferation of vacuoles occurs preferentially in the interphase. Warren and Wickner (1996) reviewed the two inheritance strategies used by organelles; stochastic inheritance and ordered inheritance. Stochastic inheritance is based on the law of probability, while in ordered inheritance, organelles are inherited using certain machineries. It has been reported with mammalian Madin-Darby canine kidney (MDCK) cells that lysosomes are dispersed throughout the cytoplasm in the early prophase, but in a juxtanuclear position just after cytokinesis (Bergeland et al. 2001). In tobacco BY-2 cells, which contain large developed vacuoles, it has been reported that the central part of the vacuole becomes tubular and finally segregated into two smaller vacuoles, each of which is inherited by daughter cells (Kutsuna et al. 2003). In S. cerevisiae, the vacuolar membrane extends into the emerging bud via a tubular and vesicular structure, which then fuses into a new vacuole in the daughter cell (Catlett and Weisman 2000). These observations indicate that certain mechanisms exist to actively control vacuole/lysosome partitioning. In C. merolae, vacuoles changed their localization dynamically and partitioned almost evenly (Figs. 5, 6, Table 3). The change in localization did not seem to be directly related to the outer structure of the vacuoles since both types of vacuoles were observed throughout the cell cycle. The percentage of vacuoles with a layer increased and vacuoles without a layer decreased in the later stages of the cell cycle (Table 4). Although the physiological meaning of the difference in outer structure is unknown, this change might be caused by a variation in metabolism during the cell cycle. We propose the following model of vacuolar partitioning in the cell cycle of C. merolae (Fig. 9a–g). During interphase, the vacuoles are distributed throughout the cytoplasm, where proliferation occurs (Fig. 9a). When the chloroplast starts to elongate, the vacuoles begin to move to the upper side of the mitochondrion whereas the microbody localizes at the center of the mitochondrion (Fig. 9b). Most vacuoles are located in close proximity to the mitochondrion (Fig. 9c). After division of the chloroplast, the mitochondrion, nucleus and microbody divide, in this particular order. The vacuoles are distributed almost evenly to either side of the cell as the division and partitioning of organelles proceeds (Fig. 9d). During cytokinesis, the vacuoles begin to move close to the division plane of the cell, where some start to leave the vicinity of mitochondria (Fig. 9e). After cytokinesis, all vacuoles are redistributed in the cytoplasm via the former division plane (Fig. 9f). We speculated that the vacuoles are bound to the mitochondria and distributed together with them. In some peripheral region of the mitochondria, the mitochondrial membrane bulges outward and moves into direct contact with the vacuoles (Figs. 6, 7). Thus, at least several vacuoles seem to bind to the mitochondria in a positive manner. Filamentous structures were observed between vacuoles and mitochondria (Fig. 6) and might be involved in the binding of the two organelles (Fig. 9g).

Schematic model of the partitioning of vacuoles during cell cycle. During interphase, the vacuoles are distributed in the cytoplasm (a). When the chloroplast starts to elongate, vacuoles and the microbody begin to move to the upper side of the mitochondrion (b). Most of the vacuoles are located in close proximity to the mitochondrion (c). Vacuoles are mostly distributed evenly to either side of the cell as the division and partitioning of organelles proceeds (d). During cytokinesis, vacuoles begin to move close to the division plane of the cell and leave the vicinity of mitochondria (e). After cytokinesis, all vacuoles are redistributed in the cytoplasm via the former division plane (f). Vacuoles and mitochondria are speculated to be bound with specific structures (g)
Localization of vacuoles in oryzalin-treated cells
What triggers this cell cycle-dependent change in localization and how vacuoles move and attach to the mitochondria remains to be determined. Subcellular localization of vacuoles in interphase does not depend on microtubules since α-tubulin is expressed only in cells just before and during mitosis (Nishida et al. 2005). The spindle does not seem necessary in the movement over the mitochondria since the vacuoles changed their localization in the presence of oryzalin (Fig. 8). This result is consistent with the partitioning of mammalian lysosomes, which are excluded from and independent of the mitotic spindle (Dunster et al. 2002). Actin filaments and class V myosin Myo2p are needed for the inheritance of vacuoles in S. cerevisiae (Hill et al. 1996; Catlett and Weisman 2000). C. merolae genome codes for one actin and six actin-related proteins, but there are no proteins that appear to contain the motor domain of dynein or myosin (Matsuzaki et al. 2004). Although the transcript of actin gene has been detected by Northern hybridization (Takahashi et al. 1995), actin protein or filaments were not detected by immunoblot analysis or by staining with rhodamine-conjugated phalloidin or anti-actin antibodies (Kuroiwa 1998). Further, cytochalasin B, an actin depolymerization drug, did not arrest the cell cycle of C. merolae (Kuroiwa 1998). We also used latrunculin B and cytochalasin D, other inhibitors of actin polymerization, and observed no significant effect on the cell cycle, viability, or the relocation of vacuoles (data not shown). Therefore C. merolae is considered to be lacking typical actin filaments and the partitioning of vacuoles is likely to involve other, so far undiscovered, mechanisms.
In conclusion, the present study confirmed the existence of vacuoles in C. merolae. Our results show that vacuoles are distributed to each daughter cell in a specific manner. Since vacuoles of C. merolae are low in number per cell, have a distinct shape, and are easily detected by several dyes, they are thought to provide an excellent system to further understand the specialization, proliferation and distribution of vacuoles and lysosomes.
Abbreviations
- CMAC-Arg:
-
7-Amino-4-chloromethylcoumarin l-arginine amide
- DAPI:
-
4′, 6-Diamidino-2-phenylindole
- Pi:
-
Phosphate
- TMA-DPH:
-
1-(4-Trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate
- V-ATPase:
-
Vacuolar H+-ATPase
- V-PPase:
-
Vacuolar H+-pyrophosphatase
References
Allen MB (1959) Studies with Cyanidium caldarium, an anomalously pigmented Chlorophyta. Arch Microbiol 32:270–277
Allan RA, Miller JJ (1980) Influence of S-adenosylmethionine on DAPI-induced fluorescence of polyphosphate in the yeast vacuole. Can J Microbiol 26:912–920
Babst M (2005) A protein’s final ESCRT. Traffic 6:2–9
Bergeland T, Widerberg J, Bakke O, Nordeng TW (2001) Mitotic partitioning of endosomes and lysosomes. Curr Biol 11:644–651
Bethke PC, Jones RL (2000) Vacuoles and prevacuolar compartments. Curr Opin Plant Biol 3:469–475
Bolte S, Talbot C, Boutte Y, Catrice O, Read ND, Satiat-Jeunemaitre B (2004) FM-dyes as experimental probes for dissecting vesicle trafficking in living plant cells. J Microsc 214:159–173
Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell 11:467–480
Catlett NL, Weisman LS (2000) Divide and multiply: organelle partitioning in yeast. Curr Opin Cell Biol 12:509–516
Cramer CL, Vaught LE, Davis RH (1980) Basic amino acids and inorganic polyphosphates in Neurospora crassa vacuoles: independent regulation of vacuolar pools. J Bacteriol 142:945–952
Dunster K, Toh BH, Sentry JW (2002) Early endosomes, late endosomes, and lysosomes display distinct partitioning strategies of inheritance with similarities to Golgi-derived membranes. Eur J Cell Biol 81:117–124
Hill KL, Catlett NL, Weisman LS (1996) Actin and myosin function in directed vacuole movement during cell division in Saccharomyces cerevisiae. J Cell Biol 135:1535–1549
Holtzman E (1989) Lysosomes. Plenum Press, New York
Illinger D, Poindron P, Fonteneau P, Modollel M, Kuhry JG (1990) Internalization of the lipophilic fluorescent probe trimethylamino-diphenylhexatriene follows the endocytosis and recycling of the plasma membrane in cells. Biochim Biophys Acta 1030:73–81
Kornberg A (1995) Inorganic polyphosphate: toward making a forgotten polymer unforgettable. J Bacteriol 177:491–496
Kruckeberg AL, Ye L, Berden JA, van Dam K (1999) Functional expression, quantification and cellular localization of the Hxt2 hexose transporter of Saccharomyces cerevisiae tagged with the green fluorescent protein. Biochem J 339:299–307
Kulaev IS, Vagabov VM, Kulakovskaya TV (2004) The biochemistry of inorganic polyphosphates, 2nd edn. Wily, West Sussex
Kuroiwa T (1998) The primitive red algae: Cyanidium caldarium and Cyanidioschyzon merolae as model system for investigating the dividing apparatus of mitochondria and plastids. BioEssays 20:344–354
Kuroiwa T, Kawazu T, Takahashi H, Suzuki K, Ohta N, Kuroiwa H (1994) Comparison of ultrastructures between the ultra-small eukaryote Cyanidioschyzon merolae and Cyanidium caldarium. Cytologia 59:149–158
Kutsuna N, Kumagai F, Sato MH, Hasezawa S (2003) Three-dimensional reconstruction of tubular structure of vacuolar membrane throughout mitosis in living tobacco cells. Plant Cell Physiol 44:1045–1054
Maeshima M (2000) Vacuolar H+-pyrophosphatase. Biochem Biophys Acta 1465: 37–51
Marty F (1999) Plant vacuoles. Plant Cell 11:587–599
Matile P (1969) Vacuoles as lysosomes of plant cells. Biochem J 111:26–27
Matsuzaki M, Misumi O, Shin-I T, Maruyama S, Takahara M, Miyagishima S, Mori T, Nishida K, Yagisawa F, Nishida K, Yoshida Y, Nishimura Y, Nakao S, Kobayashi T, Momoyama Y, Higashiyama T, Minoda A, Sano M, Nomoto H, Oishi K, Hayashi H, Ohta F, Nishizaka S, Haga S, Miura S, Morishita T, Kabeya Y, Terasawa K, Suzuki Y, Ishii Y, Asakawa S, Takano H, Ohta N, Kuroiwa H, Tanaka K, Shimizu N, Sugano S, Sato N, Nozaki H, Ogasawara N, Kohara Y, Kuroiwa T (2004) Genome sequence of the ultra-small unicellular red alga Cyanidioschyzon merolae 10D. Nature 428:653–657
Misumi O, Matsuzaki M, Nozaki H, Miyagishima SY, Mori T, Nishida K, Yagisawa F, Yoshida Y, Kuroiwa H, Kuroiwa T (2005) Cyanidioschyzon merolae genome. A tool for facilitating comparable studies on organelle biogenesis in photosynthetic eukaryotes. Plant Physiol 137:567–585
Miyagishima S, Itoh R, Toda K, Kuroiwa H, Nishimura M, Kuroiwa T (1999) Microbody proliferation and segregation cycle in the single microbody in alga Cyanidioschyzon merolae. Planta 208:326–336
Nelson N, Perzov N, Cohen A, Hagai K, Padler V, Nelson H (2000) The cellular biology of proton-motive force generation by V-ATPases. J Exp Biol 203:89–95
Nishida K, Yagisawa F, Kuroiwa H, Nagata T, Kuroiwa T (2005) Cell cycle-regulated, microtubule-independent organelle division in Cyanidioschyzon merolae. Mol Biol Cell 16:2493–2502
Nozaki H, Matsuzaki M, Takahara M, Misumi O, Kuroiwa H, Hasegawa M, Shin-i T, Kohara Y, Ogasawara N, Kuroiwa T (2003) The phylogenetic position of red algae revealed by multiple nuclear genes from mitochondria-containing eukaryotes and an alternative hypothesis on the origin of plastids. J Mol Evol 56:485–497
Peverly JH, Adamic J, Parthasarathy MV (1978) Association of potassium and some other monovalent cations with occurrence of polyphosphate bodies in Chlorella pyrenoidosa. Plant Physiol 62:120–126
Pick U, Weiss M (1991) Polyphosphate hydrolysis within acidic vacuole in response to amin-induced alkaline stress in the halotolerant alga Dunaliella salina. Plant Physiol 97:1234–1240
Pisoni RL, Lindley ER (1992) Incorporation of [32P] orthophosphate into long chains of inorganic polyphosphates within lysosomes of human fibroblasts. J Biol Chem 267:3626–3631
Rojo E, Zouhar J, Kovaleva V, Hong S, Raikhel NV (2003) The AtC-VPS protein complex is localized to the tonoplast and the prevacuolar compartment in Arabidopsis. Mol Biol Cell 14:361–369
Sanderfoot AA, Raikhel NV (1999) The specificity of vesicle trafficking: coat proteins and SNAREs. Plant Cell 11:629–642
Shirahama K, Yazaki Y, Sakano K, Wada Y, Ohsumi Y (1996) Vacuolar function in the phosphate homeostasis of the yeast Saccharomyces cerevisiae. Plant Cell Physiol 37:1090–1093
Suzuki K, Ehara T, Osafune T, Kuroiwa H, Kawano S, Kuroiwa T (1994) Behavior of mitochondria, chloroplasts and their nuclei during the mitotic cycle in the ultramicroalga Cyanidioschyzon merolae. Eur J Cell Biol 63:280–288
Suzuki T, Oiso N, Gautam R, Novak EK, Panthier JJ, Suprabha PG, Vida T, Swank RT, Spritz RA (2003) The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc Natl Acad Sci USA 100:1146–1150
Takahashi H, Takano H, Yokoyama A, Hara Y, Kawano S, Toh-e A, Kuroiwa T (1995) Isolation, characterization and chromosomal mapping of an actin gene from the primitive red alga Cyanidioschyzon merolae. Curr Genet 28(5):484–490
Takasu A, Nakanishi Y, Yamauchi T, Maeshima M (1997) Analysis of the substrate binding site and carboxyl terminal region of vacuolar H+-pyrophosphatase of mung bean with peptide antibodies. J Biochem 122:883–889
Tanaka K, Oikawa K, Ohta N, Kuroiwa H, Kuroiwa T, Takahashi H (1996) Nuclear encoding of a chloroplast RNA polymerase sigma subunit in a red alga. Science 272:1932–1935
Tijssen JPF, Beeks HW, Steveninck JV (1982) Localization of polyphosphate in Saccharomyces fragilis, as revealed by 4′-6-diamidino-2-phenylindole fluorescence. Biochem Biophys Acta 721:394–398
Toda K, Takano H, Matsunaga S, Miyagishima S, Kuroiwa H, Kuroiwa T (1998) Characterization of chloroplast isoform of serine acetyltransferase from the thermo-acidiphilic red alga Cyanidioschyzon merolae. Biochim Biophys Acta 1403:72–84
Vorisek J, Knotkova A, Kotyk A (1982) Fine cytochemical localization of polyphosphates in the yeast Saccharomyces cerevisiae. Zbl Mikrobiol 137:421–432
Warren G, Wickner W (1996) Organelle inheritance. Cell 84:395–400
Weisman LS, Bacallao R, Wickner W (1987) Multiple methods of visualizing the yeast vacuole permit evaluation of its morphology and inheritance during the cell cycle. J Cell Biol 105:1539–1547
Weiss M, Bental M, Pick U (1991) Hydrolysis of polyphosphate and permeability changes in response to osmotic shocks in cells of the halotolerant alga Dunaliella. Plant Physiol 97:1241–1248
Winter V, Hauser MT (2006) Exploring the ESCRTing machinery in eukaryotes. Trends Plant Sci 11:115–123
Acknowledgments
The authors wish to acknowledge Dr. M. Maeshima (Nagoya University, Japan) for the generous gift of anti-V-PPase antibodies and Drs. Y. Ohsumi and K. Suzuki (National Institute for Basic Biology, Okazaki, Japan) for their kind advice on staining vacuoles. This work was supported by a grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (no. 17051029 to T. K.) and a grant-in-aid from the Promotion of Basic Research Activities for Innovative Biosciences (ProBRAIN to T. K).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yagisawa, F., Nishida, K., Kuroiwa, H. et al. Identification and mitotic partitioning strategies of vacuoles in the unicellular red alga Cyanidioschyzon merolae . Planta 226, 1017–1029 (2007). https://doi.org/10.1007/s00425-007-0550-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-007-0550-y