
Abstract
Resurrection plants differ from other species in their unique ability to survive desiccation. In order to understand the mechanisms of desiccation tolerance, proteome studies were carried out using leaves of the resurrection plant Boea hygrometrica to reveal proteins that were differentially expressed in response to changes in relative water content. This opportunity was afforded by the rare ability of excised B. hygrometrica leaves to survive and resume metabolism following desiccation in a manner similar to intact plants. From a total of 223 proteins that were reproducibly detected and analyzed, 35% showed increased abundance in dehydrated leaves, 5% were induced in rehydrated leaves and 60% showed decreased or unchanged abundance in dehydrated and rehydrated leaves. Since the induction kinetics fall into clearly defined patterns, we suggest that programmed regulation of protein expression triggered by changes of water status. Fourteen dehydration responsive proteins were analyzed by mass spectrometry. Eight proteins were classified as playing a role in reactive oxygen species scavenging, photosynthesis and energy metabolism. In agreement with these findings, glutathione content and polyphenol oxidase activity were found to increase upon dehydration and rapid recovery of photosynthesis was observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drought is an environmental stress that reduces crop yield and restricts plant distribution. Depending on the extent of water availability, plants can be affected in different ways from the inhibition of normal physiological activities and cellular damage to cell death. Many land plants have evolved structural and physiological mechanisms to protect against mild drought stress. In contrast, only a small number of species, called resurrection plants, have evolved the ability to tolerate extreme desiccation by assuming an inactive stage for an indefinite period and rapidly resuming growth upon rehydration (Gaff 1971; Deng et al. 1999; Oliver et al. 2000; Phillips et al. 2002). Resurrection plants are therefore excellent model systems for the study of mechanisms by which plants survive dehydration and the identification of genes that could potentially enhance drought tolerance of crops by biotechnological methods (Bartels and Salamini 2001).
To date, efforts have been focused on structural, physiological and molecular aspects of desiccation tolerance (Gaff 1997; Neale et al. 2000; Bartels and Salamini 2001; Ramanjulu and Bartels 2002; Cooper and Farrant 2002; Vicré et al. 2004). Specific genes are associated with the acquisition of desiccation tolerance based on RNA expression studies; however, little is known about the expression of the encoded proteins. Transcriptome analysis is a powerful tool to understand desiccation tolerance (Kuang et al. 1995; Gaff et al. 1997; Blomstedt et al. 1998a, b; Collett et al. 2004); however, mRNA abundances may only represent putative function since there is still a questionable correlation between mRNA and protein levels (Futcher et al. 1999; Gygi et al. 1999). This is further highlighted by a survey of the proteomes of the model legume Medicago truncatula (Watson et al. 2003). In this study, it was revealed that approximately 50% of the proteins appear to be correlated with their corresponding mRNA levels. In contrast, proteomics provides a more physiologically accurate snapshot of biochemical processes by revealing the actual protein constituents performing the enzymatic, regulatory, and structural functions encoded by the genome and transcriptome at a given point in time. Furthermore, proteomics approaches provide additional information on gene regulation, especially important for the cases that mRNAs may not be translated, or that changes in protein level or enzyme activity have occurred without any detectable change in transcript abundance due to translational or other levels of control (Gygi et al. 1999).
Boea hygrometrica is a small dicotyledenous, homiochlorophyllous resurrection plant that is widespread in China, inhabiting shallow rock crevices from humid tropical regions to arid temperate zones (Deng et al. 1999). A wild population of this species was found in Beijing city, where monthly precipitation fluctuates from 2.7–8.3 mm (November to March) to 185.2–159.7 mm (July to August). (Average monthly precipitation data from 1971 to 2000 provided by Beijing Meteorological Bureau). Even in summer, these plants also experience rapid changes of water deficit due to dramatic changes in daily air humidity. A remarkable feature of these plants is the ability of detached leaves to possess a similar level of desiccation tolerance as that of whole plants (Deng et al. 2003). This feature is not common to all resurrection angiosperms. Craterostigma plantagineum, Myrothamnus flabellifolia, Craterostigma nanum are other notable examples that share this ability (Gaff and Loveys 1984; Bartels et al. 1990; Sherwin 1995). Since the desiccation and rehydration of detached leaves avoid interference from developmental regulation and long-distance signaling from other organs, it has been used as a simplified system to investigate the molecular mechanisms desiccation tolerance (Deng et al. 1999, 2003).
Here, we report a detailed study of the changes in protein expression that occur in dehydrated and rehydrated B. hygrometrica detached leaves using two-dimensional polyacrylamide gel electrophoresis (2DE) and the identification of 14 dehydration-responsive proteins by mass spectrometry. In doing so, our study represents the first reported analysis of the desiccation responsive proteome in a resurrection plant. Analysis of the identified proteins reveals that detoxification mechanisms mediated through glutathione and remodeling of the photosynthetic apparatus are important for desiccation tolerance.
Materials and methods
Chemicals and equipments
Urea, Tris and glycine were purchased from USB (Cleveland, OH, USA) and SDS from Pharmacia (Uppsala, Sweden). Ampholyte and Acrylamide/Bis solutions were obtained from Bio-Rad (Hercules, CA, USA). The molecular weight (MW) and isoelectric focusing (IEF) 3–10 markers were purchased from Tianwei (Beijing, China) and SERVA (Heidelberg, Germany), respectively.
Plant materials and stress treatments
Boea hygrometrica plants were collected from their natural habitat in Beijing (shallow rock crevices, 100 m) and maintained in a greenhouse with regular irrigation. Leaves detached from the fully hydrated plants were dried under 50% relative humidity and moderate illumination at 25°C for 0 h (unt), 0.5 h (d0.5), 8 h (d8), 48 h (d48) in a climate chamber. For rehydration, the 48 h dried leaves were placed in Petri dishes with wet filter papers for 8 h (r8) and 48 h (r48). For the measurement of glutathione, leaves were dehydrated for 8 days and subsequently rehydrated for 3 days.
Estimation of water content
The untreated detached leaves (unt) were weighed to obtain the fresh weight (W f), and the dehydrated and rehydrated leaves were weighed (W t). The dry weight (W d) of leaves was estimated after heating at 105°C for 15 min, followed by incubation at 80°C for 24 h. The relative water content (RWC) was defined as follows:
Three repetitions were performed and the average RWC at each time point was calculated.
Electrolyte leakage
The extent of electrolyte leakage was measured in detached and intact (treated as whole plants) leaves that were dehydrated and rehydrated as described by Farrant et al. (1999), in comparison with untreated controls and heat-killed (HK) leaves that were rehydrated at 42°C for 48 h following dehydration. Four replicates of individual leaves were used for each treatment. Leaves were placed in 30 ml Milli-Q (Millipore) ultra-pure water and conductivity was monitored at 5 min intervals for 30 min using a Hanna EC 215 conductivity meter (Hanna instruments, Padova, Italy). Leakage rate was calculated (the slope of the line generated from the time course of leakage) and leakage values (μS g dry mass−1 min−1) were obtained from leakage rate corrected by leaf dry mass.
Protein extraction
Protein extraction was performed according to Damerval et al. (1986) and Kamo et al. (1995), with minor modifications. Leaves were grounded in liquid nitrogen, resuspended in a pre-cooled acetone solution containing 10% TCA and 0.07% 2-mercaptoethanol and then incubated for overnight at −20°C. Samples were centrifuged at 20,000 × g for 15 min at 4°C and the precipitates were resuspended and washed three times in pre-chilled acetone solution containing 0.07% 2-mercaptoethanol to remove the remaining TCA. The protein pellets were then air-dried and 50 mg powder of each sample were dissolved in 1 ml of Lysis buffer (8 M urea, 2% NP−40, 2% Triton X-100, 5% PVP-40, 5% 2-mercaptoethanol, 0.8% Ampholyte 3–10) at room temperature. After precipitation at 25,000 × g for 15 min, the supernatants were loaded onto IEF gels immediately or stored in aliquots at −80°C.
Two-dimensional polyacrylamide gel electrophoresis
Two-dimensional polyacrylamide gel electrophoresis (2DE) was performed basically as described by O’Farrell (1975). Briefly, 50 μg protein samples, as quantified by the Coomassie brilliant blue G250 assay (Sedmak and Grossberg 1977) were loaded onto the alkaline end of an 8-cm long and 2-mm diameter tube gel (4% acrylamide, 8 M urea, 2% NP-40, 1% Ampholyte 3–10, 1% Ampholyte 5–8) and overlaid with 15 μl of 1/2 Lysis buffer, IEF gel electrophoresis was carried out at 200 V for 0.5 h, 400 V for 17 h and 800 V for 1 h. After focusing, the gels were equilibrated twice for 15 min in equilibration buffer (62.5 mM Tris-HCl, pH 6.8, 2.5% SDS, 10% glycerol, 5% 2-mercaptoethanol) and then fixed on top of 4% stacking and 12.5% separating polyacrylamide gels with 1% agarose gel melted in equilibration buffer. Secondary dimension SDS-PAGE was carried out using a Bio-Rad Mini-PROTEAN II system (Hercules, CA, USA) at 15 mA constant current until the tracking dye reached the bottom of the gel. Gels were stained in CBB solution (1% Coomassie blue R-250, 25% ethanol and 7% acetic acid). The MW and isoelectric point (pI) of each protein spot was calibrated by co-electrophoresis of MW markers and IEF markers 3–10. For each sample, 2DE was repeated at least three times with protein extracted from different sample pools. A representative gel for each sample was selected for image and data analysis.
Image and data analysis
The CBB stained gels were scanned using the flatbed scanner UMAX Powerlook 1120 (Dallas, TX, USA). The data were analyzed using ImageMaster 2D Platinum 5.0 software (Amersham Bioscience). Relative volume (%vol) used to quantify and compare the spots was calculated as follows:
where vols is the volume of spot S in a gel containing n spots.
The criteria for defining the expression patterns were determined as follows: up-regulated, %vol increased at least twofold; down-regulated, %vol decreased at least twofold; unchanged, %vol varied within twofold.
Protein identification
The selected protein spots were excised from the gels manually. Protein digestion and MALDI-TOF-MS analysis were performed according to Yang et al. (2006), using an AXIMA-CFR Plus MALDI-TOF mass spectrometer (Shimadzu Biotech, Kyoto, Japan). The proteins were identified by PMF search in NCBI database as described by Yang et al. (2006). Nanospray ESI Q-TOF MS/MS (Micromass) analysis was performed according to Dai et al. (2006). The MS/MS data were processed using MassLynx 3.5 and searched against NCBI nr protein sequence databases with the MS/MS ion searching program MASCOT (http://www.matrixscience.com). Ions score is −10 * log(P), where P is the probability that the observed match is a random event. Individual ions scores indicate identity or extensive homology, * indicates that P < 0.05; Protein scores are derived from ions scores as a non-probabilistic basis for ranking protein hits.
Chlorophyll fluorescence
Quantum efficiency (F v/F m), the minimal fluorescence yield (F 0, PSII fully opened) and the maximum fluorescence yield (F m, PSII fully closed) of dark adapted leaves at various stages of dehydration and rehydration were obtained according to the fast chlorophyll a fluorescence induction kinetics curves generated using PAM-101 (Walz, Germany), with a saturating light intensity of approximate 800 μmol m−2 s−1 and a duration of 4.5 s. The experiment was performed twice with at least five independent leaves for each treatment at each time point.
Glutathione determination
Leaf materials were homogenized in ice-cold 5% trichloroacetic acid (w/v) and centrifuged at 12,000 × g for 15 min. The supernatant was then used for the determination of the reduced glutathione using a TaKaRa (Japan) GSH assay Kit.
Polyphenol oxidase (PPO) activity determination
Leaves were grounded in liquid nitrogen with pre-cooled pestle and mortar and homogenized with 4 ml of 50 mM potassium phosphate (pH 6.4) containing 1% of polyvinyl pyrrolidone (PVP). After filtration the homogenate was centrifuged at 12,000 × g for 30 min at 4°C and the resulting supernatant was used in enzyme assays. Polyphenol oxidase (PPO) activity was measured by incubating 0.1 ml of leaf extract in 2.9 ml of buffered substrate (50 mM potassium phosphate, pH 6.4 and 500 mM catechol) at 30°C and monitoring the change of absorbance at 398 nm for 10 s with 4-tert-butylcatechol (TBC) (ε398 = 1,150 M−1 cm−1), as described by Wang et al. (2004). One unit of enzyme was defined as the amount of the enzyme that produced 1 μmol of tert-butyl-oquinone per minute per mg protein.
Results
Detached leaves of B. hygrometrica are desiccation tolerant
Leaves detached from fully hydrated B. hygrometrica plants were subjected to dehydration and subsequent rehydration under the conditions described in Materials and methods. During dehydration, the RWC of the detached leaves decreased rapidly from 100% (untreated; unt) to a stable, dried state of 2.4% after 24 h and thereafter. During rehydration, the RWC of the detached leaves regained turgor to 100.2% after 48 h and the leaves resumed broadly the original morphology (Fig. 1). Accordingly, three points representing early phase (dehydration for 0.5 h when the RWC dropped to 79.3%; d0.5), mid-phase (dehydration for 8 h when RWC dropped to 6.7%; d8); and late phase (dehydration for 48 h when RWC dropped to 2.4%; d48), as well as two time points representing partially rehydrated leaves (rehydrated for 8 h subsequently after 48 h of dehydration, when the RWC resumed to 77.2%; r8) and fully rehydrated leaves (rehydrated for 48 h subsequently after 48 h of dehydration, when the RWC resumed to 100.2%; r48) were sampled.

Dehydration and resurrection of detached leaves of B. hygrometrica. Leaves detached from fully hydrated plants were dried under 50% relative humidity and moderate illumination at 25°C for 0 h (100.0% RWC) (unt), 0.5 h (79.3% RWC) (d0.5), 8 h (6.7% RWC) (d8), 48 h (2.4% RWC) (d48) in a climate chamber and subsequently rehydrated for 8 h (77.2% RWC) (r8) and 48 h (100.2% RWC) (r48) in Petri dishes with wet filer papers on both lid and bottom, respectively, as indicated
To test cell viability of dehydrated and rehydrated detached leaves, electrolyte leakage assays were performed. Broadly no difference in ion leakage was observed in untreated and intact leaves from dehydrated and rehydrated whole plants (Fig. 2), thus demonstrating that B. hygrometrica survives drying of excised leaves. Further proof that excised leaves were capable of reviving full metabolism is demonstrated in Fig. 5, where the activity of photosystem II was shown to rapidly recover upon rehydration.

Electrolyte leakage in leaves of B. hygrometrica untreated (unt), dehydrated (d0.5, d8, d48) and rehydrated (r8, r48) as detached (gray) and as intact (white), in comparison with the leaves that were heat-killed (HK) by rehydration at 42°C after dehydration normally
Protein separation by two-dimensional electrophoresis
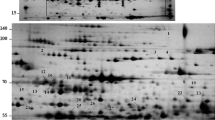
Fifty microgram of proteins from the six detached leaf samples, including the untreated control, were resolved using 2DE. Flatbed gel scanning and image analysis software enabled over 400 spots to be detected and numbered on a 2D gel map. Due to the lower resolution at the anodal and cathodal ends of the first dimension tube gels, only the gel region where the pI ranged from 4.2 to 6.9 was further analyzed (Fig. 3a). In total, 223 protein spots were reproducibly detected in this region in three independent experiments. Proteins displayed different specific profiles when the expression levels in the five samples were compared with the untreated control. Representative examples of all six different profiles of proteins have been enlarged in Fig. 3b.


a Comparison of 2D gel maps of proteins isolated from detached leaves dehydrated and rehydrated for different periods as indicated. The pH range is indicated along the top of each gel, and the sizes of MW markers (kDa) are indicated down the left-hand side. b Different gel regions were numbered and enlarged to show examples of differential expression patterns. (i) Proteins showing increased expression since early dehydration; (ii) Proteins showing increased expression in mid or late dehydration stage; (iii) Proteins showing decreased expression in dehydrated leaves and resumed in rehydrated leaves; (iv) Proteins showing decreased expression in dehydrated leaves but did not resumed in rehydrated leaves; (v) Proteins showing unchanged expression in dehydrated but increased in rehydrated leaves; (vi) Proteins showing unchanged expression in both dehydrated and rehydrated leaves
Protein expression changes during dehydration and rehydration processes
The 223 proteins depicted in Fig. 3a were quantified using ImageMaster 2D Platinum 5.0 software (Amersham Bioscience). The relative volume (%vol) of each protein spot at various stages of dehydration and rehydration was compared to the protein level in untreated leaves. The results are summarized in Table 1 together with a representative curve that describes each protein accumulation pattern. Broadly, six types of pattern can be recognized according to the expression alteration in response to dehydration:
Seventy-eight proteins (35%) increased in abundance in response to dehydration. A subgroup of these proteins was transiently expressed during the early phase of dehydration (d0.5) (30 proteins; pattern i). The remaining 48 proteins accumulated to the highest level when the leaves were further dehydrated (pattern ii).
Forty-three proteins (19%) were repressed by dehydration. Among them, 34 proteins showed a decrease in the expression that correlated with the loss in RWC (pattern iii), while the remaining nine proteins were repressed in both dehydrated and rehydrating tissues (pattern iv).
A total of 102 proteins (46%) were broadly unchanged during dehydration. A subset of 10 proteins showed increased abundance at 8 or 48 h after rehydration (pattern v), whereas the remaining 92 displayed constitutive levels of expression in all samples tested (pattern vi).
A detailed description of the expression kinetics of all dehydration/rehydration up-regulated proteins is listed in Supplementary Tables 1 and 2.
Proteins identified through MALDI-TOF-MS and ESI Q-TOF MS/MS
To further understand the proteins that may function in dehydration tolerance of B. hygrometrica leaves, representative proteins showing increased expression (patterns i and ii, Fig. 4) were chosen and subjected to trypsin digestion and MALDI TOF-MS or ESI Q-TOF MS/MS analysis to obtain peptide mass fingerprint (PMF) data. Five out of 14 proteins analyzed showed significant matches (P < 0.05) to previously reported proteins associated with photosynthesis, energy metabolism and unknown function (Table 2). Three further proteins with relatively high scores and pI/MW close to known proteins in the database are also reported (Table 2). These proteins are associated with glutathione and polyphenol metabolism, which prompted analysis of the glutathione levels and polyphenol oxidase enzyme activity during desiccation/rehydration.

Different gel regions were enlarged to show expression patterns of protein spots indicated
Photosynthesis
MS analysis identified two proteins that significantly matched to chloroplast-located proteins. Protein no. 373 was 20 kDa and found to transiently accumulate during the mid to late stages of dehydration. The protein significantly matched to a fragment of the 52 kDa Rubisco large subunit from Pinguicula caerulea (accession no. P28440) (Albert et al. 1992). The second protein (no. 348) was found to accumulate in desiccated and partially rehydrated tissues and was 24 kDa. This protein was identified as precursor of the oxygen-evolving complex (OEC) of photosystem II, similar to that reported from sunflower (Helianthus paradoxus; accession no. CF079724).
Based on these findings, the effects of dehydration/rehydration on photosynthesis were investigated. Chlorophyll a induction kinetics was used as an indicator of photosystem II activity during dehydration and rehydration. The analysis showed that the quantum efficiency (F v/F m) was not significantly affected until 8 h after dehydration when RWC dropped to 6.7% (Fig. 5a). However, F 0 and F m, regarding to the minimal fluorescence yield when PSII fully opened and the maximum fluorescence yield when PSII fully closed, respectively, of dark adapted leaves started to decrease as soon as 0.5 h after leaves were exposed to dehydration (Fig. 5b, c).

Quantum efficiency (F v/F m) (a), the minimal fluorescence yield (F 0, PSII fully opened) (b) and the maximum fluorescence yield (F m, PSII fully closed) (c) with various treatments. Detached leaves were dehydrated and rehydrated for different periods as described in Fig. 1
Energy metabolism
Two proteins were identified by MS analysis that match proteins involved in energy metabolism. Levels of a 70 kDa protein (spot no.78) correlated with the RWC of the leaves, with the highest levels observed in the desiccated sample. This protein matched significantly with a 36 kDa vacuolar H+-ATPases from Citrus unshiu (accession no. AB036926). A second protein (spot no. 105) with a dehydration and rehydration responsive profile was identified as having a putative ATPase function. This protein was 62 kDa and matched significantly to a 28 kDa ATP-binding cassette (ABC) transporter ATPase from Chlamydia trachomatis (accession no. NC_000117).
Unknown protein
Protein no. 176 was also found to be both dehydration and rehydration responsive. This protein was 44 kDa and matched a 30 kDa protein of unknown function from Arabidopsis thaliana (accession no. NP_567979). Little is known about this protein; however, there is an indication that the protein may function as a cyclase.
Glutathione metabolism
A 17 kDa protein with a pI of 5.1 (no. 391) was found to be transiently expressed since the early response to water deficit. MS analysis revealed that the protein probably matched with a 19 kDa glutathione peroxidase protein from barley (accession no. AJ238744) (Churin et al. 1999), an enzyme that is suggested to play a role in catalyzing glutathione-mediated reactive oxygen species (ROS) scavenging. A second protein (no. 322) with an MW of 29 kDa (pI 6) accumulated to the highest level in dried tissues that had high score with a 26 kDa glutathione S-transferase protein (ATGSTU6) from A. thaliana.
The identification of glutathione peroxidase and possibly glutathione S-transferase as dehydration-responsive proteins indicated that glutathione may serve as a major antioxidant in B. hygrometrica. To provide more insight on this hypothesis, the contents of glutathione were also measured during a prolonged dehydration and rehydration (Fig. 6). Data showed that glutathione markedly increased during the early stages of dehydration and reached the highest level after 48 h, when the leaves were desiccated. A relatively high level of glutathione was maintained until full turgor was restored. These data strongly support a role for glutathione in the acquisition of desiccation tolerance of B. hygrometrica leaves.

Reduced glutathione (GSH) contents in B. hygrometrica leaves during dehydration and rehydration. The treatments were same as in Fig. 1. Error bars represent S.E. Number of observations was 3. Leaves were dehydrated for 8 days before rehydration for the subsequent 3 days
Polyphenol metabolism
A 57 kDa protein with a pI of 5.6 (no. 122) was found to increase in abundance during the early stages of water deficit. MS analysis indicated that the protein was similar to a 71 kDa PPO precursor protein from tomato (accession no. gi|22729), a chloroplast-targeted enzyme that is predicted to catalyze the oxidation of mono- and o-diphenols to o-diquinones. Enzyme activity assays were performed to measure the activity of PPO at different stages of RWC (Fig. 7). PPO activity increased significantly during the early stages of water loss (d0.5–d8) and a second activity peak was seen during early rehydration (r8). The increase in the PPO protein levels therefore broadly correlates with an increase in PPO enzyme activity.

Polyphenol oxidase (PPO) activities in B. hygrometrica leaves during dehydration and rehydration. The treatments were same as in Fig. 1. Error bars represent S.E. Number of observations was 3. Leaves were dehydrated for 2 days before rehydration for the subsequent 2 days
Discussion
Here, we present a study of the responses of the resurrection plant B. hygrometrica to stages of dehydration and rehydration at the protein level. B. hygrometrica belongs to a group of resurrection plants that can survive desiccation as detached leaves. Although all resurrection plants survive dehydration as whole plants (including B. hygrometrica), there is diversity in the ability of detached leaves to survive desiccation. For example, Sporobolus stapfianus leaves cannot survive dehydration if detached from fully hydrated parent plants (Blomstedt et al. 1998a); B. hygroscopica leaves are tolerant to equilibration to 65–80% relative humidity but sensitive to equilibration to very low (close to 0%) relative humidity (Bochicchio et al. 1998); whereas detached leaves of C. plantagineum and M. flabellifolia survive desiccation and especially, detached leaves of C. nanum survive rapid desiccation by equilibration to low relative humidity down to a RWC lower than 1% (Gaff and Loveys 1984; Bartels et al. 1990; Sherwin 1995). Although different mechanisms may be used in different types of resurrection plants, the viability of the resurrected cells (showed by both electrolyte leakage and Chlorophyll a induction kinetics assays) clearly demonstrated that the detached leaves of B. hygrometrica possess all the fundamental mechanisms pre-required for desiccation tolerance and thus represent a simplified system to investigate the basis of desiccation tolerance, taking advantage of avoidance of possible developmental regulation and long-distance signaling from other organs.
Of the analyzed 223 protein spots reproducibly detected in the gel region where the pI ranged from 4.2 to 6.9, 41% (Table 1, pattern vi) were constitutively expressed, indicating that protective mechanisms existing in the resurrection plant B. hygrometrica lead to an overall stabilization of protein metabolism. Proteins showing consistent level during both dehydration and rehydration are likely to be structural proteins or proteins associated with the basic metabolic activities that are important to keep the minimum viability in cells under all kinds of conditions. They may not directly play roles in desiccation tolerance; however, they appear necessary for survival.
The majority of proteins (59%; Table 1, patterns i–v) were differentially expressed, suggesting that a complex program of events are involved in the acquisition of desiccation tolerance. Among them, 19% (Table 1, patterns iii and iv) of the analyzed proteins showed decreased abundance in dehydrated leaves. The reduction in expression level may have resulted from repression of gene expression and/or increased protein turnover. Proteins showing these patterns are likely to be involved in metabolic pathways that were active in well-watered tissues but inactivated by the decrease in osmotic potential. These proteins may not be directly associated with stress tolerance per se; however, it cannot be ruled out that the reduction in expression was actively triggered by dehydration for purposes such as resource reallocation for activating physiological mechanisms that are necessary for desiccation tolerance.
Thirty-five percent (Table 1, patterns i and ii) of the analyzed proteins were transiently induced during dehydration with differing kinetics and consequently represent proteins that function at specific stages during the program that leads to dehydration tolerance. Among these 78 detected dehydration-inducible proteins, 39% (30 proteins, subgroups 1–4, Supplementary Table 1) were induced by dehydration within 0.5 h. These included a putative ABC Transporter ATPase (protein no. 105), glutathione peroxidase (protein no. 391) and polyphenol oxidase (protein no. 122). The early appearance of new proteins is consistent with a study of protein synthesis in the desiccation tolerant grass, S. stapfianus (Kuang et al. 1995). It has been well reported that readily detectable amounts of translatable mRNA are present in air-dried leaves of many desiccation tolerant plants including monocots, eudicots, fern, moss and liverwort (Bartels et al. 1990; Oliver 1991; Reynolds and Bewley 1993; Hellwege et al. 1994; Gaff et al. 1997), although dynamic changes in protein phosphorylation were also shown to be induced in the resurrection plant (Rohrig et al. 2006). Therefore, the appearance of new proteins in early dehydration may be due to rapid de novo synthesis or the post-translational processing of pre-existing proteins that are modified when the dehydration signal is first perceived. Accumulation of 46% (36 proteins; subgroups 5–7, Supplementary Table 1) and 15% (12 proteins; subgroup 8, Supplementary Table 1) of the dehydration responsive proteins was initiated during the mid (8 h) to late (48 h) stages of dehydration, respectively. Proteins in this group included the putative Rubisco large subunit (protein no. 373), OEC of photosystem II (protein no. 348), vacuolar H+-ATPase A subunit (protein no. 78), unknown protein (protein no. 176) and glutathione S-transferase (protein no. 322). These proteins may be transcribed and translated in response to the initial dehydration signal. The existence of distinct expression profiles suggests that different sets of proteins are programmed to function in protection against dehydration.
A relatively small number (5%, Table 1, pattern v) of the analyzed proteins were up-regulated specifically during the rehydration phase. This supports the hypothesis that most changes in the gene expression occur during the dehydration phase in desiccation tolerant higher plants (Phillips et al. 2002). This phenomenon contrasts with desiccation tolerant mosses such as Tortula ruralis, where the mechanism of tolerance does not involve the synthesis of proteins induced by the onset of water deficit. Oliver (1991) showed that protein synthesis in T. ruralis was most apparent during the “repair” phase that occurred during rehydration. Examples of rehydration responsive proteins from resurrection plants do, however, exist such as the transketolase proteins (TKT7 and TKT10), which play a possible role in the conversion of sugars in C. plantagineum (Bernacchia et al. 1995).
Thirteen percent (Table 1, pattern i) of the analyzed proteins had increased expression levels in both dehydrated and rehydrated leaves, suggesting dual roles in both stages. These proteins may contribute to stabilize the cellular environment for maintenance of basic structural and physiological integrity in desiccated and rehydrated cells.
Identification of dehydration-induced proteins
Fourteen dehydration-responsive proteins were subjected to peptide mass fingerprinting. Five proteins displayed a significant score (P < 0.05) with previously reported proteins. A further three proteins also had high scores and broadly similar pI/MW values; however, the matches were not statistically significant at the 5% level. This may reflect the necessity to compare PMFs from B. hygrometrica with proteins from other plant species. According to the function of matched proteins, these identified proteins can be classified into three categories including photosynthesis, energy metabolism and detoxification.
Photosynthesis
Photosynthesis and photosynthetic apparatus are particularly vulnerable to water deficit, being the first target of photo-oxidation. In this study, two proteins that matched to a 20 kDa fragment of the Rubisco large subunit (rbcL) and a 23 kDa polypeptide of the OEC of photosystem II were identified in the dehydrated leaves of B. hygrometrica. RbcL proteins had been recorded in drought-stressed sugar beet and metal-stressed rice leaves (Salekdeh et al. 2002; Hajheidari et al. 2005). Furthermore, Desimone et al. (1998) demonstrated that ROS generated under stress conditions triggers protein degradation, specifically of rbcL in barley. ROS-treated rbcL degradation was mediated via stromal proteases and required ATP hydrolysis. Moreover, a characteristic protein fragmentation pattern was observed that consisted of four distinct peptides, including a 20 kDa fragment. It is possible, therefore, that given the conservation in structure between rbcL proteins that the appearance of the 20 kDa rbcL protein spot in B. hygrometrica may be the result of stress-induced proteolysis, probably mediated through ROS-induced chloroplast localized protease activity. This suggests that although B. hygrometrica is a homiochlorophyllous desiccation tolerant species, the induction of antioxidant systems, such as the glutathione system, to ameliorate the effects of ROS may not be sufficient to completely protect rbcL from degradation.
It is noteworthy, however, that of photosystem II activity ceased earlier than the appearance of the photosynthesis-related proteins during drying, shut down of photosynthesis during the early phase of drying has been reported in all angiosperm resurrection plants studied to date (homoiochlorophyllous or poikilochlorophyllous) and is proposed to be a mechanism to stop photosynthetically induced ROS production (Tuba et al. 1997, 1998; Sherwin and Farrant 1998; Farrant 2000; Mundree et al. 2002; Deng et al. 2003; Farrant et al. 2003). Therefore, the appearance of rbcL fragment and the oxygen-evolving protein of PSII is not immediately contributing to photosynthesis, thus suggesting a programmed proteolysis, at least in the case of rbcL.
Our data contrasts with the report that many genes related to photosynthesis, including genes encoding for OEC fragments and PSII reaction centre proteins, were down-regulated in response to dehydration in the poikilochlorophyllous monocotyledonous resurrection plant Xerophyta humilis (Collett et al. 2003, 2004). The different expression patterns of the photosynthesis-related genes in X. humilis and B. hygrometrica may reflect the different approaches employed in poikilochlorophyllous and homoiochlorophyllous resurrection plants to minimize the damage of excess light energy during dehydration. Another factor may be that in some cases the steady-state mRNA levels do not accurately reflect the changes observed at the protein level, which highlights the necessity of proteomic approaches.
Given that the 23 KDa protein of OEC in B. hygrometrica leaves closely correlated with the predicted MW of OEC proteins from other species, it is unlikely that the appearance the protein was solely due to protein degradation. Thylakoid pigment-protein complexes have been shown to be highly stable in B. hygrometrica leaves during desiccation and rehydration, despite the rapid decline in photosynthetic activity upon dehydration (Deng et al. 2003). It is proposed that certain protective mechanisms must exist in order to maintain the integrity of basal functional photosynthetic units in dried leaves in resurrection plants. The 23 kDa polypeptide of OEC of photosystem II is one of the three extrinsic proteins associate with higher plant and green algal PSII (Seidler 1996). It cannot directly bind to PSII, however, it has the function of stabilizing the Mn4O x cluster (Han et al. 2004). Therefore, the increase in its abundance in dehydrated and rehydrated leaves of B. hygrometrica may help to maintain the structure of OEC during dehydration and the regain of function during rehydration.
Energy metabolism
Two proteins matched to ATPases were also identified in the dehydrated leaves. One ABC transporter ATPase (spot no. 105) was induced since 0.5 h after dehydration, and reached a level as high as 10.5 and 12.6 folds when leaves lost >90% of water (d8 and d48). High expression was maintained during rehydration, indicating that the protein may play an important role in providing energy for protective and repair reactions in both phases. ABC transporters represent a large family of proteins that mediate the ATP-dependent transport of solutes (Higgins 1992) and are responsible for cellular detoxification processes in plants and animals (Klein et al. 2004). Members of this type of proteins have been reported to be required for aluminum tolerance in Arabidopsis (Larsen et al. 2005) or involved in guard cell hormonal signaling (Klein et al. 2003, 2004). In contrast, the expression of the other putative ATPase (spot no. 78; matching to a vacuolar H+-ATPase A subunit from Citus unshiu) was only induced in d8 and d48 leaves, suggesting a specific role during dehydration. Similar genes have also been identified in desiccated leaf of resurrection species S. stapfianus and Xerophyta viscosa (Blomstedt et al. 1998a, b; Marais et al. 2004), suggesting a link with desiccation tolerance. Vacuolar H+-ATPases are multisubunit enzymes localized to internal membranes including the tonoplast of mature vacuoles, the endoplasmic reticulum, membranes of the secretory pathway and the plasma membrane and function as proton pumps in translocating protons across membranes at the expense of ATP consumption to drive secondary transport processes across the membranes (Sze et al. 1999; Ratajczak 2000). Members of vacuolar H+-ATPase family have been shown to be required in salt tolerance and aluminum resistance in yeast (Hamilton et al. 2001, 2002). The stress-resonsive V-ATPase subunit c homologue isolated from X. viscosa is able to rescue Escherichia coli srl::Tn10 mutants subjected to a high level of sorbitol osmotic stress, indicating the involvement of this protein in osmo-protection (Marais et al. 2004). It was also reported that the ATP analogue binding to the A subunit induces conformational changes in the E subunit that involves a disulfide bond formation in plant V-ATPases (Kawamura et al. 2001).
Unknown protein
Proteins in this family are thought to be cyclase enzymes. They are found in proteins involved in antibiotic synthesis. However, they are also found in organisms that do not make antibiotics pointing to a wider role for these proteins. It is noteworthy that this class of proteins has not previously been associated with dehydration tolerance or other abiotic stresses. The proteins contain a conserved motif HXGTHXDXPXH that is likely to form a part of the active site. Genes encoding similar proteins have been isolated from other species such as rice (accession no. XM_481575) and Chinese wild grape (Vitis pseudoreticulata; accession no. DQ336281).
Detoxification
As previously stated, water stress often leads to ROS accumulation. In addition to proteolysis, ROS can damage lipids, terpenoids, carbohydrates, and nucleic acids (Monk et al. 1989; Baier and Dietz 1998). The capacity to scavenge ROS and to reduce their damaging effects is important for stress-tolerance (Tsugane et al. 1999). Free radical scavenging systems involve antioxidant compounds such as ascorbic acid, glutathione, thioredoxin and carotenoids, and proteins such as superoxide dismutase and catalase (Xiong and Zhu 2002). Genes encoding ROS scavenging enzymes have been cloned and the activity of the enzymes has been shown to increase upon stress, leading to the suggestion that higher scavenging activity may correlate with enhanced drought tolerance of the plants (Bowler et al. 1992; Roxas et al. 1997). This finding was supported by the relatively large number of antioxidant defence genes that were identified as being up-regulated by dehydration from a recent study of transcriptional profiling in X. humilis (Collett et al. 2004).
In vegetative tissues, removal of the cytotoxic products resulting from oxidative events is considered to be important for survival of drought stress because genes encoding enzymatic antioxidants become up-regulated during drying (Ingram and Bartels 1996). In C. plantagineum, an inhibitor of lipoxygenase, the activity of which results in lipid hydroperoxide formation, accumulates in the leaves during desiccation (Bianchi et al. 1992). In Craterostigma wilmsii and X. viscosa, the activity of ascorbate peroxidase increases during dehydration. During rehydration, the activity of superoxide dismutase and glutathione reductase increases (Sherwin and Farrant 1998).
Glutathione protects against oxidative stress by quenching activated oxygen species and participating in hydrogen peroxide metabolism (Neubauer and Yamamoto 1994; Sgherri and Navari-Izzo 1995). In this study, a protein matched with enzymes catalyzing glutathione-mediated detoxification and scavenging of ROS were found to be induced in the dehydrated leaves of the resurrection plant B. hygrometrica. Spot no. 391 matched with a glutathione peroxidase-like protein was induced to the highest level within 30 min. The expression level was remained high during the dehydration process but reversed after rehydration, leading to the suggestion that the glutathione cycle may play a major role in the protection from dehydration induced oxidative stress in B. hygrometrica leaves. Our observation of increased glutathione accumulation during dehydration further supports a role of glutathione in desiccation tolerance. In agreement, glutathione accumulation during desiccation was also found in the resurrection angiosperms B. hygroscopica (Navari-Izzo et al. 1997) and Ramonda serbica (Augusti et al. 2001), and the high activity of glutathione reductase and expression of glutathione transferase was found to be induced in response to dehydration in C. plantagineum (Alamillo and Bartels 2001; Smith-Espinoza 2001). Glutathione reductase activity increased, to various extents, during drying in M. flabellifolius and X. humilis (Farrant 2000). An initial increase of the glutathione reductase activity at the beginning of dehydration (70% RWC) (as well as other antioxidant enzymes) was observed in all three Eragrostis spp. with differing degrees of tolerance to water deficit; however, only the desiccation tolerant E. nindensis kept the elevated level of glutathione reductase activity when the plants were dried and desiccated, in contrast to that the two desiccation-sensitive species ceased the enzyme activities when dried below their critical water contents of 50 and 40% RWC (Illing et al. 2005), implying the important role of glutathione reductase in desiccation tolerance. Our finding that glutathione reductase and glutathione S-transferase proteins accumulate in tandem with an overall increase in glutathione levels in dehydrated and, more importantly, in desiccated leaves of B. hygrometrica also supports this concept.
Polyphenol oxidase (PPO) and o-diphenols appear to prevent proteolytic activity, which could help conserve proteins in the anabiotic state (Veljovic-Jovanovic et al. 2006; Sullivan and Hatfield 2006). Our data are in agreement with this hypothesis, since both an increase in PPO protein levels and enzyme activity was observed in drying leaf tissues. PPOs have been reported in many plant species, but many possible roles they play have not been totally resolved (Sherman et al. 1995). Because nearly all characterized PPOs appear to be localized to the chloroplast thylakoid lumen, it has been proposed that PPO may also play an indispensable role in chloroplast function with a possible involvement in a Mehler-like reaction detoxifying oxygen species (Sherman et al. 1995). Polyphenols possess ideal structural chemistry for free radical scavenging activities, and have been shown to be more effective antioxidants than vitamins C and E on a molar ratio (Rice-Evans et al. 1997).
In summary, this paper has presented a primary study of the protein expression profiles in response to dehydration and rehydration in the resurrection plant B. hygrometrica. Our direct approach to understanding biochemical processes by profiling the protein constituents and performing correlative enzymatic, metabolic and physiological assays at given RWCs is designed to complement studies that have focused primarily at the steady state mRNA level in resurrection plants. The differential expression at the protein level and the programmed regulation of protein expression in response to dehydration and rehydration is consistent with the accompanying findings at the structural and metabolic levels. MS analysis of 14 dehydration-induced protein spots resulted in the identification of proteins associated with ROS detoxification, photosynthesis and energy metabolism. The question remains, however, what makes B. hygrometrica desiccation tolerant. We propose that tolerance is likely to be a combination of (i) the quantitative effect of particularly high levels of antioxidants together with well-known osmoprotective compounds such as sugars and LEA proteins that accumulate in a specific spatial and temporal manner and (ii) the expression of novel factors, such as the proteins of unknown function reported here.
References
Alamillo JM, Bartels D (2001) Effects of desiccation on photosynthesis pigments and the ELIP-like dsp 22 protein complexes in the resurrection plant Craterostigma plantagineum. Plant Sci 160:1161–1170
Albert VA, Williams SE, ChaseMW (1992) Carnivorous plants: phylogeny and structural evolution. Science 257:1491–1495
Augusti A, Scartazza A, Navari-Izzo F, Sgherri CLM, Stevanović B, Brugnoli E (2001) Photosystem II photochemical efficiency, zeaxanthin and antioxidant contents in the poikilohydric Ramonda serbica during dehydration and rehydration. Photosynth Res 67:79–88
Baier M, Dietz KJ (1998) The costs and benefits of oxygen in photosynthetic plant metabolism. Prog Bot 60:283–314
Bartels D, Salamini F (2001) Desiccation tolerance in the resurrection plant Craterostigma plantagineum. A contribution to the study of drought tolerance at the molecular level. Plant Physiol 127:1346–1353
Bartels D, Schneider K, Terstappen G, Piatkowski D, Salamini F (1990) Molecular cloning of abscissic acid-modulated genes which are induced during desiccation of the resurrection plant Craterostigma plantagineum. Planta 181:27–34
Bernacchia G, Schwall G, Lottspeich F, Salamini F, Bartels D (1995) The transketolase gene family of the resurrection plant Craterostigma plantagineum differential expression during the rehydration phase, EMBO J 14:610–618
Bianchi G, Gamba A, Murelli C, Salamini F, Bartels D (1992) Low molecular weight solutes in desiccated and ABA-treated calli and leaves of Craterostigma plantagineum. Phytochemistry 31:1917–1922
Blomstedt CK, Gianello RD, Hamill JD, Neale AD, Gaff DF (1998a) Drought-stimulated genes correlated with desiccation tolerance of the resurrection grass Sporobolus stapfianus. Plant Growth Regul 24:153–161
Blomstedt CK, Gianello RD, Gaff DF, Hamill JD, Neale AD (1998b) Differential gene expression in desiccation-tolerant and desiccation-sensitive tissue of the resurrection grass, Sporobolus stapfianus. Aust J Plant Physiol 25:937–946
Bochicchio A, Vazzana C, Puliga S, Alberti A, Cinganelli S, Vernieri P (1998) Moisture content of the dried leaf is critical to desiccation tolerance in detached leaves of the resurrection plant Boea hygroscopica. Plant Growth Regul 24:163–170
Bowler C, van Montague M, Inzé D (1992) Superoxide dismutase and stress tolerance. Annu Rev Plant Physiol Plant Mol Biol 43:83–116
Churin Y, Schilling S, Borner T (1999) A gene family encoding glutathione peroxidase homologues in Hordeum vulgare (barley). FEBS Lett 459:33–38
Collett H, Butowt R, Smith J, Farrant J, Illing N (2003) Photosynthetic genes are differentially transcribed during the dehydrationrehydration cycle in the resurrection plant, Xerophyta humilis. J Exp Bot 54:2593–2595
Collett H, Shen A, Gardner M, Farrant JM, Denby KJ, Illing N (2004) Towards transcript profiling of desiccation tolerance in Xerophyta humilis: Construction of a normalized 11 k X. humilis cDNA set and microarray expression analysis of 424 cDNAs in response to dehyration. Physiol Plant 122:39–53
Cooper K, Farrant JM (2002) Recovery of the resurrection plant Craterostigma wilmsii from desiccation: protection versus repair. J Exp Bot 53:1805–1813
Dai S, Li L, Chen T, Chong K, Xue Y, Wang T (2006) Proteomic analyses of Oryza sativa mature pollen reveal novel proteins associated with pollen germination and tube growth. Proteomics 6:2504–2529
Damerval C, Vienne D, Zivy M, Thiellement H (1986) The technical improvements in two-dimensional electrophoresis increase the level of genetic variation detected in wheat-seedling proteins. Electrophoresis 7:52–54
Deng X, Hu Z, Wang H (1999) mRNA differential display visualized by silver staining tested on gene expression in resurrection plant Boea hygrometrica. Plant Mol Biol Rep 17:279
Deng X, Hu Z, Wang H, Wen X, Kuang T (2003) A comparison of photosynthetic apparatus of the detached leaves of the resurrection plant Boea hygrometrica with its non-tolerant relative Chirita heterotrichia in response to dehydration and rehydration. Plant Sci 165:851–861
Desimone M, Wagner E, Johanningmeier U (1998) Degradation of active oxygen modified Rubisco by chloroplastic proteases requieres ATP-hydrolysis. Planta 205:459–466
Farrant JM (2000) A comparison of mechanisms of desiccation tolerance among three angiosperm resurrection plant species. Plant Ecol 151:29–39
Farrant J, Cooper K, Kruger LA, Sherwin HW (1999) The effect of rate of drying on three different resurrection plants. Ann Bot 84:371–379
Farrant JM, Vander Willigen C, Loffell DA, Bartsch S, Whittaker A (2003) An investigation into the role of light during desiccation of three angiosperm resurrection plants. Plant Cell Environ 26:1275–1286
Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI (1999) A sampling of the yeast proteome. Mol Cell Biol 19:7357–7368
Gaff DF (1971) Desiccation-tolerant flowering plants in Southern Africa. Science 25:1033–1034
Gaff DF (1997) Mechanism of desiccation tolerance in resurrection vascular plants. In: Basra AS, Basra RK (eds) Mechanisms of environmental stress resistance in plants. Harwood Academic, Amsterdam, pp 43–58
Gaff DF, Loveys BR (1984) Abscisic acid content and effects during dehydration ofdetached leaves of desiccation tolerant plants. J Exp Bot 35:1350–1358
Gaff DF, Bartels D, Gaff JL (1997) Changes in gene expression duringdrying in a desiccation-tolerant grass Sporobolus stapfianus and a desiccation-sensitive grass Sporobolus pyramidalis. Aust J Plant Physiol 24:617–622
Gygi SP, Rochon Y, Franza BR, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19:1720–1730
Hajheidari M, Abdollahian-Noghabi M, Askari H, Heidari1 M, Sadeghian S, Ober E, Salekdeh G (2005) Proteome analysis of sugar beet leaves under drought stress. Proteomics, 5:950–960
Hamilton CA, Good AG, Taylor GJ (2001) Induction of vacuolar ATPase and mitochondrial ATP synthase by aluminum in an aluminum-resistant cultivar of wheat. Plant Physiol 125:2068–2077
Hamilton CA, Taylor GJ, Good AG (2002) Vacuolar H+-ATPase, but not mitochondrial F1F0-ATPase, is required for NaCl tolerance in Saccharomyces cerevisiae. FEMS Microbiol Lett 208:227–232
Han G, Li S, Tang C, Li L, Kang T (2004) Progress on the structure of photosynthetic oxygen evolving complex and the mechanism of photosynthetic water oxidation. Prog Chem 16:184–195
Hellwege EM, Dietz KJ, Volk OH, Hartung W (1994) Abscisic acid and the induction of desiccation tolerance in the extremely xerophilic liverwort Exormotheca holstii. Planta 194:525–531
Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113
Illing N, Denby K, Collett H, Shen A, Farrant JM (2005) The signature of seeds in resurrection plants: a molecular and physiological comparison of desiccation tolerance in seeds and vegetative tissues. Integr Comp Biol 45:771–787
Ingram J, Bartels D (1996) The molecular basis of dehydration tolerance in plants. Annu Rev Plant Physiol Plant Mol Biol 47:377–403
Kamo M, Kawakami T, Miyatake N, Tsugita A (1995) Separation and characterization of Arabidopsis thaliana proteins by two-dimensional gel electrophoresis. Electrophoresis 16:423–430
Kawamura Y, Arakawa K, Maeshima M, Yoshida S (2001) ATP analogue binding to the A subunit induces conformational changes in the E subunit that involves a disulfide bond formation in plant V-ATPase. Eur J Biochem 268:2801–2809
Klein M, Perfus-Barbeoch L, Frelet A, Gaedeke N, Reinhardt D, Mueller-Roeber B, Martinoia E, Forestier C (2003) The plant multidrug resistance ABC transporter AtMRP5 is involved in guard cell hormonal signalling and water use. Plant J 33:119–129
Klein M, Geisler M, Suh S, Kolukisaoglu HÜ, Azevedo L, Plaza S, Curtis MD, Richter A, Weder B, Schulz B, Martinoia E (2004) Disruption of AtMRP4, a guard cell plasma membrane ABCC-type ABC transporter, leads to deregulation of stomatal opening and increased drought susceptibility. Plant J 39:219–236
Kuang J, Gaff DF, Gianello RD, Blomstedt CK, Neale AD, Hamill JD. (1995) Changes in in vivo protein complements in drying leaves of the desiccation-tolerant grass Sporobolus stapfianus and the desiccation-sensitive grass Sporobolus pyramidalis. Aust J Plant Physiol 22:1027–1034
Larsen PB, Geisler MJ, Jones CA, Williams KM, Cancel JD (2005) ALS3 encodes a phloem-localized ABC transporter-like protein that is required for aluminum tolerance in Arabidopsis. Plant J 41:353–363
Marais S, Thomson JA, Farrant JM, Mundree SG (2004) VATP1XV—a novel stress responsive V-ATPase subunit c homologue isolated from the resurrection plant Xerophyta viscosa Baker. Physiol Plant 122:54–61
Monk LS, Fagerstedt KV, Crawford RMM (1989) Oxygen toxicity and superoxide dismutase as an antioxidant in physiological stress. Physiol Plant 76:456–459
Mundree SG, Baker B, Mowla S, Peters S, Marais S, Vander Willigen C, Govender K, Maredza A, Muyanga S, Farrant JM, Thomson JA (2002) Physiological and molecular insights into drought tolerance. Afri J Biotechnol 1:28–38
Navari-Izzo F, Meneguzzo S, Loggini B, Vazzana C, Sgherri CLM (1997) The role of the glutathione system during dehydration of Boea hygroscopica. Physiol Plant 99:23–30
Neale AD, Blomstedt CK, Bronson P, Le TN, Gutteridge K, Evans J, Gaff DF, Hamill JD (2000) The isolation of genes from the resurrection grass Sporobolus stapfianus, which are induced during severe drought stress. Plant Cell Environ 25:265–277
Neubauer C, Yamamoto HY (1994) Membrane barriers and Mehler-peroxidase reaction limit the ascorbate available for violaxanthin de-epoxidase activity in intact chloroplasts. Photosynth Res 39:137–147
O’Farrell PH (1975) High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250:4007–4021
Oliver MJ (1991) Influence of protoplasmic water loss on the control of protein synthesis in the desiccation-tolerant moss Tortula ruralis: Ramifications for a repair-based mechanism of desiccation tolerance. Plant Physiol 97:1501–1511
Oliver MJ, Tuba Z, Mishler BD (2000) The evolution of vegetative desiccation tolerance in land plants. Plant Ecol 151:85–100
Phillips JR, Oliver MJ, Bartels D (2002) Molecular genetics of desiccation tolerant systems. In: Black M, Pritchard HW (eds) Desiccation and survival in plants: drying without dying. CABI Publishing, Wallingford, pp 319–341
Ramanjulu S, Bartels D (2002) Drought- and desiccation-induced modulation of gene expression in plants. Plant Cell Environ 25:141–151
Ratajczak R (2000) Structure, function and regulation of the plant vacuolar H(+)-translocating ATPase. Biochim Biophys Acta 1465:17–36
Reynolds TL, Bewley JD (1993) Abscisic acid enhances the ability of the desiccation-tolerant fern Polypodium virginianum to withstand drying. J Exp Bot 44:1771–1779
Rice-Evans CA, Miller NJ, Paganga G (1997) Antioxidant properties of phenolic compounds. Trends Plant Sci 2:152–159
Roxas VP, Smith RK, Allen ER, Allen RD (1997) Overexpression of glutathione S-transferase/glutathione peroxidase enhances the growth of transgenic tobacco seedlings during stress. Nat Biotechnol 15:988–991
Rohrig H, Schmidt J, Colby T, Brautigam A, Hufnagel P, Bartels D (2006) Desiccation of the resurrection plant Craterostigma plantagineum induces dynamic changes in protein phosphorylation. Plant Cell Environ 29:1606–1617
Salekdeh GH, Siopongco J, Wade LJ, Ghareyazie B, Bennett J (2002) Proteomic analysis of rice leaves during drought stress and recovery. Proteomics 2:1131–1145
Sedmak JJ, Grossberg SE (1977) A rapid, sensitive, and versatile assay for protein using Coomassie brilliant blue G250. Anal Biochem 79:544–552
Seidler A (1996) The extrinsic polypeptides of Photosystem II. Biochim Biophys Acta 1277:35–60
Sgherri CLM, Navari-Izzo F (1995) Sunflower seedlings subjected to increasing water deficit stress: oxidative stress and defence mechanisms. Physiol Plant 93:25–30
Sherman TD, Gardeur TL, Lax AR (1995) Implications of the phyogenetic distribution of polyphenol oxidase in plants. In: Lee CY, Whitaker JR (eds) Enzymatic browning and its prevention. American Chemical Society, Washington, DC, pp 103–119
Sherwin HW (1995) Desiccation tolerance and sensitivity of vegetative plant tissue. PhD thesis, University of Natal, Durban, South Africa
Sherwin HW, Farrant JM (1998) Protection mechanism against excess light in the resurrection plants Craterostigma wilmsii and Xerophyta viscosa. Plant Growth Regul 24:203–210
Smith-Espinoza CJ (2001) Analysis of ABA and drought stress mediated gene expression in the desiccation tolerant resurrection plant Craterostigma plantagineum. PhD thesis, University of Cologne, Germany
Sullivan ML, Hatfield RD (2006) Polyphenol oxidase and o-diphenols inhibit post-harvest proteolysis in red clover and alfalfa. Crop Physiol Met 46:662–670
Sze H, Li XH, Palmgren MG (1999) Energization of plant cell membranes by H+-pumping ATPases. Regulation biosynthesis. Plant Cell 11:677–689
Tuba Z, Proctor MCF, Csintalan Zs (1998) Ecophysiological responses of homoichlorophyllous and poikilichlorophyllous desiccation tolerant plants: a comparison and an ecological perspective. Plant Growth Regul 24:211–217
Tuba Z, Smirnoff N, Csintalan Zs, Nagy Z, Szente K (1997) Respiration during slow desiccation of the poikilochlorophyllous desiccation tolerant plant Xerophyta scabrida at present-day CO2 concentrations. Plant Physiol Biochem 35:381–386
Tsugane K, Kobayashi K, Niwa Y, Ohba Y, Wada K, Kobayashi H (1999) A recessive Arabidopsis mutant that grows photoautotrophically under salt stress shows enhanced active oxygen detoxification. Plant Cell 11:1195–1206
Veljovic-Jovanovic S, Kukavica B, Stevanovic B, Navari-Izzo F (2006) Senescence- and drought-related changes in peroxidase and superoxide dismutase isoforms in leaves of Ramonda serbica. J Exp Bot 57:1759–1768
Vicré M, Lerouxel O, Farrant J, Lerouge P, Driouich A (2004) Composition and desiccation-induced alterations of the cell wall in the resurrection plant Craterostigma wilmsii. Physiol Plant 120:229–239
Wang Y, Tian S, Xu Y, Qin G, Yao H (2004) Changes in the activities of pro- and anti-oxidant enzymes in peach fruit inoculated with Cryptococcus laurentii or Penicillium expansum at 0 or 20°C. Postharvest Biol Technol 34:21–28
Watson BS, Asirvatham VS, Wang L, Sumner LW (2003) Mapping the proteome of Barrel Medic (Medicago truncatula). Plant Physiol 131:1104–1123
Xiong L, Zhu J (2002) Plant responses to osmotic stress. Plant Cell Environ 25:131–139
Yang P, Liang Y, Shen S, Kuang T (2006) Proteome analysis of rice uppermost internodes at the milky stage. Proteomics 6:3330–3338
Acknowledgments
We thank Dr. Shihua Shen, Institute of Botany, Chinese Academy of Sciences and Ms. Anne Bräutigam, Max-Planck-Institut für Züchtungsforschung, Germany, for their excellent discussion and aids on establishment of proteomic platform, and Professor Dr. Dorothea Bartels, IMBIO (Molekulare Physiologie und Biotechnologie der Pflanzen), University of Bonn for critical reading and valuable suggestions. The project was supported by National Natural Science Fundation of China (no. 30400027) and National Basic Research Program of China (973 Programs) (no. 2006CB403206).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Jiang, G., Wang, Z., Shang, H. et al. Proteome analysis of leaves from the resurrection plant Boea hygrometrica in response to dehydration and rehydration. Planta 225, 1405–1420 (2007). https://doi.org/10.1007/s00425-006-0449-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-006-0449-z