
Abstract
A plant lectin was isolated from barley (Hordeum vulgare) coleoptiles using acidic extraction and different chromatographic methods. Sequencing of more than 50% of the protein sequence by Edman degradation confirmed a full-length cDNA clone. The subsequently identified open reading frame encodes for a 15 kDa protein which could be found in the soluble fraction of barley coleoptiles. This protein exhibited specificity towards mannose sugar and is therefore, accordingly named as Horcolin (Hordeum vulgare coleoptile lectin). Database searches performed with the Horcolin protein sequence revealed a sequence and structure homology to the lectin family of jacalin-related lectins. Together with its affinity towards mannose, Horcolin is now identified as a new member of the mannose specific subgroup of jacalin-related lectins in monocot species. Horcolin shares a high amino acid homology to the highly light-inducible protein HL#2 and, in addition to two methyl jasmonic acid-inducible proteins of 32.6 and 32.7 kDa where the jasmonic acid-inducible proteins are examples of bitopic chimerolectins containing a dirigent and jacalin-related domain. Immunoblot analysis with a cross-reactive anti-HL#2 antibody in combination with Northern blot analysis of the Horcolin cDNA revealed tissue specific expression of Horcolin in the coleoptiles. The function of Horcolin is discussed in the context of its particular expression in coleoptiles and is then compared to other lectins, which apparently share a related response to biotic or abiotic stress factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants have developed strategies to cope with stress and pathogens by expressing a wide range of proteins that belong to various families of the pathogenesis-related (PR) proteins (Datta and Muthukrishnan 1999). In addition to these PR-proteins, lectins are a class of intensely studied proteins that are thought to play a role in both plant defense (Peumans and Van Damme 1995) and stress response (Zhang et al. 2000). Lectins are found in a wide range of species within the plant kingdom and have different functions, structures, tissue localisations and carbohydrate-binding specificities.
Van Damme et al. (1998) classified the plant lectins into seven families according to their evolutionary and structural characteristics. One of these families are jacalin-related lectins (JRL) where the jacalin from the seeds of jack fruit (Artocarpus integrifolia) gives this class its name (Skea et al. 1988) and binds to T-antigen and galactose. Many lectins were identified with a broad carbohydrate-binding affinity/properties that share sequence similarities to jacalin. Based on the occurrence of JRL in separate plant families with different sugar specificity and biosynthesis reactions, Peumans et al. (2000a) were able to subdivide the JRL into two subgroups: the galactose- (gJRL) and the mannose-specific (mJRL) jacalin- related lectins. The latter subgroup include Calsepa from Calystegia sepium (Van Damme et al. 1996), Heltuba from Helianthus tuberosus (Bourne et al. 1999), BanLec from Musa acuminata L. (Peumans et al. 2000b) and the protein Orysata from the gene salT from Oryza sativa (Claes et al. 1990; Garcia et al. 1998; Zhang et al. 2000). It has previously been suggested that BanLec and Orysata JRLs are expressed in response to abiotic or biotic stresses present in monocotyledonous plants because they respond to jasmonic acid treatment (Garcia et al. 1998; Peumans et al. 2000b; Zhang et al. 2000).
A light stress regulated protein in barley plants (Hordeum vulgare) was characterized as HL#2 by Potter et al. (1996). However, sequence analysis showed that HL#2 (17 kDa) shared sequence homologies to JRLs and, more importantly, had both an affinity towards sugar and exhibited a response to methyl jasmonate. To learn more about HL#2, an antibody was used against the entire recombinant protein HL#2 and, after Western-blot analysis, the presence of HL#2 was revealed in leaves and in coleoptiles, however not in roots or in young etiolated coleoptiles of barley (Menhaj et al. 1999). Instead, there was a cross-reaction between a coleoptile-specific protein of 15 kDa (Horcolin, former name HL#2*) and two JIPs of 32.6 and 32.7 kDa (Mishra et al. 1999).
This investigation describes the isolation, molecular cloning and partial characterization of the 15 kDa protein, which is to date the first identified tissue-specific member of the mannose-binding jacalin-related lectin subgroup in barley.
Materials and methods
Plant growth and harvesting
Barley (Hordeum vulgare L. cv. Apex, Lochow-Petkus GmbH, Bergen, Germany) was grown for 3–6 days on Vermiculite (Deutsche Vermiculite, Sprockhoevel, Germany) at 25°C either under constant irradiance from 100 μmol photons in a 12 h light/12 h dark cycle or in the dark to collect etiolated coleoptiles. Plants were manually separated into roots, (primary) leaves and coleoptiles. Collected materials were then frozen in liquid nitrogen and stored at −80°C or were directly used for Horcolin purification.
Extraction and purification of Horcolin from coleoptiles
Frozen coleoptiles (100 g) were homogenized in extraction buffer (50 mM citric acid, pH 3.1, 200 mM NaCl, 1 mM MgCl2, 1 mM PMSF) at 4°C and mixed vigorously. After settlement of larger cell debris, the supernatant was centrifuged for 30 min at 5,000 g, filtered through a 0.2 μm syringe filter (Sartorius AG, Goettingen, Germany), concentrated with ultra concentrators (Viviaspin, cut off 5 kDa, Sartorius) and subsequently the buffer was changed by dialysis against a 10 mM phosphate buffer (pH 6.8). Subsequently, the sample was passed through a hydroxyl apatite column adjusted with 10 mM phosphate buffer pH 6.8 (Macro-Prep CHT, 40 μm type I, Bio-Rad) and the eluent containing the Horcolin protein was collected while most of the other soluble proteins in the extract were bound to the column matrix. In the next chromatographic step, the eluent from the hydroxyl apatite column was dialyzed against 20 mM piperidin buffer (pH 12.0) containg 20 mM NaCl and then applied on a equilibrated MonoQ HR5/5 column (GE Healthcare, Munich, Germany) connected to a FPLC system (GE Healthcare). Afterwards, Horcolin was eluted using a linear salt gradient of 50–500 mM NaCl in 20 mM piperidin buffer (pH12) and a flow rate of 1.5 ml/min. Fractions were assayed for Horcolin using both silver stained gel and Western-blot analysis with a Horcolin specific antibody. Finally, fractions containing Horcolin were pooled, concentrated and then separated in SDS-PAGE (6% stacking, 12% spacer and 17% separating gel) according to Schägger and von Jagow (1987). Proteins were blotted using the method of Dunn (1986) on PVDF membranes. After Coomassie staining of the membrane and destaining, the Horcolin containing protein band was excised and used for Edmann degradation of the whole protein. Alternatively, the protein was digested in a gel containing the protease LysC as described in Bökenkamp et al. (1994).
Isolation of intercellular washing fluid (IWF)
Approximately 100 primary leaves of Hordeum (7 days after germination) were selected for the isolation of IWF. This was performed according to Hogue and Asselin (1987) with slight modifications. Leaves were first removed from the coleoptiles and infiltrated for 5 min with a 100 mM Tris–HCl buffer (pH 7.5) or with a 100 mM citrate–phosphate buffer (pH 3.0) under vacuum. In addition, both buffers contained 1 mM MgCl2, 100 mM NaCl and 0.005% Triton X-100. The infiltrated leaves were subsequently inserted into homemade holders between two brackets that contained a depression in the shape of the leaves. The leaves were orientated with the cut area to the bottom of the centrifuge tube and then centrifuged for 1 min at 130 g in a T40 swing-out rotor (Jouan CR3i) to remove the excess liquid. The IWF was collected by a second centrifugal step for 15 min at 200 g and, finally, stored at −20°C.
Analytical SDS-PAGE and Western-blot analysis
For the extraction of total soluble proteins, either 6 day-old barley coleoptiles or a combination of barley coleoptiles and leaves (for Western-blot analysis) or barley roots were homogenised in liquid nitrogen followed by the addition of 5 volumes of different extraction buffers [100 mM citric acid (pH 1.9–5.8) or 50 mM Tris-HCl (pH 6.8–9.4)] containing 1 mM MgCl2, 200 mM NaCl, 1 mM PMSF, 1 mM benzamidine and 5 mM aminocaproic acid to test for the optimal pH. Cellular debris was removed by centrifugation (10 min at 10,000 g) and the supernatant was then removed and either stored for 15 min on ice (4°C) or boiled for 15 min and again centrifuged.
SDS-PAGE and sample denaturation were carried out as described in Schägger and von Jagow (1987). Silver staining was done according to Blum et al. (1987). Immunoblotting was performed using 0.45 μm pore size nitrocellulose filters (Towbin et al. 1979). Blots were blocked with 5% skim milk and incubated with antibodies (1:500 dilution) against anti HL#2 or Horcolin. These antibodies were raised in goats or chickens, respectively (Grunwald et al. 2003). The detection of primary antibodies was performed using anti-goat-IgG from rabbit labelled with alkaline phosphatase or using anti-chicken-IgG from rabbit labelled with alkaline phosphatase (dilution 1:20,000, Sigma).
Lectin-binding assay
Barley seedlings (Hordeum vulgare cv. Apex) were grown for 5 days in the dark on Vermiculite and then irradiated for 12 h at 100 μmol m−2 s−1. Primary leaves (30 g) containing the coleoptiles were homogenised with 80 ml extraction buffer (50 mM citric acid, 200 mM NaCl and 1 mM MgCl2 adjusted to pH 2.9) in a commercial mixer on ice. The extract was centrifuged twice (15 min at 5,000 g) to remove all insoluble cell material. The supernatant was concentrated using an ultra concentrator and adjusted to pH 7.2 with NaOH.
Meanwhile, a mannose-agarose column (Sigma) was washed three times with 10 bed vol of binding-buffer (100 mM Tris–HCl (pH 7.2), 100 mM NaCl and 1 mM MgCl2). Afterwards, the concentrated supernatant was removed and 3.7 ml of the barley extract was applied to the mannose-agarose column. For the binding step, the column was placed on a shaker at room temperature for 30 min. The gel matrix was centrifuged for 1 min at 5,000 g. After removal of the supernatant, the mannose-agarose was washed several times with the binding buffer until no protein could be detected in the washing buffer (confirmed by Bradford protein determination 1976).
For the elution of Horcolin, the mannose matrix was first incubated for 20 min at room temperature with one bed volume of elution-buffer (50 mM citric acid, 100 mM NaCl, 1 mM MgCl2 and 300 mM mannose adjusted to pH 2.4) and then, for the second elution step, at 90°C for 5 min with SDS-sample buffer to remove quantitatively all bound Horcolin from the matrix. The supernatants were collected and precipitated by addition of acetone (final concentration 80%) overnight at −20°C. After centrifugation for 30 min at 15,000 g, the pellet was dissolved in 30 μL sample buffer and then separated in SDS-PAGE. Horcolin was detected by Western analysis using the anti-HL#2 antibody (1:500 dilution).
Immunocytochemistry
Green leaves, coleoptiles and etiolated grown coleoptiles from Hordeum vulgare were sliced in small pieces and prepared as previously described by Michel et al. (1998). For the detection of both Horcolin and HL#2, the primary antibodies were diluted 1:100.
Molecular cloning, over expression and purification of Horcolin
Coleoptiles (6 days after germination) were ground in liquid nitrogen and the total RNA was isolated using the Invisorb Spin Plant-RNA Mini Kit (Invitek, Berlin, Germany). To circumvent DNA contamination, the samples were treated with DNase. Based on the identified peptides in addition to using an EST-sequence (Genebank accession number BF630621), the following primers were designed: forward primer GCA CGA GCT AAG TCA CTC TTC and reverse primer GCA GCG TCA CAC ATT ATC AC. For RT-PCR, the Qiagen “OneStep RT-PCR Kit” was used according to the manufacturer’s directions. Reaction products were cloned into pTAdv (Clontech) plasmid and subsequently sequenced in both the forward and reverse directions.
For over expression of Horcolin, a 445 bp fragment was PCR amplified using primers corresponding to the mature protein (forward primer: CGCGGATCCATGAGCAAGCCTGTGAAGATTGG and reverse primer CGCAAGCTTTGGAGTAATATAGAACCCAATCG) by BIO-X-Act-DNA (Bioline, London, UK) polymerase from the Horcolin cDNA clone and then cloned into the BamHI and HindIII cloning sites of the pQE30 expression vector (Qiagen) containing a N-terminal histidine tag. For expression of Horcolin in E. coli M15, production was induced using an optical density of 0.6 (600 nm) with 1 mM IPTG and then incubated for 2 h at 37°C. Subsequently, 1 l of the E. coli cell suspension was harvested by centrifugation at 4,000 g. The cells were broken using a French press operating at 15,000 p.s.i. The protein concentration was determined using a Bio-Rad protein assay kit (Bio-Rad). The filtered and centrifuged supernatant was purified by histidine affinity chromatography according to the manufacturer’s (Qiagen) batch purification procedure. Fractionation of the recombinant protein by SDS-PAGE and protein elution from the gel (gel eluter from Bio-Rad) revealed a single protein species. Western blot experiments using the anti-His tag antibody (Qiagen) and the anti-HL#2 antibody confirmed that the purified protein was His tagged Horcolin.
Hemagglutination assay
Rabbit blood was diluted with PBS buffer to a final solution concentration of 2%. Afterwards, 50 μl of the 2% blood was gently mixed with either 50 μl of 10 μg/ml recombinant Horcolin solution (dialysed against PBS buffer) or a concanavalin A (Sigma) solution (30 μg/ml in PBS buffer) and then incubated for 2 h at RT under gentle shaking.
Isolation of total RNA and Northern blot
Total RNA was isolated from leaves and coleoptiles, separated by electrophoresis on agarose gels containing formaldehyde and then transferred onto Biodyne-B transfer membranes (Pall GmbH, Dreieich, Germany) as described by Menhaj et al. (1999). Each lane was loaded with 10 μg RNA. Northern blots were performed as described by Potter et al. (1996) with 32P-labelled Horcolin cDNA as the probe.
Sequence alignments, database search and molecular homology modeling
The amino acid sequence alignments were carried out using the clustalW program (Thompson et al. 1994) with the amino acid sequences of Heltuba (Genbank accession number AAD11575), BanLec (Genbank accession number AAB82776), Orysata/salT (Genbank accession number AAB53810), HL#2 (Genbank accession number CAB40792), Calsepa (Genbank accession number AAC49564) and Jacalin (Genbank accession number A46630). Conserved domain database (CDD) searches (Marchler-Bauer et al. 2003) were performed using protein-protein blast at NCBI (Altschul et al. 1990) using the sequences of JIP 32.6 (Genbank accession number U43497/ AAA87041), BanLec, Orysata (salT) and Horcolin. For molecular modelling of Horcolin, pdb files were generated using the FAMS program from Kitasato University (Ogata et al. 1998) using the Horcolin- AAK54458 amino acid sequence. For Horcolin, the atomic coordinates from Heltuba were automatically used in the program for homology modelling. A second modelling program, Geno3D, produced results similar to those from the FAMS program (http://www.geno3d-pbil.ibcp.fr).
Results
Previous investigations have shown that the antibody against the HL#2 protein (17 kDa) also recognised a second protein band with an apparent molecular mass of 15 kDa (SDS-PAGE) in tissue extracts of coleoptiles. This protein was formerly named as HL#2* (Menhaj et al. 1999) and, in recognition of its mannose binding capacity, was renamed as Horcolin (Hordeum vulgare coleoptile lectin). For further characterisation of Horcolin, this protein was purified, sequenced and partially analyzed.
Purification of Horcolin
Since Horcolin is present in the coleoptiles of etiolated plants, the purification started with dark-grown coleoptiles. For identification of Horcolin during the purification procedure the antibody against HL#2 was used.
Horcolin remains soluble at pH 3.0 when the cells were broken at either 4°C or at an elevated temperature of 100°C (Fig. 1). It is evident that only a relatively small amount of Horcolin is precipitated during the heating step, especially at lower pH values. Horcolin fractions obtained by purification under acidic conditions at pH 3.0 showed, after neutralisation, the capacity to bind to mannose-agarose. Boiled fractions were not tested for their binding characteristics.

Analyses of different extraction conditions for immunoblotting. Horcolin was extracted from etiolated coleoptiles using different pH values and temperatures. Analysis was performed extraction buffers with different pHs and two temperatures. Each lane was loaded with an equal amount of the Horcolin containing supernatant
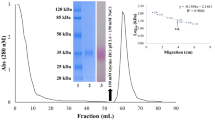
The neutralised pH 3.0 fraction was then applied to a hydroxyl-apatite column where the protein was obtained in the void volume (Fig. 2a, lane 1). This preparation was then subjected to anion exchange chromatography as described in “Materials and methods”. Fractions containing Horcolin were pooled and analysed in SDS-PAGE followed by silver staining (Fig. 2a, lane 2) and immunoblotting with the anti-HL#2 antibody (Fig. 2a, lane 3). Starting from 100 g coleoptiles approximately 11 μg of Horcolin could be obtained after all purification steps. Since the NH2-terminus of Horcolin was blocked, endogenous peptide sequences were obtained after electroblotting onto a PVDF membrane followed by staining with Coomassie and digestion with the LysC protease. Fragments were separated by HPLC and sequenced by Edmann degradation. The endogenous peptide sequences obtained in this manner corresponded to 55% of the predicted apparent molecular mass for Horcolin by SDS-PAGE. Moreover, SDS-PAGE under reduced and non-reduced conditions yielded a single band of 15 kDa. The native molecular mass of Horcolin has been determined as 58 kDa with gel filtration experiments on a Superose 12 column (column flow 0.5 ml/min) in presence of the extraction buffer on the basis of protein standards for gel chromatography. Therefore we concluded a tetrameric size of the purified Horcolin (data not shown).

Identification of Horcolin during different purification steps and the lectin binding assay. a SDS-PAGE and immunoblot, HA chromatography (lane 1), the major fraction after MonoQ column anion exchange chromatography was analysed by silver gel (lane 2), and corresponding immunoblot using the anti-HL#2 antibody in 1:500 dilution (lane 3). Each lane was loaded with 5 μg total protein. b Extracts of Horcolin were applied to mannose agarose and eluted by mannose application under acidic conditions. Binding of Horcolin was analysed by immunoblotting with the anti-HL#2-antibody (1:500 dilution). Lane 1 extract before binding, lane 2 extract after binding to mannose agarose, lane 3 washing fraction, lane 4 elution fraction from mannose-agarose
Identification of the Horcolin DNA sequence
TBLASTN searches for EST-clones in the NCBI database with Horcolin peptide sequences were performed and resulted in identifying an EST clone (Genebank accession number BF630621). Based on this EST-clone, PCR primer pairs were designed and used in RT-PCR. The PCR product with 665 bp was then cloned into a pTAdv vector and sequenced. Sequence analysis showed an open reading frame from 76 to 516 bp meaning that it encoded for translated protein of 145 amino acids with a calculated molecular mass of 15.1 kDa and an isoelectric point of 7.9. The DNA sequence is available under Genebank accession number AY033628. All identified peptides can be matched with parts of the deduced amino acid sequence. The obtained sequence shares 97% similarity to deduced amino acid sequence from BF630621. In contrast, the previously identified HL#2 has only 65% similarity to Horcolin. The alignment of HL#2 and Horcolin is shown in Fig. 3.

Amino acid sequence comparison between Horcolin and HL#2. The deduced amino acid sequences were obtained from the corresponding Horcolin (AAK54458, Genbank) and HL#2 (AAP87359, Genbank) ORFs. Peptide sequences obtained by N-terminal sequencing according to Edman degradation are shown in bold letters. Identical residues are marked with “*”, conserved substitutions with “:” and semi-conserved substitutions with “.”
Database search, sequence alignment and molecular modelling
However, using a Blastp (BLASTP 2.2.8) search, higher similarities were found to the lectins Orysata/salT from Orysata sativa (Genebank accession number AAB53810), BanLec from Musa acuminata (Genebank accession number AAB82776), Heltuba from Helianthus tuberosus (Genebank accession number AAD11575) and Calsepa from Calystegia sepium (Genebank accession number AAC49564). The multiple sequence alignment of Horcolin with the amino acid sequences of proteins like BanLec, Heltuba, Orysata and Calsepa revealed a highly conserved GPWGGNGG amino acid motif in the N-terminal region and, especially in the C-terminal region, other strong similarities.
The database search also indicated high homologies to two 32.6 and 32.7 kDa JIPs (Lee et al. 1996; Genebank accession number AAA87041 and AAA87042). Interestingly, these JIPs consist of a bitopic structure containing a dirigent-like protein domain and a jacalin domain. It is thought that proteins containing this dirigent-like domain are formed during a disease response in plants (see Pfam accession number PF03018 at http://www.pfam.wustl.edu). The three-dimensional structure of Horcolin based on modelling of the homologous lectin Heltuba reveals a threefold symmetric β-prism fold consisting of three four-stranded β sheets, which is overall very similar to Heltuba. For example, the bundles are orientated parallel to the axis of the β-prism fold (data not shown) in a similar way to those in Heltuba (Bourne et al. 1999) or Orysata (Zhang et al. 2000).
Lectin-binding and hemagglutination assay
To investigate the ability of Horcolin to react with carbohydrates, several different sugars that were immobilised on agarose beads were tested for protein binding. These experiments were monitored by Western-blot analysis with the HL#2 and Horcolin specific antibody.
Out of all the carbohydrates tested (galactose, maltose, mannan, and mannose), Horcolin binds only specific to mannose-agarose (see Fig. 2b). The Horcolin can be eluted from the mannose matrix with mannose, whereas the best elution is achieved using mannose in combination with an acidic elution buffer. Since the binding of Horcolin to mannose clearly matches to the definition of a lectin. Therefore it can be classified into the mannose-binding JRLs.
To confirm the lectin character of Horcolin, a hemagglutination assay in the presence of recombinant Horcolin was performed (data not shown). Specifically, an agglutination assay in the presence of a protein solution of recombinant Horcolin showed a hemagglutination of rabbit erythrocytes. The hemagglutination titre of the recombinant Horcolin was calculated with approximately 10 μg/ml meaning that the recombinant Horcolin showed a relatively weak activity compared to other JRLs.
Tissue specific expression of Horcolin
For a detailed analysis of tissue specific expression of Horcolin, etiolated and light-grown barley seedlings were separated into roots, leaves and coleoptiles (Fig. 4) and analysed by Northern- and Western-blotting techniques. As shown in Fig. 4a and b, Horcolin was not found in either the roots or leaves. However, Horcolin was exclusively expressed in both light- or dark-grown coleoptiles while HL#2 could be detected in light-grown coleoptiles and leaves. In contrast, etiolated grown barley plants showed no HL#2 protein in the coleoptiles by the fifth day but instead expressed it in the leaves (Fig. 4a, b, c). Furthermore, HL#2 could be detected in the spikes (ears) of barley. Using the anti-HL#2 antibody, other cross-reacting proteins with the same size as Horcolin (15 kDa) and HL#2 (17 kDa) could be found in Avena sativa (oat), Oryza sativa (rice), Triticum vulgare (wheat) and Secale cereale (rye), but not in Zea mays (maize) and Ipomoea batata (sweet potato; data not shown).

Tissue specific localization of Horcolin by Northern- and Western-blot analyses. Seedlings were grown either in darkness or under light. a Ethidium bromide stained RNA gel from different tissues (Co- coleoptiles, L- leaves, R- roots). b Northern blot with total RNA from different tissues using a 440 bp Horcolin cDNA fragment as probe. Each lane was loaded with 10 μg total RNA. c Western-blot analysis of Horcolin with anti-HL#2 antibody. Each lane was loaded with 10 μg of the soluble protein fraction from coleoptiles, leaves, and roots
Localisation of Horcolin in the plant cell
Sequence evaluation of Horcolin for targeting prediction using different bioinformatic tools available on the internet (e.g., ESLpred, TargetP and Wolfpsort) revealed no general consensus on the sequence. Therefore localisation studies were performed by immuno-blotting and immuno-histological electron microscopy. The results are presented in Figs. 5, 6. The intercellular washing fluid (IWF) for Hordeum vulgare obtained by an infiltration-centrifugation technique at pH 3.0 and 7.5 were separated on SDS-PAGE and analyzed with different antibodies against constitutively expressed, cytoplasmatic located, heat-shock cognate protein (HSC), apoplast located chitinase (pathogenesis related protein 3, PR-3) and Horcolin. As the force of gravity on the Hordeum cell wall during centrifugation can break the wall and contaminate the sample, a careful analysis was performed according to Lohaus et al. (2001) to determine the maximum allowable centrifugal force, which was found to be 1,500 g. The absence of cytosolic contamination in the IWF was confirmed by immunoblotting with an antiserum against the constitutively expressed cytosolic HSC70 protein (Anderson et al. 1994; Fig. 5) and revealed that HSC70 was only detectable in crude extract from leaves. For further characterisation, the IWF from Hordeum was blotted against the apoplast-located and constitutively expressed chitinase (PR-3, see Fig. 5). Immunblotting with the antibodies against HL#2 as well as the Horcolin specific peptide antibody showed that Horcolin was exclusively localized in the apoplast (Fig. 5).

Localisation of Horcolin by immunoblotting. Intercellular washing fluid (IWF) was isolated from barley and was subsequently analysed using antibodies against the heat shock cognate protein (HSC70, 1:1,000 dilution), chitinase (PR-3, 1:2,000 dilution), HL#2 (1:500 dilution) and Horcolin (1:1,000 dilution). Each lane was loaded with 2 μg protein. Samples include pH 3.0; extraction of IWF at pH 3.0; pH 7.5; extraction of IWF at pH 7.5 and T: total cell extract
Based on these results, further investigations over the localisation of Horcolin were performed by immune histology and electron microscopy (Fig. 6). Electron microscopical immunological investigations of etiolated coleoptiles with peptide antibodies against both Horcolin and HL#2, highlighted in Fig. 6b, revealed that Horcolin was localized in the cell wall. Etiolated coleoptiles showed no expression of HL#2 (see Fig. 6c). This finding was subsequently confirmed by immunoblotting against a crude extract of green leaves and analysing the single 17 kDa band for HL#2 (data not shown).

Electron microscopical immuno-localisation of Horcolin in etiolated coleoptiles of Hordeum vulgare. a Control microgaph showing staining with pre-immune serum (dilution 1:100). b Immune localisation of Horcolin with the Horcolin peptide antibody (dilution 1:100). c Immunostaining of etiolated coleoptiles with a peptide antiserum against HL#2 (dilution 1:100). Cw cell wall
Discussion
Studying the expression of the high light-induced protein HL#2 using a polyclonal antibody against this protein leads to the discovery of a coleoptile specific 15 kDa protein that has the capacity to bind mannose. Cloning of this protein revealed an obvious sequence homology to proteins of the jacalin related lectin family that have an affinity towards mannose-sugars. Based on these findings, the protein was named Horcolin for “Hordeum coleoptile lectin” to describe both its character and function.
Horcolin was extracted from etiolated coleoptiles using acidic extraction buffers and both HA and anion exchange chromatography. In a similar manner, other groups (Nakagawa et al. 1996; Zhang et al. 2000) have successfully applied acidic extraction methods for the purification of lectins. As discussed in Peumans et al. (2000b), this is a key feature in the purification of certain lectins from other plant species due to the reversible loss of sugar-binding at an acidic pH. In fact, Horcolin displayed a much weaker binding to the mannose-agaroses tested here when extracted in neutral pH buffers, but had a high affinity when extracted under acidic conditions.
After analysing Horcolin using different bioinformatics programs, e.g., PSORT (Nakai and Kanehisa 1991), no obvious targeting sequence was observed in silico, which is in contrast to storage lectins with a putative defensive role (van Damme et al. 1998). However, it was possible to demonstrate by immune-histological electron microscopy that Horcolin was predominately localised in the cell wall of etiolated coleoptiles. The electron microscopical localisation studies were carried out by two independent research groups with different antibodies against Horcolin (raised in either goats or chickens) and their results were shown to be free from preparation artefacts through a series of IWF studies.
Van Damme et al. (2002) classified the jacalin related lectin into two subgroups: the gJRL and mJRL. Based on its primary and tertiary structure and its affinity towards mannose, the data presented here suggests that Horcolin belongs to the mJRL subgroup for mannose-specific jacalin related lectins. Van Damme et al. (2004a) introduced a different classification system for lectins based on their sugar binding characteristics. They classified lectins as inducible or classical where classical lectins are not involved in specific lectin-carbohydrate interactions within the plant cells itself. However, the induced lectins are expressed by exogenous or endogenous stimuli, e.g., abiotic/biotic stress factors or plant hormones and might be involved in cellular regulation and signalling (Van Damme et al. 2004c).
The homologous members of the mannose-specific JRLs, such as Orysata and BanLec, are putative candidates functioning in response to stresses (Peumans et al. 2000b; Zhang et al. 2000) because they were induced by jasmonic acid treatment or, in the case of Orysata, by salt, drought, abscisic acid treatment and, especially, after infection with pathogens (Garcia et al. 1998; Qin et al. 2003). Together with the recently identified lectin Nictaba (Chen et al. 2002), which is located in the nucleus and cytoplasma and is thought to play a role in signal transduction, the group of inducible lectins appears to function within the context of biotic/abiotic stress signalling in monocots and dicots (Van Damme et al. 2004b).
The expression of the Horcolin homologous protein HL#2 in barley is influenced by light (Potter et al. 1996) and methyl jasmonate treatment (Menhaj et al. 1999). In etiolated coleoptiles, light treatment leads to a de novo synthesis of the HL#2 protein. Preliminary experiments have shown that the Horcolin mRNA is up-regulated under salt-stress conditions (data not shown). The protein is detectable three days after germination until the coleoptile becomes senescent. The characteristic property of Horcolin as a member of the mannose-specific JRL is its tissue specificity. The protein is present only in the coleoptile while the most closely related protein in barley, HL#2, is expressed in all leafy tissues including the light-grown coleoptile. Therefore it appears that barley plants possess two closely related lectins, which are regulated and expressed independently of each other.
Monocotyledonous plants, like grasses, use coleoptiles for the protection of the primary leaf during seed germination. After passing through the soil and being exposed to light, the growth of these specialised embryonic structures is inhibited, while the primary leaf continues to grow and emerges through a pore located at the apex of the coleoptile (O’Brien and Thimann 1965). In this way, the coleoptile helps the plant to cope with any present abiotic and biotic stress factors. Future studies will evaluate whether Horcolin plays a role in the perception or transduction of biotic/abiotic stress induced signals or in the handling of altered environmental conditions.
Abbreviations
- CDD:
-
Conserved domain database
- gJRL:
-
Galactose-specific JRL
- HA:
-
Hydroxyl apatite
- IWF:
-
Intercellular washing fluid
- JIP:
-
Jasmonic acid induced protein
- JRL:
-
Jacalin-related lectin
- mJRL:
-
Mannose- specific JRL
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Anderson JV, Haskell DW, Guy CL (1994) Differential influence of ATP on native spinach 70 kilodalton heat-shock cognates. Plant Physiol 104:1371–1380
Blum H, Beier H, Gross HJ (1987) Improved silver staining of plant proteins RNA and DNA in polyacrylamide gels. Electrophoresis 8:93–99
Bökenkamp D, Jungblut PW, Thole HH (1994) The C-terminal half of the porcine estradiol receptor contains no post-translational modification: determination of the primary structure. Mol Cell Endocrinol 104:163–172
Bourne Y, Zamboni V, Barre A, Peumans WJ, Van Damme EJ, Rouge P (1999) Helianthus tuberosus lectin reveals a widespread scaffold for mannose-binding lectins. Struct Fold Des 7:1473–1482
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen Y, Peumans WJ, Hause B, Bras J, Kumar M, Proost P, Barre A, Rouge P, Van Damme EJM (2002) Jasmonic acid methyl ester induces the synthesis of a cytoplasmic/nuclear chito-oligosaccharide binding lectin in tobacco leaves. FASEB J 16:905–907
Claes B, Dekeyser R, Villarroel R, Van den Bulcke M, Bauw G, Van Montagu M, Caplan A (1990) Characterization of a rice gene showing organ-specific expression in response to salt stress and drought. Plant Cell 2:19–27
Datta SK, Muthukrishnan S (1999) Pathogenesis-related proteins in plants. CRC, Boca Raton
Dunn SD (1986) Effects of the modification of transfer buffer composition and the renaturation of proteins in gels on the recognition of proteins on Western blots by monoclonal antibodies. Anal Biochem 157:144–153
Garcia AB, Engler Jde A, Claes B, Villarroel R, Van Montagu M, Gerats T, Caplan A (1998) The expression of the salt-responsive gene salT from rice is regulated by hormonal and developmental cues. Planta 207:172–180
Grunwald I, Rupprecht I, Schuster G, Kloppstech K (2003) Identification of guttation fluid proteins: the presence of pathogenesis-related proteins in non-infected barley plants. Physiol Plant 119:192–202
Hogue R, Asselin A (1987) Detection of 10 additional pathogenesis-related B proteins in intercellular fluid extracts from stressed Xanthi tobacco leaf tissue. Can J Bot 65:476–481
Lee J, Parthier B, Lobler M (1996) Jasmonate signalling can be uncoupled from abscisic acid signalling in barley: identification of jasmonate-regulated transcripts which are not induced by abscisic acid. Planta 199:625–632
Lohaus G, Pennewiss K, Sattelmacher B, Hussmann M, Muehling KH (2001) Is the infiltration-centrifugation technique appropriate for the isolation of apoplastic fluid? A critical evaluation with different plant species. Physiol Plant 111:457–465
Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej T, Marchler GH, Mazumder R, Nikolskaya AN, Panchenko AR, Rao BS, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y, Yamashita RA, Yin JJ, Bryant SH (2003) CDD: a curated entrez database of conserved domain alignments. Nucleic Acids Res 31:383–387
Menhaj AR, Mishra SK, Bezhani S, Kloppstech K (1999) Posttranscriptional control in the expression of the genes coding for high-light-regulated HL#2 proteins. Planta 209:406–413
Michel KP, Exss-Sonne P, Scholten-Beck G, Kahmann U, Ruppel HG, Pistorius EK (1998) Immunocytochemical localization in the mesophilic cyanobacterium Synechococcus PCC6301 and the thermophilic Synechoccus elongatus. Planta 205:73–81
Mishra SK, Menhaj AR, Bezhani S, Kloppstech K(1999) In: Argyroudi JH, Argyroudi SH (eds) The chloroplast: from molecular biology to biotechnology. Kluwer, The Netherlands, pp 107–112
Nakai K, Kanehisa M (1991) Expert system for predicting protein localization sites in gram-negative bacteria. Proteins 11:95–110
Nakagawa R, Yasokawa D, Ikeda T, Nagashima K (1996) Purification and characterization of two lectins from callus of Helianthus tuberosus. Biosci Biotech Biochem 60:259–262
O’Brien TP, Thimann KV (1965) Histological studies on the coleoptile. I. Tissue and cell types in the coleoptile tip. Am J Bot 52:910–918
Ogata K, Ohya M, Umeyama H (1998) Amino acid similarity matrix for homology modeling derived from structural alignment and optimized by the Monte Carlo method. J Mol Graph Model 16:178–189, 254
Peumans WJ, Van Damme EJ (1995) Lectins as plant defense proteins. Plant Physiol 109:347–352
Peumans WJ, Hause B, Van Damme EJ (2000a) The galactose-binding and mannose-binding jacalin-related lectins are located in different sub-cellular compartments. FEBS Lett 477:186–192
Peumans WJ, Zhang W, Barre A, Houles AC, Balint-Kurti PJ, Rovira P, Rouge P, May GD, Van Leuven F, Truffa-Bachi P, Van Damme EJ (2000b) Fruit-specific lectins from banana and plantain. Planta 211:546–554
Potter E, Beator J, Kloppstech K (1996) The expression of mRNAs for light-stress proteins in barley: inverse relationship of mRNA levels of individual genes within the leaf gradient. Planta 199:314–320
Qin Q, Zhang Q, Zhao W, Wang Y, Peng Y (2003) Identification of a lectin gene induced in rice in response to Magnaporthe grisea infection. Acta Bot Sin 45:76–81
Schägger H, von Jagow G (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379
Skea DL, Christopoulous P, Plaut AG, Underdown BJ (1988) Studies on the specificity of the IgA-binding lectin, jacalin. Mol Immunol 25:1–6
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Van Damme EJM, Barre A, Verhaert P, Rouge P, Peumans WJ (1996) Molecular cloning of the mitogenic mannose/maltose-specific rhizome lectin from Calystegia sepium. FEBS Lett 397:352–356
Van Damme EJM, Peumans WJ, Barre A, Rouge P (1998) Plant lectins: a composite of several distinct families of structurally and evolutionary related proteins with diverse biological roles. Crit Rev Plant Sci 17:575–692
Van Damme EJM, Hause B, Hu J, Barre A, Rouge P, Proost P, Peumans WJ (2002) Two distinct jacalin-related lectins with a different specificity and subcellular location are major vegetative storage proteins in the bark of the black mulberry tree. Plant Physiol 130:757–769
Van Damme EJM, Lannoo N, Fourquart E, Peumans W (2004a) The identification of inducible cytoplasmic/nuclear carbohydrate-binding proteins urges to develop novel concepts about the role of plant lectins. Glycoconj J 20:449–460
Van Damme EJM, Barre A, Rouge P, Peumans WJ (2004b) Potato lectin: an updated model of a unique chimeric plant protein. Plant J 37:34–45
Van Damme EJM, Barre A, Rouge P, Peumans WJ (2004c) Cytoplasmic/nuclear plant lectins: a new story. Trends Plant Sci 9:484–489
Zhang W, Peumans WJ, Barre A, Astoul CH, Rovira P, Rouge P, Proost P, Truffa-Bachi P, Jalali AA, Van Damme EJ (2000) Isolation and characterization of a jacalin-related mannose-binding lectin from salt-stressed rice (Oryza sativa) plants. Planta 210:970–978
Acknowledgments
This work was supported by grants from the DFG (Deutsche Forschungsgemeinschaft). We express our gratitude to Dr. M. Legrand (IBMP, Strasbourg, France) who kindly provided the antibody against the PR-3 protein (chitinase) and to Dr. M. Koutb and Dr. J. Dean for a critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grunwald, I., Heinig, I., Thole, H.H. et al. Purification and characterisation of a jacalin-related, coleoptile specific lectin from Hordeum vulgare . Planta 226, 225–234 (2007). https://doi.org/10.1007/s00425-006-0467-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-006-0467-x