
Abstract
The developmental expression pattern and localization of calreticulin were studied in Nicotiana tabacum L. anthers, pollen and pollen tubes. High transcript and protein levels were detected throughout anther development. Immunolocalization of calreticulin in the anthers showed particular dense label in tapetum and pollen at developmental stage 2, when the tapetum is highly active and the pollen tetrads are formed. Much lower transcript and protein levels were detected in dry and hydrated pollen and in pollen tubes. Immunofluorescence labeling of both chemically fixed and cryo-fixed and freeze-substituted pollen tubes showed the presence of calreticulin in Golgi apparatus and endoplasmic reticulum (ER). Calreticulin was seen throughout the stacks in the Golgi apparatus and in the areas with coated-Golgi vesicles but much less so in the ER. Calreticulin was not found in the secretory vesicles. A relatively intense label was occasionally seen adjacent to the wall of the tube. No significant label was observed in mitochondria, vacuoles, generative cells, cell wall or callose plugs. The present results are consistent with a role of calreticulin in Ca2+-dependent folding of secreted glycoproteins in tapetum, pollen and pollen tubes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soluble proteins residing in the lumen of the endoplasmic reticulum (ER) are called reticuloplasmins. One of the more abundant and highly conserved reticuloplasmins is calreticulin, which has been shown to play an important role in Ca2+ binding (Michalak et al. 1992) and may function in Ca2+ sequestration and Ca2+-dependent protein folding (Michalak et al. 1999). Calreticulin occurs ubiquitously in all eukaryotic organisms, and in the complex system of higher plants it is constitutively expressed in all meristematic and mature cell types (Mariani et al. 2003). Its expression level appears to be related to the abundance of ER membranes. As a molecular chaperone and Ca2+ sensor within the ER, plant calreticulin shares a similar molecular structure and basic functions with its animal counterpart (Mariani et al. 2003).
Although the precise roles of calreticulin in plants have not yet been fully ascertained, increased levels occur during the reproductive cycle in the ovaries of barley shortly after pollination in the first stages of embryogenesis (Chen et al. 1994), in somatic embryos and fertilized ovules of Nicotiana plumbaginifolia (Borisjuk et al. 1998), in sperm cells (Williams et al. 1997), zygotes and immature embryos of maize (Dresselhaus et al. 1996) and in Arabidopsis flowers (Nelson et al. 1997). High levels also occur in pollen of Liriodendron tulipifera (Navazio et al. 1998) and Ginkgo biloba (Nardi et al. 1998). In addition to the sequestration of Ca2+, these high levels of calreticulin have been implicated in secretion events (Denecke et al. 1995). Calreticulin in plant cells has also been suggested to be involved in cell contact via the plasmodesmata (Baluška et al. 1999) and in pollen–pistil interaction (Lenartowska et al. 2002).
In the investigation reported here, we have addressed the expression and distribution of calreticulin in anthers, pollen and pollen tubes of N. tabacum L. Immunolocalizations at both the light microscopic and ultrastructural level were performed to determine the distribution patterns of calreticulin. The results are discussed within the framework of secretory events and Ca2+ sequestration during pollen formation and pollen tube tip growth.
Materials and methods
Plant material
Plants of N. tabacum L. cv. Petit Havana (Botanical Garden Leiden, The Netherlands; Nijmegen accession no. 904750309) were grown under standardized greenhouse conditions (19–22°C; photoperiod: 18/6 h [light/dark]). Pollen was collected at anther dehiscence and used either as fresh material or air-dried and stored in a dessicator for use within a few days. For extraction purposes, young anthers (stage 1–11, according to Koltunow et al. 1990) were frozen in liquid nitrogen immediately after harvesting and kept at −80°C. For RNA extraction, 100-mg aliquots of pollen were mixed with 10 ml sterile liquid medium (Read et al. 1993) and maintained in suspension under slow agitation at 28°C for various lengths of time. For protein extraction, hydrated pollen was obtained by placing the fresh or air-dried pollen in a moist chamber for 1 h at room temperature followed by mixing with 10 ml culture medium for 10 min at room temperature. For the immunocytological studies, small samples of pollen were germinated for at least 2 h on discs of dialysis membrane placed on top of a solid agar (1%)/sucrose (10%)/boric acid (0.01%) medium, as described by Derksen et al. (1995b).
Chemicals and antibodies
Unless stated otherwise, all chemicals (grade: P.A.) were from Merck (Darmstadt, Germany), and the commercial antibodies were from Nordic (Nordic Immunological Lab., Tilburg, The Netherlands). An antiserum against spinach calreticulin (Navazio et al. 1995) was used for immunoblot analysis and immunolocalization on both the light and electron microscopic preparations. Spinach calreticulin was used as a positive control in SDS-PAGE and Western analysis.
Northern analyses
RNA was isolated as described by van Eldik et al. (1995). RNA and DNA extracts were quantified by absorbance measurements at 260 nm. Ten-microliter aliquots of RNA were separated on 1% agarose gels containing 0.4 M formaldehyde and 0.1 μg/ml ethidium bromide. The probes were obtained following the restriction of part of the cDNA of tobacco calreticulin (Denecke et al. 1995) with the endonuclease BamHI (Fermentas, St Leon-Rot, Germany), which released a 599-nucleotide cDNA fragment that was subsequently labeled with [32P] (Costa Mesa, CA, USA) by random primed labeling. Following the electrophoresis, the gels were photographed under UV illumination to document equal loading of each lane and then blotted to nylon Hybond N+ filters (Amersham, UK) overnight in 20× SSC (3 M NaCl, 0.3 M sodium citrate, pH 7.0). The blots were subsequently pre-hybridized for 1 h at 65°C in a solution containing 6× SSC, 5× Denhardt’s reagent, 0.1% SDS and 100 μg/ml of denatured herring sperm DNA (Promega) and then hybridized overnight under the same conditions but in the presence of the [32P]-labeled probe. After two washes for 15 min at 65°C in 2× SSC and 0.1% SDS and two additional washes at the same temperature in 0.2× SSC and 0.1% SDS, the blots were exposed using X-omat AR X-ray films (Eastman Kodak) at −80°C for 5 days with an intensifying screen. Following development, the blots were stripped, re-hydrated and re-hybridized with a tobacco ribosomal [32P]-labeled DNA probe homologous to the tobacco 28s ribosomal RNA for quantitative analysis. [32P] labeling was performed by random primed labeling.
Biochemical analyses
Tobacco pollen was suspended in 10 ml/g of 0.1 M potassium phosphate buffer, pH 7.1, 2.66 M (NH4)2SO4, 1 mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride (PMSF), 0.5 mM benzamidine and broken up (5×1.5 min, each period followed by 30 s on ice) in a bead beater (BioSpec Products, Bartlesville, OK, USA) with 1.5 g/ml acid-washed 500-μm glass beads (Sigma). Homogenates were removed from the settled beads, and the beads were washed three times with the same volume of extraction buffer. Calreticulin was isolated and purified from the total pollen homogenates by a selective ammonium sulfate precipitation procedure followed by chromatography on DEAE-cellulose (Whatman, Maidstone, UK) using a linear elution gradient (0.050–1 M NaCl, pH 7.1) as described by Milner et al. (1991).
Protein extracts from tobacco anthers were prepared by grinding the material to a fine powder under liquid nitrogen and re-suspending it with 4 ml/g of extraction buffer (10 mM Hepes pH 7.4, 0.32 M sucrose, 0.5 mM PMSF, 0.5 mM benzamidine). The homogenates were centrifuged at 20,000 g for 10 min at 4°C and the supernatant collected for SDS-PAGE analysis.
To prepare protein extracts from the pollen tubes, pollen was grown in vitro for varying lengths of time and then centrifuged for 2 min at 150 g. After rinsing and re-suspension in extraction buffer, the samples were homogenized in a French Pressure Cell (American Instruments Company, Silver Spring, Md) at 1,500 psi for 10 s (10 cycles).
Proteins were quantified following the method of Bradford (1976). SDS-PAGE electrophoresis was carried out on 10% or linear (7.5–10%) gradient gels. Molecular weights were determined with standard molecular markers (BDH, Dorset, UK). Staining was with Coomassie Brillant Blue R-250 (Sigma) or Stains All (BioRad). With Stains All, proteins that either bind Ca2+ or can be phosphorylated or glycosylated with sialyc acid stain blue; all other proteins stain pink-purple (Campbell et al. 1983).
The protein gels were blotted to nitrocellulose membranes according to Towbin et al. (1979) and used in a number of assays. Western blotting to detect calreticulin was performed by incubating the blotted membrane with anti-spinach calreticulin antiserum (Navazio et al. 1995) at a 1:2,000 dilution. The secondary antibody was a goat anti-rabbit IgG antibody conjugated to alkaline phosphatase, and the substrate was 5-bromo-4-chloro-3-indolyl phosphate and nitro-blue-tetrazolium (Sigma).
The presence of high-mannose N-glycans was detected using overlays with horseradish peroxidase-conjugated concanavalin A (Con A; 1 μg/ml [Sigma], as described by Navazio et al. 1995).
For Ca2+ binding analysis, [45Ca2+]-overlay assays were performed on nitrocellulose membranes in a medium containing 5 mM MgSO4, 60 mM KCl, 5 mM imidazole (pH 7.4) and 10 μM [45Ca]Cl2 (Amersham Pharmacia Biotech, Piscataway, NJ, USA), as described by Maruyama et al. (1984). Visualization occurred by autoradiography on Hyperfilm MP films (Amersham Pharmacia Biotech).
In vitro phosphorylation was performed according to Baldan et al. (1996). Briefly, samples of calreticulin were incubated for 10 min at 37°C in 50 mM Tris–HCl, pH 7.5, 12 mM MgCl2 with 20 μM β-[32P]-ATP (ICN) in the presence of 10 mU purified (Meggio et al. 1994) casein kinase-2 (CK2; kindly provided by F. Meggio, Padova, Italy). The reaction was blocked in an ice bath and the samples separated on 10% SDS-PAGE. After staining with Coomassie Brilliant Blue R-250, the gels were analyzed with a Camberra-Packard Instant Imager (Santa Clara, CA, USA).
Immunocytochemistry of anthers
Anthers were first fixed in 3% paraformaldehyde (PFA) and 0.5% glutaraldehyde in 50 mM K-phosphate buffer, pH 7.2, for 2 h at room temperature, rinsed, embedded in paraplast (Sigma), sectioned and once again hydrated according to standard procedures. After a 2 h incubation at room temperature with anti-spinach calreticulin antiserum (Navazio et al. 1995) it was diluted in the ratio 1:1,000 in 50 mM Tris–saline buffer, pH 7.4 with 1% BSA, the sections were rinsed (three times) in buffer containing BSA and incubated for 1 h in horseradish peroxidase-conjugated goat anti-rabbit (GAR-HRP, Pierce Rockford, IL, USA) (1:100 dilution in buffer with BSA). The peroxidase reaction was carried out at room temperature for 10 min with 0.5 mg/ml 3,3-diaminobenzidine (DAB; Sigma) and 0.01% H2O2 in a buffer containing 2.5 mg/ml nickel ammonium sulfate (Brunschwig Chemie, Amsterdam) as substrate (Adams 1981). Following washing in double-distilled water, the sections were transferred to xylene via a water/ethanol/xylene series and mounted in entellan. Eosine (0.01%) was added to the 50% ethanol dehydration step as a counterstain. Photography occurred with a Leitz Orthoplan (Leica) microscope equipped with a Color Coolsnap digital camera (Roper Scientific, Tucson, AZ, USA) and MetaVue (Universal Imaging Corporation, West Chester, PA, USA) software.
Fluorescence microscopy on pollen tubes
Pollen tubes were fixed with 3% PFA in one of two buffers (50 mM K-phosphate or 50 mM Pipes with 1 mM CaCl2, both at pH 7.4 and 10% sucrose) for 2 h at room temperature. For whole mounts, the samples were washed in buffer (three times), treated with 1% Macerozyme (Serva) in buffer for 60 s and washed again. A portion of the samples was treated with 0.5% Triton X-100 in buffer for 10 min to improve permeability. The samples were blocked with 1% BSA in buffer for 10 min, washed in buffer (three times) and incubated overnight at room temperature with the anti-spinach calreticulin antiserum (Navazio et al. 1995) diluted 1:400 in buffer containing 1% BSA. The samples were then washed again, incubated for 2 h at 4°C with Alexa 488-conjugated goat anti-rabbit IgG (Molecular Probes) diluted 1:200 in buffer containing 1% (BSA) and, after three additional washes, mounted in 1:1 (v/v) glycerol/water. For sectioning, the pollen tubes were dehydrated, embedded in butyl methyl methacrylate (BMM) and processed as described by Baskin et al. (1992). The sections were used immediately as even short-term storage appeared to be detrimental to the label intensity. The preparations were post-fixed with 3% PFA in PBS for 30 min and rinsed in PBS. Aldehydes were blocked with 0.1 M ammonium chloride in PBS for 10 min. After one rinse and further blocking with 1% bovine serum albumin acetylated (BSAc) (Aurion, Wageningen, The Netherlands) in PBS for 30 min, the preparations were incubated overnight at 4°C with anti-spinach calreticulin antiserum diluted 1:400 in PBS containing 0.1% BSAc. Following three rinses in PBS, the preparations were incubated with Alexa 488 goat anti-rabbit IgG diluted 1:200 in 0.1% BSAc containing PBS for 1 h at room temperature, rinsed in PBS and mounted in 1:1 (v/v) glycerol/water. In the control experiments, the primary antibody was either replaced by pre-immune serum or was completely omitted. Preparations were examined in a Zeiss Axiovert TV135 microscope (Zeiss,) using the METAFLUOR software (Universal Image Corp, Downingtontown, PA, USA). Differential interference contrast (DIC) images were made for the identification of cellular structures. For photographic purposes, a Coolsnap fx camera (Roper Scientific, Duluth, GA, USA) was used. Monochrome images were printed in black and white. Preparations from at least three independent experiments were used.
Electron microscopy of pollen tubes
Pollen tubes were grown on dialysis-membrane disks, cryo-fixed and freeze-substituted as described by Derksen et al. (1995b), but omitting uranyl acetate and OsO4 from the substitution medium. Disks with pollen tubes were embedded in LR White (London Resin Cy, Reading, UK) and cured at 60°C. Sections were cut using a Sorvall MT-2 ultra-microtome (Sorvall) and mounted on nickel grids (Stork-Veco, Eerbeek, The Netherlands). A 50-mM K-phosphate, pH 7.0, was used as the buffer. For immunodetection, grids with sections were floated on a drop of buffer containing 1% BSA for 15 min and then incubated for 2 h with the 1:50 diluted anti-calreticulin antiserum or with the pre-immune serum. After three washes of 10 min each in buffer containing 1% BSA, the sections were incubated with 1:100 diluted goat anti-rabbit IgG bound to 10-nm colloidal-gold particles (Aurion, Wageningen, The Netherlands) for 1 h and rinsed in buffer (three times). After three additional rinses in double-distilled water, the sections were stained with 2% uranyl acetate in double-distilled water for 15 min, washed three times (5 min each time) in double-distilled water and air-dried. The preparations were examined in a Jeol 100 CX (Jeol, Tokyo, Japan) transmission electron microscope. For quantitative evaluation, particle counts were made on images of five randomly chosen organelles or wall areas per pollen tube printed at a magnification of 5×7,500. The total surface area was always 225 μm2. All results were obtained from at least three independent experiments.
Results
Isolation and characterization of calreticulin
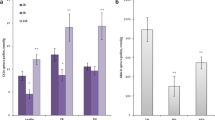
Calreticulin was purified and biochemically characterized from tobacco pollen. Protein fractionation by DEAE-cellulose chromatography produced fractions containing putative calreticulin at elution concentrations between 0.2 and 0.3 M NaCl. In the immunoblotting analysis, antibodies against spinach calreticulin (Navazio et al. 1995) specifically recognized a 60-kDa protein band (Fig. 1a), which stained blue with Stains All in the SDS-PAGE (data not shown). No other bands were detected, demonstrating the specificity of the antiserum that was subsequently used for additional immunological studies. No label was observed with the pre-immune serum (data not shown).

Identification of purified tobacco pollen calreticulin. a Immunoblot with anti-spinach calreticulin antiserum. Left lane (Sp) Spinach calreticulin (2 μg), right lane (To) tobacco pollen extract fraction eluted from DEAE-cellulose between 0.2 and 0.3 M NaCl (20 μg). A 60-kDa band is recognized by the anti-calreticulin antiserum. b [45Ca2+]-overlay assay. Left lane (Sp) spinach calreticulin (30 μg), right lane (To) purified tobacco pollen extract (40 μg). c In vitro phosphorylation by protein kinase CK2. Left lane (Sp) Spinach calreticulin (2 μg), right lane (To) purified tobacco pollen extract (20 μg). d Affinoblot with Con A. Left lane (Sp) spinach calreticulin (2 μg), right lane (To) purified tobacco pollen extract (20 μg). The labeled bands in b, c and d correspond with the bands detected with the anti-calreticulin antiserum in a. The arrow indicates the 60-kDa position
The tobacco 60-kDa protein detected by anti-spinach calreticulin antiserum (Fig. 1a) was found to bind Ca2+ in a [45Ca2+]-overlay assay (Fig. 1b) and to undergo in vitro phosphorylation by protein kinase CK2 (Fig. 1c). In addition, the presence of glycan chains, probably of the high-mannose type, was detected by affinoblotting with lectin Con A (Fig. 1d). Taken together, the biochemical properties of the calreticulin that we obtained from tobacco pollen match well with those of all plant calreticulins.
Calreticulin expression
The expression profile of calreticulin was obtained by Northern blot analysis (Fig. 2a) of total RNA with the 599-nucleotide cDNA fragment from tobacco calreticulin (Denecke et al. 1995) and by protein blotting (Fig. 2b, c) with the anti-spinach calreticulin antiserum (Navazio et al. 1995). The results show relatively high levels of calreticulin and its transcripts at all stages in the developing anthers. In dry and hydrated pollen, the signal was much lower, and this low level of expression remained stable in germinating and growing pollen tubes (Fig. 2a). Immunoblot analysis of crude protein extracts from anthers at different developmental stages and germinating pollen tubes showed the same trend at the protein level (Fig. 2b, c). Therefore, we conclude that calreticulin transcripts and protein are present at high levels in the anthers but that they occur at low, although constant, levels in dry, hydrated and germinating pollen as well as in the growing pollen tubes.

Expression of calreticulin in anthers, pollen and pollen tubes. a Northern blotting of calreticulin transcripts from anthers during pollen development and from pollen and pollen tubes growing in vitro. A [32P]-labeled 599-nucleotide cDNA fragment (see text) from the tobacco calreticulin (CR) DNA sequence was used (row 1). All lanes were equally loaded (10 μg of total RNA). Post hybridization with the [32P]-labeled DNA fragment homologous to 28S rRNA as a probe (row 2) shows equal loading. RNA from anthers (A) at stages 1–11 (Koltunow et al. 1990) showed a strong reaction. Dry (D) and hydrated (H) pollen and pollen tubes grown for 1, 2, 4, 12 and 24 h show much lower but constant calreticulin transcript levels. b Western blotting of total anther protein extracts using anti-spinach calreticulin antiserum. Protein was extracted from anthers at stages 2, 6, 8 and 11. In all lanes, 120 μg of protein was loaded. The label appears largely equal in all extracts. c Western blotting of total pollen-tube extracts (taken at 1, 2, 4, 8 and 24 h of growth) using anti-spinach calreticulin antiserum. In all lanes, 120 μg of protein were loaded. The label appears to be largely equal in all extracts. In the last lane some aspecific cross-reacting bands of a higher Mr are present. The arrow in b and c indicates the 60-kDa position
Immunolocalization of calreticulin
Light and fluorescence microscopy
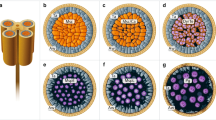
Calreticulin immunolocalization of sectioned and fixed anthers revealed the labeling at all stages of anther development (e.g, stage 2 in Fig. 3a). In contrast, immunolocalization with pre-immune serum did not reveal significant labeling (Fig. 3b). A particularly dense label was observed in the active tapetum and in pollen at the early stages of development, especially at the tetrad formation (stage 2, Fig. 3a, c). The tapetum began to degenerate after stage 2 (Fig. 3c), and by stage 5 (Fig. 3d) no significant label was discernable. Pollen also became less strongly labeled between stages 2 and 5 (Figs. c, d), but the label increased once more in intensity upto stage 8 and then remained stable between stages 8 and 11. In the other parts of the anther, the intensity of the label remained unchanged between stages 2 and 11 (Fig. 3c–f), indicating that there was no decrease in the level of calreticulin during tissue degeneration.

Immunolocalization of calreticulin in anthers. a Anther section showing label in all tissues at stage 2. b, c Sections of the same anther as in a viewed at a higher magnification, showing pollen, tapetum and some surrounding tissue. No significant label could be detected with the pre-immune serum (b). In particular, the pollen tetrads (pointers) and tapetum (arrows) are densely labeled by the anti-spinach calreticulin antiserum (c). d–f Similar sections of anthers labeled with anti-spinach calreticulin antiserum at stages 5, 8 and 11, respectively. The tapetum degenerated between stages 2 (c) and 5 (d), and no significant amount of label remained at stage 5 (d). The strength of the label diminished at the same time (between stages 2 and 5) in the pollen; after stage 5 (d) labeling of the pollen increased once again and remained stable between stages 8 (e) and 11 (f). In the other anther tissues the label remained unchanged after stage 2. Bars: 50 μm
Fluorescence microscopy of the pollen tubes clearly showed a speckled label throughout the cytoplasm of the vegetative cell, but the corresponding structures could not be identified unambiguously in the pollen tubes viewed by DIC (Fig. 4a, d). BMM-embedded material (Fig. 4b, e) and whole mounts fixed in 3% PFA in Pipes buffer with 0.01 M CaCl2 (Fig. 4c, f) revealed similar patterns. In addition, some larger spots emitting less fluorescence were observed (Fig. 4c). The fluorescence was slightly less distinct following the Triton X-100 treatments and became even diffuse after fixation in phosphate buffer (not shown). No label was found in the hyaline zone with the secretory vesicles (SV) in the tip (Fig. 4b, c, e). The most intense labeling often appeared at the borders of the hyaline zone (Fig. 4b) and in the cortical area behind the tip (Fig. 4f).

Immunofluorescence localization of calreticulin in pollen tubes. a-c Pollen tubes viewed along the longitudinal median plane of the tip region. a, b Section from a BMM-embedded pollen tube labeled with anti-spinach calreticulin antiserum: a viewed in DIC, b fluorescent image. c Optical section of a labeled whole mount following fixation in Pipes buffer with 1 mM CaCl2. d, e Section from a BMM- embedded pollen tube viewed along the median plane throughout the part shown: d viewed in DIC, e fluorescent image. f Cortical optical section of a labeled whole mount following fixation in Pipes buffer containing 1 mM CaCl2. A distinct, speckled label is visible in all of the fluorescent images, but no clear correlation with the structures in the DIC images could be made. b A more intense label (arrows) was seen occasionally at the border of the hyaline zone in the tip. c Labeled organelles were sometimes detected in the tip. Larger, weakly stained structures (pointers) and fluorescent streaks (arrow) were also visible. e Strong label (pointers) was occasionally seen adjacent to the wall of the pollen tube. f In cortically viewed whole mounts, the label may appear to be more intense in the area behind the tip. All magnifications: bar in f = 5 μm
Faint streaks of label were occasionally observed in the cytoplasm of the pollen tubes (Fig. 4c), while a relatively strong label was occasionally found adjacent to the wall of the tube (Fig. 4e). No label was ever recorded in the generative cell, the nuclei and the vacuoles or in the callose plugs (not shown). In pollen tubes fixed with phosphate buffer, the tip areas often became densely labeled, whereas the speckled label on the cytoplasm was much less distinct (not shown). The non-specific signal that emerged after omission of the second antibody or after the use of pre-immune serum was always negligible (not shown). These results show that calreticulin is mainly associated with organelles that occur throughout the pollen tubes. Immunolocalization on preparations for the electron microscope was used to identify the labeled structures.
Electron microscopy
The cytoplasmic organization and preservation of cryo-fixed pollen tubes was the same as shown in previous studies, with a clear zone at the tip that is essentially free of organelles, with the exception of SV and, perhaps, some putative smooth ER (Derksen et al. 1995a, b; Lancelle et al. 1987). Immunocytochemistry with the anti-calreticulin serum revealed label in the Golgi stacks at both the cis and trans-sides of the stacks (Fig. 5a). Label was also present in the area of the coated-Golgi vesicles (Fig. 5b). Less intense labeling occurred in the ER (Fig. 5c). Gold particle counts were made on the Golgi apparatus, rough ER, mitochondria, vacuoles, wall and callose plugs (Table 1). With the exception of the ER and Golgi apparatus, no significant label was found. Low levels of irregularly distributed label were found in the cytoplasm. No significant label was recorded using the pre-immune serum (Fig. 5d). These observations clearly show that the brightly labeled organelles observed by fluorescence microscopy (Fig. 4) must be the Golgi apparatus. The less brightly labeled structures (Fig. 4c) probably represent the ER. Thus, calreticulin appears to be present in higher concentrations in the Golgi apparatus than in the ER.

Immunogold localization of calreticulin in electron microscopic preparations of pollen tubes grown in vitro. a Strong labeling occurred in the Golgi stacks and the area surrounding the coated-Golgi vesicles. b Golgi apparatus sectioned through the trans surface showing label in the area containing the coated-Golgi vesicles. c In the ER, distinct, but less intense label can be found. d No labeling was found following incubation with the pre-immune serum. All magnifications, bar in d = 0.1 μm
Discussion
The protein that we detected in tobacco anthers and pollen with the anti-spinach calreticulin antiserum has all the properties of a typical calreticulin molecule. Its apparent molecular weight (60 kDa) falls within the usual mobility range (50–60 kDa) shown by calreticulins run on SDS-PAGE and corresponds to the M r detected for calreticulin isolated from tobacco germinating seeds (Denecke et al. 1995). Since tobacco calreticulin carries two consensus sites for N-linked glycosylation (Denecke et al. 1995), the observed size difference between spinach (54 and 56 kDa) and tobacco (60 kDa) calreticulins may be partly attributed to the presence of two high-mannose glycan chains on the tobacco calreticulin (Crofts et al. 1999).
Both Northern blotting with the [32P]-labeled 599-nucleotide cDNA fragment from the tobacco calreticulin DNA sequence (Denecke et al. 1995) and protein blotting with the anti-spinach calreticulin antiserum (Navazio et al. 1995) revealed a strong expression of calreticulin in the anthers. Immunolocalizations on anther sections showed distinct labeling, especially in the tapetum and pollen tetrads at stage 2; subsequent to stage 2, in maturating pollen, expression of the label was relatively much weaker. In dry pollen, the levels of the transcripts and protein were much lower that those maintained after germination and for the first 24 h of pollen tube growth. If we compare the situation in tobacco with observations made on barley aleuron tissue (Denecke et al. 1995), the high abundance of calreticulin present in the fertilized ovaries of N. plumbaginifolia (Borisjuk et al. 1998) can be related to a specific function of calreticulin: a chaperone of protein folding that is required for the high secretion activity of the inner cell layer of the ovule integument, the endothelium. Likewise, the high level of calreticulin in the tapetum and pollen tetrads at stage 2 may relate to the high level of chaperone activity required for the high secretory activity of the tapetum and the wall synthesis of the pollen (Bhandari 1984). Nelson et al. (1997) correlated high levels of calreticulin to secretion in the various flower parts during A. thaliana flower development. Although these same researchers (Nelson et al. 1997) also detected high calreticulin levels at anther dehiscence in Arabidopsis, in tobacco no such specific high levels have been recorded prior to (Fig. 3f) or at dehiscence (not shown). Since no data on calreticulin distribution in Arabidopsis anther tissues are available, the possible role of calreticulin remains unclear. One possible explanation is that the high level of calreticulin found in Arabidopsis anthers originates from their mature, tri-cellular pollen. An additional function of calreticulin in Ca2+ binding/storage in the tobacco anthers, especially at stage 2, should not be excluded and might reflect Ca2+ exchanges between the tapetum and pollen, as presumably is the case in Gasteria verrucosa at the tetrad stage (Willemse 1993).
Fluorescence microscopy of the pollen tubes revealed a distinctive speckled calreticulin label in the cytoplasm of the vegetative cell with occasional streaks in the cytoplasm. Based on the immunolocalization results with the electron microscopic preparations, it is clear that this speckled label derives from calreticulin in the Golgi apparatus. The larger spots with less fluorescence may represent flatly viewed rough ER or locally accumulated smooth ER; however, it may just be out-of-focus Golgi apparatus. Sometimes the more intense labeling at the borders of the hyaline zone in the tip is probably caused by the Golgi apparatus and ER reversing their motion at the tip (Derksen et al. 1995a, b). The faint fluorescence streaks detected in the pollen tubes probably represent rough ER, but they could also represent Golgi apparatus lined up around the actin filaments over which they presumably move (Derksen et al. 1995b; de Win et al. 1999). The dense label seen in the tip following fixation in phosphate buffer was not reproducible in the cryo-fixed and BMM-embedded preparations, indicating an artifact. Possible effects of fixation on the specific cytoplasmic organization of tip growing cells have been shown to occur in lily pollen tubes (Miller et al. 1996) and Saprolegnia hyphae (Kaminskyj et al. 1992). We also found some irregular background label in the cytoplasm in our fluorescence and electron microscopic preparations. This label, however, may derive from irregularly distributed smooth ER, partly sectioned and obliquely viewed rough ER and Golgi apparatus that cannot be identified unambiguously due to the omission of OsO4 fixation.
The relatively strong label that was occasionally found adjacent to the cell wall of the tube may represent a plasma membrane-associated ER rich in calreticulin (Borisjuk et al. 1998; Baluška et al. 1999), but it obviously cannot be related to the plasmodesmata. While in vivo studies revealed a clear localization of calreticulin in the vacuoles, cell walls and callose plugs of Petunia hybrida (Lenartowska et al. 2002), this could not be confirmed by our in vitro observations on tobacco pollen tubes (see also Table 1).
Calreticulin is a component of the Golgi stacks (Borisjuk et al.1998) and Golgi-coated vesicles (Pimpl et al. 2000), and it has been shown to be transported from the cis- to trans-Golgi side based on protein glycosylation with complex glycans (Navazio et al. 2002). The localization of calreticulin in the Golgi apparatus, including Golgi-coated vesicles and, at a lower abundance, in the ER that we found in tobacco pollen tubes may reflect Golgi–ER protein recycling if we take into account that tobacco calreticulin also carries two glycan chains of the typical ER high-mannose form (Crofts et al. 1999).
The presence of high levels of calreticulin in pollen tubes would appear to be related to its role as a chaperone of protein folding; as such, it would expediate the high rate of protein synthesis required for fast growth (Derksen et al. 1995b; Lancelle et al. 1987). This is most likely the case for the tapetum and pollen tetrads in the tobacco anthers at stage 2 and also for the female reproductive system of the closely related species N. plumbaginifolia (Borisjuk et al. 1998). The present findings do not indicate that the putative accumulation of smooth ER in the tip (Derksen et al. 1995a) is involved in Ca2+ sequestration to maintain the tip-focused Ca2+ gradient (Pierson et al. 1994). However, as ER and Golgi endomembrane compartments may well act as releasable intracellular Ca2+ stores in the pollen tubes (Holdaway-Clarke and Hepler 2003), calreticulin with its Ca2+-buffering properties may contribute to the maintenance of cytoplasmic Ca2+-homeostasis.
Abbreviations
- BMM:
-
Butyl methyl methacrylate
- CK2:
-
Casein kinase-2
- BSAc:
-
Bovine serum albumin acetylated
- Con A:
-
Concanavalin A
- DIC:
-
Differential interference contrast
- PFA:
-
Paraformaldehyde
- PMSF:
-
Phenylmethanesulfonyl fluoride
- SD:
-
Standard deviation
References
Adams JC (1981) Heavy metal intensification of DAB-based HRP reaction product. J Histochem Cytochem 29:775
Baldan B, Navazio L, Friso A, Mariani P, Meggio F (1996) Plant calreticulin is specifically and efficiently phosphorylated by protein kinase CK2. Biochem Biophys Res Commun 221:498–502
Baluška F, Šamaj J, Napier R, Volkmann D (1999) Maize calreticulin localizes preferentially to plasmodesmata in root apex. Plant J 19:481–488
Baskin TI, Busby CH, Fowke LC, Sammut, Gruber (1992) Improvements in immunostaining samples embedded in methacrylate: localization of microtubules and other antigens throughout developing organs in plants and diverse taxa. Planta 187:405–413
Bhandari NN (1984) The microsporangium. In: Johri BM (ed) Embryology of angiosperms. Springer, Berlin Heidelberg New York, pp 53–121
Borisjuk N, Sitailo L, Adler K, Malysheva L, Tewes A, Borisjuk L, Manteuffel R (1998) Calreticulin expression in plant cells: developmental regulation, tissue specificity and intracellular distribution. Planta 206:504–514
Bradford MM (1976) A rapid and sensitive method of the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Campbell KP, MacLennan DH, Jorgensen AO (1983) Staining of the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C and S-100 with the cationic carbocyanine dye Stains-All. J Biol Chem 258:11267–11273
Chen F, Hayes PM, Mulrooney DM, Pan A (1994) Identification and characterization of cDNA clones encoding plant calreticulin in barley. Plant Cell 6:835–843
Crofts AJ, Leborgne-Castel N, Hillmer S, Robinson DG, Phillipson B, Carlsson LE, Ashford DE, Denecke J (1999) Saturation of the endoplasmic reticulum retention machinery reveals anterograde bulk flow. Plant Cell 11:2233–2247
Denecke J, Carlsson LE, Vidal S, Hoglund AS, Ek B, van Zeijl MJ, Sinjorgo KMC, Palva ET (1995) The tobacco homolog of mammalian calreticulin is present in protein complexes in vivo. Plant Cell 7:391–406
Derksen J, Rutten T, van Amstel T, de Win A, Doris F, Steer M (1995a) Regulation of pollen tube growth. Acta Bot Neerl 44:93–119
Derksen J, Rutten T, Lichtscheidl IK, de Win AHN, Pierson ES, Rongen G (1995b) Quantitative analysis of the distribution of organelles in tobacco pollen tubes: implications for exocytosis and endocytosis. Protoplasma 188:267–276
De Win AHN, Pierson ES, Derksen J (1999) Rational analysis of organelle trajectories in tobacco pollen tubes reveal characteristics of the actomyosin cytoskeleton. Biophys J 76:1648–1658
Dresselhaus T, Hagel C, Lorz H, Kranz E (1996) Isolation of a full length cDNA encoding calreticulin from a PCR library of in vitro zygotes of maize. Plant Mol Biol 31:23–34
Holdaway-Clarke TL, Hepler PK (2003) Control of pollen tube growth: role of ion gradients and fluxes. New Phytol 159:539–563
Kaminskyj SGW, Jackson SL, Heath IB (1992) Fixation induces differential polarized translocations of organelles in hyphae of Saprolegnia ferax. J Microsc 167:153–168
Koltunow AM, Truettner J, Cox KH, Wallroth M, Goldberg RB (1990) Different temporal and spatial gene expression patterns occur during anther development. Plant Cell 2:1201–1224
Lancelle SA, Cresti M, Hepler PK (1987) Ultrastructure of the cytoskeleton in freeze-substituted pollen tubes of Nicotiana. Protoplasma 140:141–150
Lenartowska M, Karaš K, Marshall J, Napier R, Bednarska E (2002) Immunocytochemical evidence of calreticulin-like protein in pollen tubes and styles of Petunia hybrida Hort. Protoplasma 219:23–30
Mariani P, Navazio L, Zuppini A (2003) Calreticulin and the endoplasmic reticulum in plant cell biology. In: Michalak M, Eggleton P (eds) Calreticulin, 2nd edn. Landes Bioscience, Georgetown, pp 94–101
Maruyama K, Mikawa T, Ebashi S (1984) Detection of calcium binding proteins by 45Ca autoradiography on nitrocellulose membrane after sodium dodecyl sulfate gel electrophoresis. J Biochem 95:511–519
Meggio F, Boldyreff B, Issinger OG, Pinna L (1994) Casein kinase 2 down- regulation and activation by polybasic peptides are mediated by acidic residues in the 55–64 region of the beta-subunit. A study with calmodulin as phosphorylatable substrate. Biochemistry 33:4336–4342
Michalak M, Milner R, Burns K, Opas M (1992) Calreticulin. Biochem J 285:681–692
Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M (1999) Calreticulin: one protein, one gene, many functions. Biochem J 344:281–292
Miller DD, Lancelle SA, Hepler PK (1996) Actin MFs do not form a dense meshwork in Lilium longiflorum pollen tube tips. Protoplasma 196:21–33
Milner RE, Baksh S, Shemanko C, Carpenter MR, Smillie L, Vance JE, Opas M, Michalak M (1991) Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J Biol Chem 266:7155–7165
Nardi MC, Giacomelli E, Dainese P, Fitchette-Laine AC, Faye L, Baldan B, Navazio L, Mariani P (1998) Ginkgo biloba L. expresses calreticulin, the major calcium-binding reticuloplasmin in eukaryotic cells. Bot Acta 111:66–70
Navazio L, Baldan B, Dainese P, James P, Damiani E, Margreth A, Mariani P (1995) Evidence that spinach leaves express calreticulin but not calsequestrin. Plant Physiol 109:983–990
Navazio L, Sponga L, Dainese P, Fitchette-Laine AC, Faye L, Baldan B, Mariani P (1998) The calcium binding protein calreticulin in pollen of Liriodendron tulipifera L. Plant Sci 131:35–42
Navazio L, Miuzzo M, Royle L, Baldan B, Varotto S, Merry AH, Harvey DJ, Dwek RA, Rudd PM, Mariani P (2002) Monitoring endoplasmic reticulum-to-Golgi traffic of a plant calreticulin by protein glycosylation analysis. Biochemistry 41:14141–14149
Nelson DE, Glausinger B, Bohnert HJ (1997) Abundant accumulation of the calcium-binding molecular chaperone calreticulin in specific floral tissues of Arabidopsis thaliana. Plant Physiol 114:29–37
Pierson ES, Miller DD, Callaham DA, Shipley AM, Rivers BA, Cresti M, Hepler PK (1994) Pollen tube growth is coupled to the extracellular calcium ion flux and the intracellular calcium gradient: effect of BAPTA-type buffers and hypertonic media. Plant Cell 6:1815–1828
Pimpl P, Movafeghi A, Coughlan S, Denecke J, Hillmer S, Robinson DG (2000) In situ localization and in vitro induction of plant COPI-coated vesicles. Plant Cell 12:2219–2235
Read SM, Carke AE, Bacic A (1993) Requirements for division of the generative nucleus in cultured pollen tubes of Nicotiana. Protoplasma 174:101–115
Towbin H, Stahelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Van Eldik GJ, Vriezen WH, Wingens M, Ruiter RK, van Herpen MMA, Schrauwen JAM, Wullems GJ (1995) A pistil specific gene of Solanum tuberosum is predominantly expressed in the stylar cortex. Sex Plant Reprod 8:173–179
Willemse MTM (1993) Calcium and calmodulin distribution in the tapetum of Gasteria verrucosa during anther development. Plant Syst Evol 7(Suppl):107–115
Williams CM, Zhang G, Michalak M, Cass DJ (1997) Calcium-induced protein phosphorylation and changes in levels of calmodulin and calreticulin in maize sperm cells. Sex Plant Reprod 10:83–88
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nardi, M.C., Feron, R., Navazio, L. et al. Expression and localization of calreticulin in tobacco anthers and pollen tubes. Planta 223, 1263–1271 (2006). https://doi.org/10.1007/s00425-005-0175-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-005-0175-y