
Abstract
Pectin methylesterases (PME, EC. 3.1.1.11) are enzymes that demethylesterify plant cell wall pectins in muro. In Arabidopsis thaliana, putative PME proteins are thought to be encoded by a 66-member gene family. This study used real-time RT-PCR to gain an overview of the expression of the entire family at eight silique developmental stages, in flower buds and in vegetative tissue in the Arabidopsis. Only 15% of the PMEs were not expressed at any of the developmental stages studied. Among expressed PMEs, expression data could be clustered into five distinct groups: 19 PMEs highly or uniquely expressed in floral buds, 4 PMEs uniquely expressed at mid-silique developmental stages, 16 PMEs highly or uniquely expressed in silique at late developmental stages, 16 PMEs mostly ubiquitously expressed, and 1 PME with a specific expression pattern, i.e. not expressed during early silique development. Comparison of expression and phylogenetic profiles showed that, within phylogenetic group 2, all but one PME belong to the floral bud expression group. Similar results were shown for a subset of one of the phylogenetic group, which differed from others by containing most of the PMEs that do not possess any PRO part next to their catalytic part. Expression data were confirmed by two promoter:GUS transgenic plant analysis revealing a PME expressed in pollen and one in young seeds. Our results highlight the high diversity of PME expression profiles. They are discussed with regard to the role of PMEs in fruit development and cell growth.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The plant cell wall is a highly complex structure consisting of cellulose, hemicellulose, pectins and proteins, which provide cell shape and modulate growth. Among these polysaccharides, pectins represent nearly 35% of the total dry weight in dicotyledonous cell walls (Cosgrove 2001). Pectins, which consist of a backbone of homogalacturonan and rhamnogalacturonan, are polymerised in the cis Golgi apparatus. Homogalacturonans are subsequently methylesterified in the medial Golgi apparatus by pectin methyltransferases (PMT). The addition of side chains to the backbones gives rhamnogalacturonan I, rhamnogalacturonan II and xylogalacturonan. It occurs in the trans Golgi apparatus, before exocytosis of pectins to the cell wall where they contribute to the structuration of the middle lamella and the primary cell wall. Pectin synthesis and metabolism is a series of complex enzymatic processes where PME plays a central role. PMEs (EC. 3.1.1.11) are enzymes belonging to the class 8 of the carbohydrate esterases (CAZY: http://www.afmb.cnrs-mrs.fr/CAZY/ and Coutinho et al. 2003). They are involved in the demethylesterification of pectins in muro, creating carboxyl groups which play a role in the complexation of calcium ions, increasing cell wall firmness (Willats et al. 2001). In addition, action of PMEs is required prior to the action of polygalacturonases (PG), which are involved in the degradation of the pectic matrix during fruit ripening (Wakabayashi et al. 2003). Thus, PMEs contribute both to the firming and softening of the cell wall.
The crystalline structure of a plant PME has been determined and compared with bacterial PMEs, which allowed the identification of conserved amino acids residues (Johansson et al. 2002). On this basis, in the Arabidopsis genome, 66 genes have been suggested to potentially encode pectin methylesterases. PME sequences consist of two types: the type I, which has a long N-terminal PRO region (pre-pro-proteins) and the type II which has a short or absent PRO region (Micheli 2001). Several functions for the PRO region have been suggested, including targeting of PME to the cell wall, correct folding of the enzyme and inhibition of the PME activity (Micheli 2001). Only the mature part of the protein can be extracted from the cell wall, but the exact mechanism of the PRO-mature cleavage remains unknown. Numerous studies (see Giovane et al. 2004 for review) have shown that the PRO region shows similarities with PME inhibitors (PMEI), and it was recently demonstrated that the PRO region plays a role in preventing an early demethylesterification of pectins in the Golgi apparatus (Bosch et al. 2005).
So far, very few PME sequences have been shown to code proteins biochemically active as carbohydrate esterases. The high number of putative PME sequences in the Arabidopsis genome suggests that these proteins could be involved in different developmental processes and/or act on different classes of pectins. PMEs have been notably shown to play a role in such diverse processes as cambial cell differentiation (Micheli et al. 2000), hypocotyl elongation (Al-Qsous et al. 2004), microsporogenesis (Lacoux et al. 2003) and as host receptor for the tobacco mosaic virus movement protein (Chen and Citovsky 2003). Recently, VANGUARD1, encoding a pectin methylesterase homologous gene, has been shown to be specifically localised in Arabidopsis pollen grains and to play a central role in pollen tube growth in the style (Jiang et al. 2005). NtPPME1 was shown to have the same function in tobacco (Bosch et al. 2005). In Medicago sativa and Medicago truncatul, eight PMEs genes have been isolated, two of them showing a flower-specific expression pattern, while another one was shown to be expressed in roots during the nodulation process (Rodriguez-Llorente et al. 2004). Among all developmental processes studied so far, the main highlight has been put on the implication of PME in fruit development. PME have notably been shown to be involved in fruit maturation in tomato (Eriksson et al. 2004), Phaseolus (Stolle-Smits et al. 1999), strawberries (Castillejo et al. 2004), berries (Barnavon et al. 2001), bananas (Nguyen et al. 2002). The results show either an increase or a decrease in PME activity during fruit maturation.
Silique formation in Arabidopsis is a complex process where the embryo and post-fertilization carpel development occur in parallel for up to 20 days. At the embryo level, development involves three distinct phases: (1) early morphogenesis, inlcuding division and segmentation of the zygote (up to 7 days after flowering, DAF); (2) maturation phase, which includes storage of lipids (up to 17 DAF) and (3) late maturation phase (up to 20 DAF) which mainly corresponds to the acquisition to dessication (Baud et al. 2002). After ovule fertilization, cells of the carpel undergo division, expansion and differentiation to form the various tissues of the mature silique: endocarp, mesocarp and exocarp. Longitudinal growth is notably due to cell expansion in all layers of the carpel, while final silique maturation involves the formation of the dehiscence zone (DZ), a non-lignified separation layer placed between lignified cells, which will allow seeds dispersal (Ferrandiz 2002). Therefore, Arabidopsis silique development requires major changes in plant cell wall composition, structure and hydratation level. Proteins involved in those processes, such as PMEs, are likely to undergo drastic variation in the expression of their genes and/or activity levels.
A wealth of public microarray data are available for some silique developmental stages. However, only 60 out of the 66 PMEs are present on the Affymetrix ATH1 genome array, and it was shown that some discrepancies could exist between microarrays and real-time RT-PCR analysis of gene expression (Czechowski et al. 2004). The latter study notably revealed, using real-time RT-PCR, expression specificity between members of conserved gene families unseen in microarray data.
This has prompted us to use real-time RT-PCR to study the relative expression of the 66 genes encoding putative PMEs throughout eight silique developmental stages. For two of the PMEs, RT-PCR results were confirmed using promoter–GUS fusions showing the localisation of expression. The results are discussed with regard to a phylogenetic analysis and compared to freely available microarray data.
Material and methods
Phylogenetic analysis
Sixty-six putative Arabidopsis PME protein sequences were retrieved from the CAZY website (http://www.afmb.cnrs-mrs.fr/CAZY/CE_8.html) and aligned using ClustalX (Thompson et al. 1997). Phylogenetic tree was drawn with the neighbour-joining method using MEGA software (molecular evolutionary genetics analysis, Kumar et al. 2001) using pairwise deletion; 1,000 replicates were used for bootstrap analysis and the cut-off value was 50%.
Plant material and growth conditions
Arabidopsis thaliana (Col-0, obtained from the Nottingham Arabidopsis Stock Center, Nottingham, UK) wild-type plants were grown on soil in greenhouse under a 16-h photoperiod and day/night temperature of 22°C/20°C. To follow silique development, newly opened flowers were labelled daily for 20 days on 80 plants. The material (vegetative tissue, flower buds and siliques at various development stages) was harvested and frozen under liquid nitrogen before storage at −80°C. For young siliques, floral parts were dissected. Sampling was replicated once.
RNA extraction and cDNA synthesis
Tissues were ground in liquid nitrogen and after a preliminary polyphenol and polysaccharide precipitation (Gehrig et al. 2000), RNA was extracted from supernatant using a hot phenol purification protocol (Verwoerd et al. 1989). Total RNA was treated with amplification-grade DNase I (Invitrogen). Absence of genomic DNA contaminants was subsequently confirmed by PCR using primers spanning intronic sequences. RNA integrity was assessed by loading 1 μg of RNA on a 1.2% agarose gel, and RT reactions were performed using the SuperScriptII reverse transcriptase (Invitrogen) following the manufacturer’s instructions. Efficiency of RT was assessed by using control genes encoding ACTIN 2-7-8, to obtain Ct in the range of 22–23. The PME expression study was realised on these cDNAs.
Primer design
Sixty-six putative PME nucleotide sequences (excluding pseudogenes), retrieved from the CAZY web server (http://www.afmb.cnrs-mrs.fr/CAZY/CE_8.html), were used to design primers flanking, when possible, an intronic sequence using the LC Probe Design© software (Roche; Basel, Switzerland). Primers were designed to obtain an amplified product in a range of 200–500 bp, and their specificity was tested using BLASTn searches (http://www.ncbi.nlm.nih.gov/BLAST/). The specificity of the primers was subsequently tested by real-time PCR using increasing dilution of the template. For all primers, the efficiency of the PCR was in the range of 90–100%. The primer sequences are given in Supplementary material 1
Real-time PCR conditions and data analysis
Real-time PCR was performed on a Roche LightCycler® using the FastStart DNA MasterPLUS SYBR Green I kit (Roche) according to the manufacturer’s protocol. The cDNA pool was diluted to a concentration of 5 ng/μl of which 5 μl were used, together with 0.25 mM of specific primers, in a final volume of 20 μl. The following thermal profile was used for all PCR: 95°C for 10 min, 45 cycles of 95°C for 10 s, 64°C for 15 s and 72°C for 15 s. This PCR reaction was followed by a melting curve analysis from 48 to 95°C in 470 s. Crossing threshold (Ct) values, which are the PCR cycle numbers at which the accumulated fluorescent signal in each reaction crosses a threshold above background, were obtained with the LightCycler® software 3.5 (Roche) using the second derivative maximum method. Ct values are a function of the amplification efficiency of the respective PCR. PME expression was calculated using the RelQuant© software (Roche), as a relative expression, for a set 100 bp fragment; compared to the internal control ACTIN 2-7-8 (Schenk et al. 2003). For this purpose, the ΔCt (ΔCt = Ct(PME)−Ct(ACTIN 2-7-8)) was used in the following mathematic formula [PME]=2−ΔCt and was corrected by the primer efficiency, based on relative standard curves describing the PCR efficiencies of each target and reference genes. The quantifications were realised on separate cDNAs pools coming from two independent plant-sampling experiments. Data presented correspond to one of the sampling (S1), but results, checked for ten PMEs, were similar for the second experiment (S2=1.40×S1, R 2=0.956; Supplementary material 2). Data analysis and hierarchical clustering were performed according to EPClust (http://www.ep.ebi.ac.uk/EP/EPCLUST/) using the correlation measure-based distance and the average linkage (average distance, UPGMA) clustering method.
Analysis of promoter activity
The promoters of At1g69940 and At5g49180 were amplified from genomic DNA using AmpliTaq polymerase (Applied Biosystems) using gene-specific primers pairs:
PCR products were cloned into the pGEM-T Easy vector (Promega), sequenced and subcloned into the binary vector pBI101.3 upstream of the GUS coding sequence. Plant transformation, using Agrobacterium tumefaciens strain LBA4404, was according to the floral dip method (Clough and Bent 1998). Transformants were selected on kanamycin at 80 μg/ml. GUS staining was performed using 0.3 mg/ml X-Gluc in 50 mM phosphate buffer (pH 7) containing 1 mM potassium ferricyanide and 1 mM potassium ferrocyanide. Tissues were incubated for 14 h at 37°C, subsequently destained in ethanol series up to 50%, post-fixed in FAA (50% ethanol, 5% formaldehyde, 10% acetic acid) for 30 min and finally dehydrated completely in ethanol series up to 100%.
Results
Expression of PMEs during silique development
In Arabidopsis, silique development, which correlates with seed development, showed a steep increase in length and diameter up to 7–8/9–11 DAF, followed by a sole increase in diameter up to 12–14 DAF (Fig. 1). From 15 DAF, silique starts to dehydrate and prepare for dehiscence, which is concomitant with seed dehydration. This developmental pattern of seed and silique is likely to imply changes in cell wall composition and thereby in the expression and activities of enzymes involved in this process. Indeed, our analysis showed that 85% of all PMEs were expressed at one or more stages of development, and that their expression could be clustered into five distinct groups according to their kinetics and level of expression (Fig. 2). These groups were named A to E: Group A consisted of 16 PMEs mainly highly or uniquely expressed in silique, particularly during late developmental phases. Group B was restricted to one member, At1g23200, which had a very specific profile of expression, at low levels in vegetative and flower stages, not expressed during the 1–2 to 3–4 stages, and expressed at relatively high levels from stages 5–6 onwards. Group C consisted of 16 members, most of them expressed throughout silique development. However, in this latter group, some members (notably At3g05620, At5g04970, At5g47500 and At5g61680) differed from this general expression pattern. Group D was the largest, having 19 members showing high or unique expression patterns in flower buds. Group E contained 4 PMEs whose expression was restricted to early to mid-stage of silique development, and was present neither in vegetative stage nor in flower buds. These results pointed out the high diversity of expression profiles within a gene family together with putative-specific roles for various PMEs at a given stage. Moreover, within each group, relative expression levels differed between members to a very high extent (Supplementary material 3). For instance, within group A, at stage 15+, transcript expression varied between 0.021 and 424 for At5g64640 and At3g60730, respectively. In addition, summing the expression of all PMEs at a given stage brought some information on the global PME expression during silique development, as well as the relative contribution of each PME at each stage (Supplementary material 3). The PME expression level was relatively low, just above 200, in our vegetative control, which could be related to the physiological stage of the plants onto which the siliques were sampled (growth was close to an end). It is therefore likely that all PMEs expressed in young tissues, and in roots, were not taken into account. The total PME expression on an overall was very high in flower buds, mainly due to the high expression of several PMEs such as At1g69940, At2g47040, At3g62170 and At4g33220. During silique development, a biphasic PME expression pattern was shown with an increase in total expression up to stage 5–6, a drop at stage 7–8 and relatively high levels from stage 9–11 onwards. This would imply that, while specific isoforms could be involved at very specific stage of dry fruit maturation (At3g60730, At5g49180...), others are ubiquitously expressed throughout silique development and throughout the whole plant (At4g33220, At3g49220...).

Silique developmental stages used for cDNA preparation. Flowers were labelled daily on 80 plants and samples were harvested after 20 days. For stage 1–2, flower parts were dissected. FB floral bud, 1–2 to 15+: 1–2 to 15+ days after flowering (DAF)

Expression of PME was studied during silique development by real-time RT-PCR and was quantified relatively to the constitutive control ACTIN 2-7-8. All results are expressed as the ratio [PME/ACTIN 2-7-8]×1,000, and are shown as increasing red colour intensity. The cluster was realised with expression profile data clustering (EPClust) at the European Bioinformatic Institute (http://www.ep.ebi.ac.uk/EP/EPCLUST/) using the data presented in the supplementary material concerning the 56 expressed PMEs. Values represent the relative PME expression as the ratio [PME/ACTIN 2-7-8]×1,000. V vegetative control; FB floral bud; 1–2 to 15+: 1–2 to 15+ DAF. The vegetative control used in our study corresponded to leaves (rosettes and caulines) and stems harvested from the same batch of plants as the one used for the sampling of the siliques
Phylogenetic analysis of the PME family
A phylogenetic tree was built to relate sequences to expression profiles (Fig. 3). The PME family clustered in four distinct groups (named group 1 to 4). Group 3 was only composed of two members, At5g09760 and At5g64640. Apart from two sequences (At1g11370 and At4g33220), all the PMEs containing a PRO region clustered into groups 1 to 3, whereas PMEs which did not contain a PRO sequence were in group 4.

Phylogenetic analysis of the PME protein sequences. Amino-acid sequences were aligned using ClustalX and the phylogenetic tree was drawn using the molecular evolutionary genetics analysis (MEGA) package with the neighbour-joining method. Bootstrap values are indicated and are calculated for 1,000 replicates. The analysis indicates that four subgroups (1, 2, 3, 4) can be separated on the basis of the robustness of the node
PME expression in floral bud
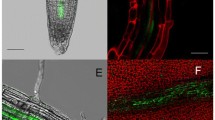
In order to relate expression patterns and phylogenetic analysis, PME expression values are shown for two groups of the phylogenetic tree (Table 1). One of the striking features of this was that, among the phylogenetic group 2 all but one PMEs were highly or uniquely expressed in flower bud, and clustered into expression group D. Similar results were obtained for a subset (bottom part) of phylogenetic group 4 (Table 1). This points out the high number of PMEs expressed in flowers in Arabidopsis. Moreover, our study revealed that At1g69940, belonging to expression group D and phylogenetic group 4 (no PRO part), showed an expression pattern restricted to floral buds, at a relatively high level (83.79). In order to localise At1g69940 promoter activity, Arabidopsis plants were transformed with a promoter:GUS construct. GUS staining showed that promoter activity was restricted to mature pollen grains in the anthers and on the stigma (Fig. 4a, b). Moreover, GUS staining was visible in pollen tubes within the style (Fig. 4c). Similar results were obtained for two additional PMEs, At5g07410 and At3g06830 (data not shown). These PME isoforms could therefore be involved in pollen maturation and/or pollen tube growth.

Promoter activity of At1g69940 and At5g49180 monitored by GUS activity. a PrAt1g69940:GUS expression showing high promoter activity in anthers and especially in pollen grains (b). c PrAt1g69940:GUS expression was sustained in pollen tubes growing within the style (arrow). d PrAt5g49180:GUS expression during silique development showing promoter activity up to stage 5–6 DAF (arrow). e, f PrAt5g49180:GUS at stage 1–2 DAF showing that promoter activity (arrow) is restricted to the central area of the newly fertilized ovule
PME expression in developing seeds
As described previously, among the phylogenetic group 2, all but one PME, At5g49180, had a preferential expression in floral bud. Real-time RT-PCR expression of At5g49180 transcripts was shown to be high in young siliques, between 1–2 and 7–8 DAF, showing a peak at 3–4 DAF (Fig. 2 and Supplementary material 3). Using a GUS fusion, we were able to confirm the pattern of At5g49180 promoter activity (Fig. 4d), which was particularly high in young seed (1–2 to 3–4 DAF, Fig. 4e). Expression was notably present in the micropyle area of the ovule just after fertilization (Fig. 4f). From this data, it is difficult to determine whether the expression is restricted to the embryo, but our data suggest that At5g49180 could be involved in the early events of seed development in Arabidopsis. Similar RT-PCR quantification and promoter–GUS staining were found for At1g11590 (data not shown), belonging to phylogenetic group 1 and expression group E.
Discussion
The expression of the entire Arabidopsis PME gene family was measured during silique development. A phylogenetic analysis of the gene family showed that Arabidopsis PMEs could be clustered into four distinct groups, and that one, group 4, was restricted to PMEs without PRO part. This sustained the common hypothesis that plant PMEs with a PRO region are likely to have evolved later (Micheli 2001). When compared to the tree of Markovic and Janecek (2004), based on 58 sequences, our analysis showed a reduced number of groups (from 7 to 4), mainly due to the fact that a bootstrap level of 50% was used. Despite these differences, group 3 was similar to group PlantX1, and very distinct from all the other groups in both studies, which could imply a specific role for the products of those two genes during plant development. This tree served as a basis for further comparisons between PMEs expression patterns and PME protein sequences.
PMEs expression showed a high diversity of profiles during silique development. Such diversity of expression patterns was obtained for 88 MADS-box transcription factors (de Folter et al. 2004) as well as for PMEs during fruit development in a wide range of species. In strawberry, FaPE1 was uniquely expressed in the ripening fruit, whereas FaPE3 was ubiquitous (Castillejo et al. 2004). In green bean, PE2 was very abundant during early pod development (Ebbelaar et al. 1996). In tomato, PE2 was a fruit-specific isoform highly expressed 25 days after anthesis (Eriksson et al. 2004) and in grape berry, specific PMEs were expressed from veraison stage onwards (Barnavon et al. 2001). Although the processes involved in fleshy and dry fruit development are different, it is likely that in both cases, PMEs could play a major role in modifying pectins leading to fruit maturation. Moreover, for a similar expression pattern, our study pointed out the high differences of expression levels between isoforms (expression group D or E for instance). Such differences were notably shown during silique development in Arabidopsis for the MADS-box family using microarray analysis, varying, for instance, between 0.46 and 11.04 for AGL89 and SEP2, respectively (de Folter et al. 2004). A discrepancy between expression ratio was shown between real-time RT-PCR and microarray approaches during the study of the expression of all transcription factors families: for instance for COL7, the shoot to root (S/R) expression ratio was 8,678 using RT-PCR and 101 using microarray (Czechowski et al. 2004). In our study, differences were obtained when comparing our RT-PCR results with some of the expression data freely available on the TAIR website (http://www.arabidopsis.org). For instance, At5g19730 had a 3–4 DAF to vegetative expression ratio of 143, whereas the globular to vegetative expression ratio was only 1.3 from microarray data. Similar discrepancies were observed, for example, for At3g17060 and At1g11590. The use of real-time RT-PCR significantly increases the sensitivity, accuracy and precision of the measurement, especially when dealing with a multigene family (Czechowski et al. 2004).
The low total PME expression at stage 7–8 was concomitant with the arrest in maximum length growth of both the silique and the embryo (Baud et al. 2002). Stage 7–8 DAF could be the PME expression shift point between isoforms involved in either length growth or silique maturation. Although a high transcript level does not necessarily translate into a high level of protein and enzyme activity, a role for PME in silique growth and differentiation could be suspected. A study on green bean did show that PME activity dropped at stage III, where the maximum length of the pod was reached, before increasing during pod maturation (Stolle-Smits et al. 1999). The increase in PME transcript quantities (notably At3g60730, At5g51490) in the late developmental stages of the silique (9–11 onwards) could increase PME activity. This would lead to a modification of the pectic network through the action of both PME and polygalacturonases (PG), enzymes involved in fruit dehiscence (Roberts et al. 2000). The analysis of homogalacturonans methylesterification patterns (blockwise or non-blockwise), and of PME activity regulation (ionic interaction, pH...) during silique development will help dissecting the role of the pectin network remodelling during fruit development.
Among the ubiquitously expressed PMEs identified in our study, microarray data anlaysis showed that At4g02330 and At3g49220 expressions in seedlings were positively regulated by brasssinosteroid and ethylene, respectively (Goda et al. 2002; De Paepe et al. 2004). This suggests a role for plant growth regulators in the signalling cascade leading to PME transcript regulation. The potential diversity of the roles of putative PMEs in modulating cell wall structure in plant development could be further illustrated by At2g45220, which was notably expressed in vegetative tissues. Microarray analysis showed that At2g45220 expression was regulated by anoxia (Loreti et al. 2005), by auxin (Goda et al. 2004), by cold (Kreps et al. 2002) as well as during the weight-induced secondary growth in Arabidopsis (Ko et al. 2004). Numerous other putative PMEs, such as At1g02810, At2g43270 and At3g10720, which were all expressed in our vegetative control, were upregulated during cell differentiation leading to wood formation in Arabidopsis (Ko et al. 2004). In a recent study, At2g43270 and At3g14310 were both shown to be highly expressed in phloem when compared to xylem and non-vascular secondary tissues of root-hypocotyl (Zhao et al. 2005), suggesting that those isoforms could play a role in conducting element differentiation.
As shown for At2g45220, several PMEs are also likely to be involved in stress response. For instance, At1g11580 expression, belonging to our expression group A, was notably shown to be induced (×5.3) by the herbicide isobaxen in suspension cultured cells (Manfield et al. 2004) as well as in distal leaf tissues after 72 h inoculation with the incompatible fungal pathogen Alternaria brassicola (Schenk et al. 2003). Such PME transcript regulation was shown in Medicago following inoculation of roots with S. meliloti (Rodriguez-Llorente et al. 2004), showing that the production of PMEs following biotic interaction might be widespread.
In our study, a correlation was shown between expression profiles and the phylogenetic tree, notably for group 2 and a subset of group 4. Such expression-to-sequence relationship was previously shown for some members of the MIKC subset of the MADS-box TF family (de Folter et al. 2004). Although the majority of the PMEs of these two groups (2 and 4) were previously shown to be highly expressed in pollen (Pina et al. 2005), our study brings a new insight into the expression profiles of those genes. For instance, At2g47040, which encodes VANGUARD1 (Jiang et al. 2005) is indeed very highly expressed in flower bud (273), but also throughout the entire silique developmental process, although with a smaller intensity (up to 26). Microarray data analysis showed a similar expression pattern, which suggests that the role of At2g47040 in pollen tube growth in the style and transmitting tract, as shown in knock-out mutant, might therefore not be the only one for this protein, since the mutant had also smaller silique with few seeds (Jiang et al. 2005). Our results suggest that this latter phenotype could either be related to the low ovule fertilization (linked to the expression of VGD1 in pollen) or to an independent phenotype due to the lack of VGD1 expression in the silique. The relative At2g47040 to At2g47030 expression ratio was 34 in our experiment, which was in the same magnitude as the one shown by Jiang et al. (2005), but could not be compared with micorarray data as At2g47030 is one of the 6 PMEs which is not available on the Affimetryx ATH1 genome array. A recent study on pollen transcriptome revealed that among the PMEs found to be expressed in floral bud in our study (expression group D), 11 out of 19 had a relative expression level above 1,000 in microarray experiments (Pina et al. 2005). Our study further revealed that At1g69940, a PME without PRO part, which is not present on the microarray data presented by Pina et al. (2005) showed an expression pattern restricted to floral bud, and more precisely in pollen grains using promoter–GUS fusions. In that respect, the localisation of expression of At1g69940 was similar to that shown for At2g47030, At2g47040 and At3g62170 (Jiang et al. 2005). It is very likely that a high redundancy of PME expression exists in pollen. Numerous studies have shown that PME or PME-like genes were expressed in pollen, notably in flax (Lacoux et al. 2003), in Zea (Wakeley et al. 1998) and in Nicotiana (Bosch et al. 2005). However, it is not until recently that the role of PME as a regulator of pollen tube growth, suspected by Parre and Geitmann (2005) in Solanum chacoense, was demonstrated in tobacco (Bosch et al. 2005). The role of At1g69940, among the other pollen-expressed PMEs, in the control of homogalacturonan esterification pattern, therefore, needs to be addressed using a reverse genetic approach. In that context, it would also be of interest to study the regulation of these numerous pollen-expressed PMEs by any of the two PME inhibitors, At-PMEI1 and At-PMEI, present in pollen (Wolf et al. 2003).
Within phylogenetic group 2, only one PME differed from the general floral bud expression pattern. At5g49180 expression clustered with that of At1g11590, At4g02300 and At4g03930, showing a specificity of expression during early stages of silique development, up to stages 7–8 DAF, and specifically in seed for At5g49180 and At1g11590 (data not shown). While microarray data analysis confirmed high expression for At5g49180 at globular/torpedo stages, no such results were available for the three other members of our expression group E (http://www.arabidopsis.org). At1g11590 and At4g03930 promoters were 87% identical, containing GARE motifs necessary for gibberelin-upregulated expression in seeds (Suto and Yamauchi 2003). At4g02300 and At5g49180 promoters contained embryo-specific expression motifs (Kim et al. 1997). Thus, cluster group E is likely to contain young seed-specific PMEs. Activities of PMEs increased in yellow cedar seeds at the onset of dormancy breakage and germination (Ren and Kermode 2000) as well as in flax during early seedling development (Alexandre et al. 1997). Moreover, in soybean seeds, pectin esterase activity had a biphasic phase with a maximum in axes at 24 DAF, a minimum at 56 DAF and an increase during desiccation phase (Koch et al. 1999). A biphasic expression was shown for PME transcripts during silique development in our study, and genes, such as At5g49180, At1g11590, At4g02300, could contribute to a first peak of PME activity which might be involved in the early event of embryo/seed development.
Conclusion
The expression kinetics of an entire cell wall gene family during silique development in Arabidopsis was determined. The expression of most PMEs was detected in flowers or siliques, and the gene family could be split into five groups according to their expression patterns. Correlations were made between expression profiles and phylogenetic analysis, showing that closely related sequences could have similar expression kinetics. Real-time RT-PCR studies, together with promoter–GUS fusions will allow the functional genomic approach to be targeted on candidates important during specific silique developmental stages. This will help the precise function of the various PMEs to be determined.
Abbreviations
- PME:
-
Pectin methylesterase
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- DAF:
-
Days after flowering
References
Alexandre F, Morvan O, Gaffe J, Mareck A, Jauneau A, Dauchel H, Balangé AP, Morvan C (1997) Pectin methylesterase pattern in flax seedlings during their development. Plant Physiol Biochem 35:427–436
Al-Qsous S, Carpentier E, Klein-Eude D, Burel C, Mareck A, Dauchel H, Gomord V, Balangé AP (2004) Identification and isolation of a pectin methylesterase isoform that could be involved in flax cell wall stiffening. Planta 219:369–378
Barnavon L, Doco T, Terrier N., Ageorges A, Romieu C, Pellerin P (2001) Involvement of pectin methyl-esterase during the ripening of grape berries: partial cDNA isolation, transcript expression and changes in the degree of methyl-esterification of cell wall pectins. Phytochemistry 58:693–701
Baud S, Boutin JP, Miquel M, Lepiniec L, Rochat C (2002) An integrated overview of seed development in Arabidopsis thaliana ecotype WS. Plant Physiol Biochem 40:151–160
Bosch M, Cheung AY, Hepler PK (2005) Pectin methylesterase, a regulator of pollen tube growth. Plant Physiol 138:1334–1346
Castillejo C, Delafuente JI, Iannetta P, Botella MA, Valpuesta V (2004). Pectin esterase gene family in strawberry fruit. Study of FaPE1, a ripening-specific isoform. J Exp Bot 55:909–918
Chen MH, Citovsky V (2003) Systemic movement of a tobamovirus requires host cell pectin methylesterase. Plant J 35:386–392
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Cosgrove DJ (2001) Wall structure and wall loosening. A look backwards and forwards. Plant Physiol 125:131–134
Coutinho PM, Stam M, Blanc E, Henrissat B (2003) Why are there so many carbohydrate-active enzyme-related genes in plants? Trends Plant Sci 8:563–565
Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK (2004) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J 38:366–379
De Folter S, Busscher J, Colombo L, Losa A, Angenent G (2004) Transcript profiling of transcription factor genes during silique development in Arabidopsis. Plant Mol Biol. 56:351–366
De Paepe A, Vuylsteke M, Van Hummelen P, Zabeau M, Van Der Straeten D (2004) Transcriptional profiling by cDNA-AFLP and microarray analysis reveals novel insights into the early response to ethylene in Arabidopsis. Plant J 39:537–559
Ebbelaar ME, Tucker GA, Laats MM, van Dijk C, Stolle-Smits T, Recourt K (1996) Characterization of pectinases and pectin methylesterase cDNAs in pods of green beans (Phaseolus vulgaris L.). Plant Mol Biol 31:1141–1151
Eriksson EM, Bovy A, Manning K, Harrison L, Andrews J, Da Silva J, Tucker GA, Seymour BG (2004) Effects of the colorless non ripening mutation on cell wall biochemistry and gene expression during tomato fruit development and ripening. Plant Physiol 136:4184–4197
Ferrandiz C (2002) Regulation of fruit dehiscence in Arabidopsis. J Exp Bot 53:2031–2038
Gehrig HH, Winter K, Cushman JC, Borland AM, Taybi T (2000) An improved RNA isolation method for succulent plant species rich in polyphenols and polysaccharides. Plant Mol Biol Rep 18:369–376
Giovane A, Servillo L, Balestrieri C, Raiola A, D’Avino R, Tamburrini M, Ciardiello MA, Camardella L (2004) Pectin methylesterase inhibitor. Biochim Biophys Acta Proteins Proteomics 1696:245–252
Goda H, Shimada Y, Asami T, Fujioka S, Yoshida S (2002) Microarray analysis of brassinosteroid-regulated genes in Arabidopsis. Plant Physiol 130:1319–1334
Goda H, Sawa S, Asami T, Fujioka S, Shimada Y, Yoshida S (2004) Comprehensive comparison of auxin-regulated and brassinosteroid-regulated genes in Arabidopsis. Plant Physiol 134:1555–1573
Jiang L, Yang S, Xie LF, Puah CS, Zhang XQ, Yang WC, Sundaresan V, Ye D (2005) VANGUARD1 encodes a pectin methylesterase that enhances pollen tube growth in the Arabidopsis style and transmitting tract. Plant Cell 17:584–596
Johansson K, El-Ahmad M, Friemann R, Jörnvall H, Markovic O, Eklund H (2002) Crystal structure of plant pectin methylesterase. FEBS Lett 514:551–555
Kim SY, Chung HJ, Thomas TL (1997) Isolation of a novel class of bZIP transcription factor that interact with ABA-responsive and embryo-specification elements in the Dc3 promoter using a modified yeast one-hybrid system. Plant J 11:1237–1251
Ko JH, Han KH, Park S, Yang J (2004) Plant body weight-induced secondary growth in Arabidopsis and its transcription phenotype revealed by whole-transcriptome profiling. Plant Physiol 135:1069–1083
Koch JL, Horbowicz M, Obendorf RL (1999) Methanol, pectin and pectin esterase changes during soybean seed maturation. Seed Sci Res 9:311–320
Kreps JA, Wu Y, Chang H-S, Zhu T, Wang X, Harper JH (2002) Transcriptome changes for Arabidopsis in response to salt, osmotic and cold stress. Plant Physiol 130:2129–2141
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Lacoux J, Gutierrez L, Dantin F, Beaudoin B, Roger D, Lainé E (2003) Antisense transgenesis of tobacco with a flax pectin methylesterase affects pollen ornamentation. Protoplasma 222:205–209
Loreti E, Poggi A, Novi G, Alpi A, Perata P (2005) A genome-wide analysis of the effects of sucrose on gene expression in Arabidopsis seedlings under anoxia. Plant Physiol 137:1130–1138
Manfield IW, Orfila C, McCartney L, Harholt J, Bernal AJ, Vibe Scheller H, Gilmartin PM, Mikkelsen JD, Knox JP, Willats WGT (2004) Novel cell wall architecture of isobaxen-habituated Arabidopsis suspension cultured cells: global transcript profiling and cellular analysis. Plant J 40:260–275
Markovic O, Janecek S (2004) Pectin methylesterases: sequence–structural features and phylogenetic relationships. Carbohydr Res 339:2281–2295
Micheli F (2001) Pectin methylesterases: cell wall enzymes with important roles in plant physiology. Trends Plant Sci 6:414–419
Micheli F, Sundberg B, Goldberg R, Richard L (2000) Radial distribution pattern of pectin methylesterases across the cambial region of hybrid aspen at activity and dormancy. Plant Physiol 124:191–199
Nguyen BL, Van Loey A, Fachin D, Verlent I, Hendrickx IM (2002) Purification, characterization, thermal and high-presure inactivation of pectin methylesterase from bananas (cv. Cavendish). Biotechnol Bioeng 78:683–691
Parre E, Geitmann A (2005) Pectin and the role of the physical properties of the cell wall in pollen tube growth of Solanum chacoense. Planta 220:582–592
Pina C, Pinto F, Jeijo JA, Becker JD (2005) Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol 138:744–756
Ren C, Kermode AR (2000) An increase in pectin methyl esterase activity accompanies dormancy breakage and germination of yellow cedar seeds. Plant Physiol 124:231–242
Roberts JA, Whitelaw CA, Gonzalez-Carranza ZH, McManus MT (2000) Cell separation processes in plants: models, mechanisms and manipulation. Ann Bot 86:223–235
Rodrıguez-Llorente ID, Perez-Hormaeche J, El Mounadi K, Dary M, Caviedes MA, Cosson V, Kondorosi A, Ratet P, Palomares AJ (2004) From pollen tubes to infection threads: recruitment of Medicago floral pectic genes for symbiosis. Plant J 39:587–598
Schenk PM, Kazan K, Manners JM, Anderson JP, Simpson RS, Wilson IW, Somerville SC, Maclean DJ (2003) Systemic gene expression in Arabidopsis during an incompatible interaction with Alternaria brassicicola. Plant Physiol 132:999–1010
Stolle-Smits T, Beekhuizen JG, Kok MTC, Pijnenburg M, Recourt K, Derksen J, Voragen AGJ (1999) Changes in cell wall polysaccharides of green bean pods during development. Plant Physiol 121:363–372
Sutoh K, Yamauchi D (2003) Two cis-acting elements necessary and sufficient for gibberelin-upregulated proteinase expression in rice seeds. Plant J 34:636–645
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Verwoerd TC, Dekker BM, Hoekema A (1989). A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res 17:2362
Wakabayashi K, Hoson T, Huber DJ (2003) Methyl de-esterification as a major factor regulating the extent of pectin depolymerization during fruit ripening: a comparison of the action of avocado (Persea americana) and tomato (Lycopersicon esculentum) polygalacturonases. J Plant Physiol 160:667–673
Wakeley PR, Rogers HJ, Rozycka M, Greenland AJ, Hussey PJ (1998) A maize pectin methylesterase-like gene, ZmC5, specifically expressed in pollen. Plant Mol Biol 137:187–192
Willats WG, Orfila C, Limberg G, Buchholt HC, van Alebeek GJ, Voragen AG, Marcus SE, Christensen TM, Mikkelsen JD, Murray BS, Knox JP (2001) Modulation of the degree and pattern of methyl-esterification of pectic homogalacturonan in plant cell walls. Implications for pectin methyl esterase action, matrix properties, and cell adhesion. J Biol Chem 276:19404–19413
Wolf S, Grsic-Rausch S, Rausch T, Greiner S (2003) Identification of pollen-expressed pectin methylesterase inhibitors. FEBS Lett 555:551–555
Zhao C, Craig JC, Petzold HE, Dickerman AW, Beers E (2005) The xylem and phloem transcriptomes from secondary tissues of the Arabidopsis root-hypocotyl. Plant Physiol 138:803–818
Acknowledgements
We thank Sylvain Jeandroz, (University of Nancy I, France) for help with the phylogenetic analysis, the CRRBM (Centre de Ressources Régionales en Biologie Moléculaire) for the use of the Roche LightCycler and the French Ministry of Research for the funding of Romain Louvet’s PhD. The technical assistance of Françoise Fournet is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Louvet, R., Cavel, E., Gutierrez, L. et al. Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana . Planta 224, 782–791 (2006). https://doi.org/10.1007/s00425-006-0261-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-006-0261-9