
Abstract
Transitory starch is stored during the day inside chloroplasts and then broken down at night for export. Recent data indicate that maltose is the major form of carbon exported from the chloroplast at night but its fate in the cytosol is unknown. An amylomaltase gene (malQ) cloned from Escherichia coli is necessary for maltose metabolism in E. coli. We investigated whether there is an amylomaltase in the cytosol of plant leaves and the role of this enzyme in plants. Two mutants of Arabidopsis thaliana (L) Heynh. were identified in which the gene encoding a putative amylomaltase enzyme [disproportionating enzyme 2, DPE2 (DPE1 refers to the plastid version of this enzyme)] was disrupted by a T-DNA insertion. Both dpe2-1 and dpe2-2 plants exhibited a dwarf phenotype and accumulated a large amount of maltose. In addition, dpe2 mutants accumulated starch and a water-soluble, ethanol/KCl-insoluble maltodextrin in their chloroplasts. At night, the amount of sucrose in dpe2 plants was lower than that in wild-type plants. These results show that Arabidopsis has an amylomaltase that is involved in the conversion of maltose to sucrose in the cytosol. We hypothesize that knocking out amylomaltase blocks the conversion from maltose to sucrose, and that the higher amount of maltose feeds back to limit starch degradation reactions in chloroplasts. As a result, dpe2 plants have higher maltose, higher starch, and higher maltodextrin but lower nighttime sucrose than wild-type plants. Finally, we propose that maltose metabolism in the cytosol of Arabidopsis leaves is similar to that in the cytoplasm of E. coli.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In leaves, transitory starch is stored during the day inside chloroplasts and then broken down at night for export. Studies have been done by several groups to investigate starch breakdown in isolated intact chloroplasts from spinach. Earlier studies showed that the principal products of starch breakdown in the presence of phosphate were triose phosphate (mainly dihydroxyacetone phosphate) and 3-phosphoglycerate (Heldt et al. 1977; Peavey et al. 1977; Stitt and ap Rees 1980). Starch was also converted to maltose and glucose in the absence of phosphate but the rates of phosphate-dependent phosphorolytic starch degradation were very much higher than those of phosphate-independent hydrolytic starch degradation (Heldt et al. 1977). Stitt and Heldt (1981) reported that the products of starch breakdown included triose phosphate, 3-phosphoglycerate (PGA), CO2, glucose, and maltose. These studies suggest that phosphorolytic starch degradation is the major pathway of starch breakdown.
However, studies done by other groups showed that both phosphorolytic and hydrolytic starch degradation were important. Levi and Gibbs (1976) reported that the major products of starch breakdown from isolated spinach chloroplasts were PGA and maltose, and, to a lesser extent, glucose phosphate — a mixture of glucose 1-phosphate (G1P) and glucose 6-phosphate (G6P). Servaites and Geiger (2002) found that glucose, maltose, and isomaltose were the principal products of starch mobilization exported from chloroplasts at night while Ritte and Raschke (2003) showed that starch breakdown in the light in guard cell chloroplasts results in substantial maltose export. Weise et al. (2003) found that maltose and glucose were the major products exported between 1 and 3 h after the lights were turned off, and no isomaltose was found in the exported products. The synthesis of sucrose is by far the major fate for the carbon released by degradation of transitory starch (Trethewey and Smith 2000). The export of maltose from chloroplasts in a significant amount indicates that there must be enzymes in the cytosol that metabolize maltose to precursors for sucrose synthesis (Servaites and Geiger 2002).
Cytosolic starch phosphorylase was found in spinach and pea leaves (Steup and Latzko 1979; Preiss et al. 1980; Steup et al. 1980) and potato plants (Mori et al. 1991; Duwenig et al. 1997). This enzyme was considered to be a starch metabolism enzyme and its presence in the cytosol has been puzzling since there is no starch in the cytosol. The chloroplastic isozyme possesses a high affinity for maltodextrin, whereas the affinity of the cytosolic isozyme for maltodextrin is much lower (Yang and Steup 1990). In contrast, the cytosolic isozyme has an extremely high affinity for a highly branched, ethanol/KCl-insoluble polysaccharide, which is a poor substrate for the plastidic counterpart (Yang and Steup 1990). The ethanol/KCl-insoluble polysaccharide isolated from spinach by Yang and Steup (1990) is a high-molecular-weight heteroglycan, with an approximate chain length range from 180 to 30,000. The heteroglycan is made up of galactose, arabinose, glucose, ribose, and, to a lesser extent, xylose and fructose. The glucose only accounts for approximately 5% of the total monosaccharide content and the non-glucose monosaccharides act as the priming groups for glucan (Yang and Steup 1990). It is generally believed that this ethanol/KCl-insoluble polysaccharide is in the cytosol due to its high affinity for cytosolic starch phosphorylase. These findings raise a question — what is the enzyme that converts maltose to the glucan part of the heteroglycan upon which the cytosolic maltodextrin phosphorylase can act? We hypothesize that amylomaltase is the enzyme that converts maltose to the glucan part of the heteroglycan in the cytosol.
Amylomaltase is a type-II 4-α-glucanotransferase (GTase; EC 2.4.1.25) that catalyzes an inter-molecular glucan transfer reaction from one 1,4-α-glucan molecule to another, or to glucose. Amylomaltase was first found in Escherichia coli as a maltose-inducible enzyme, and the amylomaltase gene (malQ) was cloned from Streptococcus pneumoniae (Lacks et al. 1982), E. coli (Pugsley and Dubrevil 1988), Clostridium butyricum (Goda et al. 1997), and Thermus aquaticus (Terada et al. 1999). According to Boos and Shuman (1998), incoming maltose and maltodextrin are metabolized to glucose and G1P by the action of three cytoplasmic enzymes: amylomaltase (MalQ), maltodextrin phosphorylase (MalP), and maltodextrin glucosidase (MalZ). Amylomaltase is essential for maltose degradation in E. coli, and malQ mutants are unable to grow on maltose; their growth is inhibited by maltose (Hofnung et al. 1971) and they accumulate large amounts of free maltose inside the cell (Szmelcman et al. 1976).
A similar type-II 4-α-GTase, disproportionating enzyme (D-enzyme), is present in plants. D-enzyme was first found in potato tubers (Peat et al. 1956; Takaha et al. 1998). Since then it has been found in many other plant organs such as carrot roots (Manners and Rowe 1969), tomato fruits (Manners and Rowe 1969), spinach leaves (Okita et al. 1979), pea leaves (Kakefuda et al. 1986), and Arabidopsis leaves (Lin and Preiss 1988). By knocking out plastidic D-enzyme, it was found that the majority of the GTase activity was in the chloroplast (Critchley et al. 2001). The plastidic D-enzyme is involved in transitory-starch breakdown, by converting short oligosaccharides into longer chains providing substrates for the plastidic starch phosphorylase and β-amylase (Kakefuda and Duke 1989). Arabidopsis plants lacking the plastidic D-enzyme (dpe1-1) accumulate maltooligosaccharides (maltotriose-maltoheptaose), but not maltose (Critchley et al. 2001).
With the completion of Arabidopsis genome sequence in the year 2000, another gene predicted to encode 4-α-GTase was found on chromosome 2 (At2g40840). This gene appears not to code for any targeting sequence as judged by Target-P (Emanuelsson et al. 2000) and the enzyme is assumed to be in the cytosol. To investigate the function of this enzyme, we identified two T-DNA knockout mutants of Arabidopsis thaliana (L) Heynh. that completely lack transcripts for cytosolic amylomaltase (DPE2). We propose that maltose metabolism in the cytosol of Arabidopsis leaves is similar to maltose metabolism in the cytoplasm of E. coli.
Materials and methods
Plants and growth conditions
Plants of Arabidopsis thaliana (L.) Heynh. ecotype Wassilewskija (WS) were grown under a 16-h photoperiod, 200 μmol photon m−2 s−1. The temperature was 22 °C during the light period and 20 °C during the dark period. Humidity was maintained at 65%. Plants used in experiments were between 3 and 5 weeks old. Wild-type and mutant plants analyzed were at the same age.
Isolation of cytosolic amylomaltase mutants
The dpe2-1, dpe2-2 mutants were identified by PCR screening of 72,960 Basta-resistant lines in ecotype WS transformed with an activation-tag vector, pSK1015 (Weigel et al. 2000), using primers specific for the DPE2 gene (5′ GCA GTT CCC ATG TTC TCT GTA AGG TCA GA 3′ and 5′ CCA AAC AGG AAA TCA GAA GGC TTA TTG CT 3′) and the T-DNA (5′ CAT TTT ATA ATA ACG CTG CGG ACA TCT AC 3′). The presence of T-DNA in the DPE2 gene was confirmed by Southern blots. The left borders of T-DNA insertions were sequenced using Big Dye Terminator technology at the UW-Madison Biotechnology Center. Candidate heterozygous dpe2 plants were backcrossed to WT three times to clean up the background and then self-crossed to produce homozygous dpe2 plants.
Preparation of RNA and northern blots
Total RNA was extracted from 2–3 g of mature Arabidopsis plants as described by Takaha et al. (1993), and electrophoresed on a formaldehyde-agarose gel (Maniatis et al. 1982) and blotted onto a Biodyne B transfer membrane (Pall Corp., East Hills, NY, USA). The membrane was pre-hybridized in Church buffer (7% SDS, 0.5 M KH2PO4, 2 mM EDTA, pH 7.0) at 65 °C for 2 h and hybridized with 32P-labeled DNA probes in Church buffer overnight at 62 °C. The membrane was washed with buffers containing 2–0.1% SSC and 2–0.1% SDS at 62 °C. Washed membrane was placed on a phosphor screen for 3 days. The exposed phosphor screen was scanned by a Cyclone storage phosphor system (Packard Instrument Corp., Meridian, CN). 32P-labeled DNA probes (derived from DPE2 cDNA and elongation factor 1 α cDNA) were made by labeling DNA fragments using the Klenow fragment of E. coli DNA polymerase I primed with random hexanucleotide primers (Feinberg and Vogelstein 1983).
Extraction and measurements of carbohydrates
Leaf samples were taken and added to microfuge tubes containing killing solution (80% ethanol, 5% formic acid) at 80 °C for 20 min. Following centrifugation, 80% ethanol was added to the pellet at 80 °C for another 20 min. The solvents in the killing solution and 80% ethanol were evaporated in a SpeedVac SC110 concentrator (Savant Instruments). The pellets were resuspended with H2O and centrifuged, and the supernatants were assayed for soluble carbohydrates. The bleached leaves were dried in the concentrator, ground with pestles and incubated in 0.2 M KOH at 95 °C for 20 min. The gelatinized starch solutions were brought to pH 5.0 by adding 1 M acetic acid. The starch solutions were then hydrolyzed by amyloglucosidase (Sigma) and α–amylase (Sigma) at room temperature overnight. The hydrolysates were centrifuged and the supernatants were assayed for glucose.
To extract the water-soluble, ethanol/KCl-insoluble polysaccharides, a method modified from Yang and Steup (1990) was used for small-scale preparation. Leaf samples taken at different time points were ground in liquid N2, suspended in chilled H2O, and centrifuged. The supernatants were deproteinized with an equal volume of chilled water-saturated phenol twice, an equal volume of chloroform once, and de-ionized with an anion- and cation-exchange resin Serdolit MB-1 (Serva). The polysaccharides were precipitated with 1% KCl and 70% ethanol, the pellets were washed twice with 75% ethanol to remove residual ethanol-soluble carbohydrates. The pellets were resuspended and hydrolyzed in 2 N HCl for 90 min at 95 °C. The hydrolysates were neutralized with 2 N NaOH and the glucose contents were determined.
Carbohydrate determination was made using NADP(H)-linked assays (Lowry and Passonneau 1972) in a Sigma ZFP 22 dual-wavelength filterphotometer (Sigma Instruments, Berlin, Germany). Phosphate buffer (pH 7.4, 50 mM KH2PO4, 20 mM KCl, 10 mM MgCl2, 2 mM EDTA) was used. Phosphate buffer (800 μl) containing 0.8 mM NADP and 0.8 mM ATP, 1.25 U (Unit=1 μmol mg−1 protein min−1) of G6P dehydrogenase and 20 U of maltose 1-epimerase was added into the cuvette before carbohydrate samples were added. Reactions were started by the addition of 1.25 U of hexokinase, and the glucose content was determined. Maltose content was determined by the addition of 10 U of maltose phosphorylase. Phosphoglucoisomerase (5 U) was added to measure fructose 6-phosphate (F6P) plus fructose. F6P without fructose was measured in a subsequent reaction so that the amount of fructose could be calculated. Sucrose in the carbohydrate samples was hydrolyzed by invertase in 50 mM citrate buffer (pH 4.5) and glucose constituent in the sucrose was determined by the same enzymatic assay. Maltose epimerase and maltose phosphorylase were purchased from the Kikkoman Corp (Tokyo). These enzymes allow accurate, precise and specific assay of maltose even in the presence of millimolar levels of sucrose (Shirokane et al. 2000). All the other enzymes used were purchased from Sigma–Aldrich (St. Louis MO, USA).
HPLC analysis
Monosaccharides in HCl-hydrolyzed polysaccharides were measured using an HPLC system consisting of a Waters 626 pump (Waters Corp., Milford, MA, USA), a Waters 717plus autosampler, a CarboPac PA100 4×50 mm guard column (Dionex Corp., Sunnyvale, CA, USA), a CarboPac PA-100 4×250 mm column, and a Waters 464 pulsed electrochemical detector with a gold electrode (Weise et al. 2003). To separate the monosaccharides, 20 mM NaOH was used isocratically. The flow rate was 1 ml min−1. The settings of the pulse amperometric detector were the following: E 1=50 mV, T 1=0.4 s, E 2=750 mV, T 2=0.2 s, E 3=−150 mV, T 3=0.4 s. The range was set to 0.2 μA.
Chloroplast isolation experiments
To isolate chloroplasts, a method modified from Weise et al. (2003) was used for Arabidopsis leaves. Rosette leaves of six wild-type Arabidopsis plants (nine dpe2 plants) were taken in the middle of the light period. Leaves were macerated in a blender using four bursts of 1 s each. Maceration buffer used was 50 ml ice-cold 50 mM Hepes buffer (pH 7.6), 330 mM mannitol, 1 mM EDTA, 5 mM MgCl2, 1 mM MnCl2, 5 mM ascorbic acid and 0.25% BSA. After maceration, the leaf slurry was filtered through four layers of cheesecloth. The filtrate was then centrifuged at 4 °C in a Beckman JS-13.1 swinging-bucket rotor (Beckman Instruments) at 1,200 g for 10 min. The pellet was gently resuspended using a soft #3 paint brush in 1 ml 50 mM Hepes buffer (pH 7.6) containing the same ingredients as maceration buffer except ascorbic acid. Resuspended solution was then placed on top of two Percoll gradients. The Percoll gradients were then centrifuged at 4 °C at 1,200 g for 7 min. The bottom band between the 60% and 80% Percoll solutions containing intact chloroplasts was removed. An equal volume of resuspension buffer was added to the chloroplast solution and centrifuged for 2 min. The chloroplast pellet was then resuspended with 0.35 ml resuspension buffer and 0.05 ml of chloroplast solution was used for chlorophyll assay, 0.25 ml was used for maltodextrin extraction and the rest was used for chloroplast intactness assays. NADP(H)-dependent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity before and after osmotic shock was measured to determine the percentage of intact chloroplasts.
Chlorophyll assays
To convert maltodextrin content from μmol mg−1 chlorophyll to μmol g−1 fresh weight, total chlorophyll from fully expanded Arabidopsis leaves was assayed according to Wintermans and De Mots (1965). Leaves were taken in the middle of the light period and ground into fine power in liquid N2. Chlorophyll was extracted with 2 ml of 96% ethanol, and the leaf slurry was further homogenized with sonication, and centrifuged for 2 min. The supernatant was used for chlorophyll assay.
Results
Isolation of cytosolic amylomaltase mutants
Arabidopsis plants containing random T-DNA insertions were screened using PCR with multiple primer combinations, for insertions in the DPE2 gene. Two insertion lines were found. DNA sequence analysis confirmed the presence of the T-DNA left border sequences. One insertion was at the end of the sixth exon of the DPE2 gene. There was a deletion of 47 bases at the 5′ end of the T-DNA. The other insertion was in the sixth intron of the DPE2 gene. There was a deletion of 17 bases at the 5′ end of the T-DNA and a rearrangement of 6 bases at the junction of the T-DNA and the DPE2 gene (Fig. 1).

Structure of the dpe2-1 and dpe2-2 loci. DPE2 gene structure is depicted from 0 (translation start) to 5412 (translation stop) base pairs. Black boxes represent exons; lines represent introns. Two vertical lines show the sites of T-DNA insertion, and nucleotide and amino acid sequences on both sides show the sequences of the insertion site. One insertion is in the sixth exon (dpe2-1); the other insertion is in the sixth intron (dpe2-2). The left end of T-DNA ends with partial left border nucleotide sequences (in capitals), and with nucleotides of unidentified origin at junction of T-DNA and DPE2 gene (italicized capitals, only in dpe2-2). The gene designation is At2g40840
Phenotype of dpe2-1 and dpe2-2 mutants
Arabidopsis dpe2-1 and dpe2-2 mutants exhibited a dwarf phenotype. They were smaller than wild-type plants but their development was not affected (Fig. 2). Both dpe2-1 and dpe2-2 mutants germinated, budded, and flowered at the same time as wild-type plants, indicating that wild-type and dpe2 plants at comparable ages are at comparable developmental stages. The dwarf phenotype of dpe2 plants was found to co-segregate with T-DNA insertion and Basta resistance, indicating that the dwarf phenotype was caused by the T-DNA insertion in the DPE2 gene. The leaves of dpe2 plants were yellowish compared with leaves of wild-type plants, suggesting that dpe2 plants might accumulate carbohydrates. However, there was no measurable difference in chlorophyll content, indicating the yellow color could result from a higher carotenoid content.

13-day-old wild-type, dpe2-1 and dpe2-2 Arabidopsis thaliana plants
Northern blot and diurnal transcription of DPE2
The transcript level of DPE2 varied between day and night. There were higher transcript levels during the light period and lower transcript levels during the dark period, suggesting that the transcription of DPE2 is light inducible (Fig. 3). The full-length DPE2 mRNA was missing in both dpe2-1 and dpe2-2 plants. A truncated DPE2 mRNA, which had a T-DNA insertion in the sixth exon, was detected during the light period in dpe2-1 plants.

Northern blot for the expression of DPE2 in wild-type and dpe2 A. thaliana plants. The transcript level of DPE2 was higher during the light period and lower during the dark period. Full-length DPE2 mRNA was not detected in either dpe2-1 or dpe2-2 plants. A truncated DPE2 mRNA, which had a T-DNA insertion in the sixth exon, was detected in dpe2-1 plants during the light period (indicated by asterisks). As a loading control, the amount of EF1α mRNA is shown. L16 indicates hours in the light period; D3 and D8 indicate hours in the dark period; L8 indicates hours in the next light period
dpe2-1 and dpe2-2 mutants accumulate a large amount of maltose and starch
The amount of maltose was 20–90 times higher in dpe2 mutants than in wild-type plants at the same age (Fig. 4). Consistent with the maltose content in bean, Phaseolus vulgaris L. (Weise et al. 2003), the maltose content in wild-type Arabidopsis was higher at night than during the day, especially early in the night. There was a peak in maltose content in the dpe2 plants 4 h after the lights came on. The starch level in both wild-type and dpe2 plants increased throughout the light period and dropped throughout the dark period. The overall change in amount was similar in wild-type and dpe2 plants. However, the dpe2 plants had a large amount of starch that was never broken down (Fig. 4).

Time course of maltose, starch, sucrose, G1P, G6P, F6P, glucose, and fructose contents of Arabidopsis leaves. The last data point is a repeat of the first data point and is shown for clarity. Data from homozygous dpe2-1 (●) and dpe2-2 (▲) plus wild-type (■) plants are shown. Values are mean ± SE (n=5). The gray area in the figure indicates the time of darkness
dpe2-1 and dpe2-2 mutants have reduced levels of G1P, G6P, and sucrose at night
The metabolites presumed to be downstream of amylomaltase were lower at night in the dpe2 plants than in wild-type plants. Specifically, levels of sucrose, G1P, and G6P in the dpe2 plants were about half those of the wild type during the dark period but during the light period they were similar. Glucose and fructose contents were high in the dpe2 plants during the day but were low in all plants at night (Fig. 4).
dpe2 mutants accumulate ethanol/KCl-insoluble maltodextrin in chloroplasts
There are two ethanol/KCl-insoluble polysaccharides in Arabidopsis, high-molecular-weight maltodextrin in chloroplasts, and a high-molecular-weight heteroglycan in the cytosol. HPLC analysis showed that the polysaccharide fraction from Arabidopsis leaves was made up of arabinose, galactose, glucose, xylose and ribose. The glucose content in the total ethanol/KCl-insoluble polysaccharides from dpe2 leaves was three times as much as that in wild-type leaves (Fig. 5). But the contents of arabinose and galactose (Fig. 5) and other monosaccharides — xylose and ribose (data not shown) — did not change as much. This is consistent with the fact that in Arabidopsis sex1 (s tarch ex cess) mutants only the portion of glucose in non-starch polysaccharides was increased (Schneider et al. 2002).

Glucose (Glc), arabinose (Ara) and galactose (Gal) contents in the ethanol/KCl-insoluble polysaccharides from dpe2 (black columns), and wild-type (white columns) A. thaliana leaves. Values are mean + SE (n=5)
To investigate whether the increased amount of glucose is from chloroplastic maltodextrin or the cytosolic heteroglycan, we extracted the high-molecular-weight maltodextrin from isolated chloroplasts. HPLC analysis showed that the chloroplast maltodextrin was made up of glucose, with little or no other monosaccharide. The amount of glucose in the ethanol/KCl-insoluble polysaccharides extracted from intact leaves (Fig. 5) was not statistically different from the amount of glucose in the maltodextrin extracted from isolated chloroplasts (Fig. 6). This indicates that most, if not all, of the increased amount of glucose observed in the dpe2 polysaccharides was in the maltodextrin in the chloroplast.

Glucose content in the maltodextrin from chloroplasts isolated from wild-type (WT) and dpe2 A. thaliana. Values are mean + SE (n=5)
Discussion
The DPE2 (cytosolic disproportionating enzyme, another name for the GTase) gene, At2g40840, is necessary for maltose metabolism in Arabidopsis. Knocking out the cytosolic GTase gene resulted in mutants having a large amount of maltose and starch and chloroplast maltodextrin compared to wild type. (T. Chia, D. Thorneycraft, S.M. Smith, and A.M. Smith reported similar findings at the 14th International Conference on Arabidopsis Research in Madison, WI.) Metabolites that would be downstream from amylomaltase (e.g. G1P, G6P, and sucrose) were lower at night but not during the day. We hypothesize that knocking out the DPE2 enzyme blocks the conversion of maltose to the cytosolic polysaccharide. This results in a large amount of maltose in dpe2 mutants. This is consistent with the fact that malQ mutants of E. coli accumulate large amounts of free maltose inside the cell (Szmelcman et al. 1976). On the other hand, the large amount of maltose may feed back to limit starch degradation reactions in chloroplasts. This would explain why dpe2 mutants have higher starch and maltodextrin contents. By extracting maltodextrin from isolated chloroplasts, we could tell that most, if not all, of the increased amount of glucose in the non-starch polysaccharides is from chloroplast maltodextrin, not from the cytosolic polysaccharide.
The dpe2 plants showed high levels of glucose and G6P 4 h after the lights came on. The reason for this is not clear to us. It could represent clearing of metabolite pools that built-up during the night but which could not be metabolized until the lights were turned on. This may explain why the dpe2 plants can break down as much starch as wild-type plants, albeit with a higher background level of starch. In dpe2 plants, starch accumulates during the first 4 h of light at least as fast as in the wild-type plants. Therefore, we believe the peak in glucose and fructose pools 4 h after the light came on represents preexisting carbon, not new photosynthate.
Type-II 4αGTase is a member of the α-amylase super-family; these enzymes have four distinct conserved regions containing proposed catalytic and binding sites (Matsuura et al. 1984; Takaha and Smith 1999). Examination of the amino acids in these regions suggested that the gene product we studied is more like the E. coli protein than other higher plant D-enzymes. To further investigate the relationships we aligned the protein sequences with MegAlign (Clustal method, gap penalty = 10, gap length penalty = 10), and generated phylogenetic trees by 1,000 bootstrap replicates under distance settings with PAUP (Fig. 7). Whether including or excluding the putative cTP from the protein sequences of 4αGTases in Arabidopsis, potato, and rice, the same tree topology and bootstrap values were obtained. The phylogenetic trees from the conserved regions (not shown) and the protein sequences both show that the cytosolic GTase in Arabidopsis is more closely related to amylomaltase in E. coli than to plastidic D-enzymes in plants (Fig. 7).

Phylogenetic relationships of the protein sequences of type-II 4αGTases from Arabidopsis thaliana, rice (Oryza sativa), potato (Solanum tuberosum), and Escherichia coli. 4αGTase from Thermus aquaticus was used to root the tree as convention. The protein sequences were aligned by MegAlign (DNAStar) and analyzed by PAUP. Numbers on the branches are bootstrap values. Whether including or excluding the putative cTP from the protein sequences of 4αGTases in Arabidopsis, potato, and rice, the same tree topology and bootstrap values were obtained. The accession numbers for the genes used are A. thaliana 1, NP_201291; S. tuberosum 1, CAA48630; O. sativa 1, BAC07076; A. thaliana 2 (DPE2), AAL91204; O. sativa 2, BAC22431; E. coli, NP_756057; T. aquaticus BAA33728. For plant enzymes, 1 refers to the plastidial form while 2 refers to the form that lacks a transit peptide
The smallest substrate that the plastidic D-enzyme recognizes is maltotriose (Palmer et al. 1976) but recent data show that maltose can act as an acceptor for amylomaltase from E. coli (Kitahata et al. 1989; Takaha and Smith 1999). We propose that the cytosolic amylomaltase in Arabidopsis leaves can act on maltose. There was no detectable difference in GTase activity towards maltotriose in dpe2 and wild-type plants (data not shown). However, preliminary assay of GTase activity with maltose showed that there was a significant decrease of maltose-dependent GTase activity in dpe2 mutants. This is consistent with the proposed cytosolic amylomaltase activity towards maltose. We are going to express the amylomaltase cDNA in E. coli and study the substrate specificity of this enzyme in greater detail.
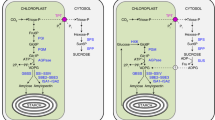
We propose that maltose metabolism in the cytosol of Arabidopsis leaves (Fig 8) is similar to maltose metabolism in the cytoplasm of E. coli (Boos and Shuman 1998). At night, starch in leaves is broken down and the carbon is exported to the cytosol in the form of maltose and glucose. After maltose is exported into the cytosol, amylomaltase transfers a polyglucan (the glucan part of the heteroglycan) onto the maltose and releases glucose, just as amylomaltase (MalQ) does in E. coli (Boos and Shuman 1998). Preliminary data using heterozygous knockout plants for a cytosolic maltodextrin phosphorylase indicate that lack of this enzyme also results in starch and maltose accumulation and low sucrose levels at night. Cytosolic maltodextrin phosphorylase recognizes maltopentaose and longer maltodextrins to form G1P and a maltodextrin that is one glucosyl unit smaller, as maltodextrin phosphorylase does in E. coli (Boos and Shuman 1998). By this cycle, maltose is converted to glucose and G1P. In the presence of hexokinase, which is attached to the outer membrane of chloroplasts (Wiese et al. 1999), glucose is converted to G6P. Subsequent steps are those well-known in sucrose synthesis: G6P is converted to fructose 6-phosphate (F6P) by the action of phosphoglucose isomerase (PGI) while G1P is converted to UDP-glucose (UDPG) by the action by UDPG pyrophosphorylase in the presence of UTP; F6P and UDPG are converted to sucrose 6-phosphate by sucrose-phosphate synthase (SPS); sucrose 6-phosphate is then converted to sucrose by the action of sucrose-phosphate phosphatase (SPP). Finally, sucrose is transported to other plant organs through phloem, or stored locally in the vacuoles.

Hypothetical pathway for transitory-starch breakdown and conversion to sucrose. We hypothesize that the glucan part of the cytosolic polysaccharide is the direct intermediate between maltose and sucrose. Therefore, this cytosolic polysaccharide is described as polyglucan in this diagram
The nature of the cytosolic polysaccharide involved in this metabolism in Arabidopsis is not clearly understood yet, but it seems significant that the heteroglycan possesses priming capacity for the phosphorylase reaction (Yang and Steup 1990). The advantages of having a cytosolic polysaccharide pool in the cytosol may include: (1) Acting as an overflow for carbohydrates (such as maltose and glucose) formed from starch degradation at night when the production of hexoses exceeds the demand for and transport of sucrose. It acts like a soluble starch pool in the cytosol just as transitory starch is a carbon overflow in chloroplasts (Stitt and Quick 1989). (2) By forming polysaccharide, the molarity of total sugar in the cytosol is kept low; as a result, the cell keeps an intracellular osmotic pressure that is good for its overall metabolic reactions in the cytosol.
In summary, the DPE2 gene is necessary for maltose metabolism in Arabidopsis. We propose that maltose metabolism in the cytosol of Arabidopsis leaves is similar to maltose metabolism in the cytoplasm of E. coli. If the maltose/maltodextrin system for maltose metabolism is the primary mechanism by which carbon in starch is converted to sucrose, then the regulation of starch metabolism may be easier to understand as the regulation of the enzymes involved are discovered.
Abbreviations
- F6P:
-
fructose 6-phosphate
- G1P:
-
glucose 1-phosphate
- G6P:
-
glucose 6-phosphate
- GTase:
-
glucanotransferase
References
Boos W, Shuman H (1998) Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol Mol Biol Rev 62:204–229
Critchley JH, Zeeman SC, Takaha T, Smith AM, Smith SM (2001) A critical role for disproportionating enzyme in starch breakdown is revealed by a knock-out mutation in Arabidopsis. Plant J 26:89–100
Duwenig E, Steup M, Willmitzer L, Kossmann J (1997) Antisense inhibition of cytosolic phosphorylase in potato plants (Solanum tubersosum L.) affects tuber sprouting and flower formation with only little impact on carbohydrate metabolism. Plant J 12:323–333
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Feinberg AP, Vogelstein B (1983) A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132:6–13
Goda SK, Eissa O, Akhtar M, Minton NP (1997) Molecular analysis of a Clostridium butyricum NCIMB 7423 gene encoding 4-alpha-glucanotransferase and characterization of the recombinant enzyme produced in Escherichia coli. Microbiology 143:3287–3294
Heldt HW, Chon CJ, Maronde D, Herold A, Stankovic ZS, Walker DA, Kraminer A, Kirk MR, Heber U (1977) Role of orthophosphate and other factors in the regulation of starch formation in leaves and isolated chloroplasts. Plant Physiol 59:1146–1155
Hofnung M, Schwartz M, Hatfield D (1971) Complementation studies in the maltose-A region of Escherichia coli K12 genetic map. J Mol Biol 61:681–694
Kakefuda G, Duke SH (1989) Characterization of pea chloroplast D-enzyme (4-α-glucanotransferase) Plant Physiol 91:136–143
Kakefuda G, Duke SH, Hostak MS (1986) Chloroplast and extrachloroplastic starch-degrading enzymes in Pisum sativum L. Planta 168:175–182
Kitahata S, Murakami H, Okada S (1989) Purification and some properties of amylomaltase from Escherichia coli IFO-3806. Agric Biol Chem 53:2653–2659
Lacks SA, Dunn JJ, Greenberg B (1982) Identification of base mismatches recognized by the heteroduplex-DNA-repair system of Streptococcus pneumoniae. Cell 31:327–336
Levi C, Gibbs M (1976) Starch degradation in isolated spinach chloroplasts. Plant Physiol 57:933–935
Lin T, Preiss J (1988) Characterisation of D-enzyme (4-α-glucanotransferase) in Arabidopsis leaf. Plant Physiol 86:260–265
Lowry OH, Passonneau JV (1972) A flexible system of enzymatic analysis. Academic Press, Orlando, FL, pp 1–291
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Manners DJ, Rowe KL (1969) Studies on carbohydrate-metabolising enzymes: Part XXI. The α-1-glucosidase and D-enzyme activity of extracts of carrots and tomatoes. Carbohydr Res 9:441–450
Matsuura Y, Kusunoki M, Harada W, Kakudo M (1984) Structure and possible catalytic residues of taka-amylase a. J Biochem Tokyo 95:697–702
Mori H, Tanizawa K, Fuku T (1991) Potato tuber type H phosphorylase isozyme: molecular cloning, nucleotide sequence, and expression of a full-length cDNA in Escherichia coli. J Biol Chem 266:18446–18453
Okita TW, Greenberg E, Kuhn DN, Preiss J (1979) Subcellular localization of the starch degradative and biosynthetic enzymes of spinach leaves. Plant Physiol 64:187–192
Palmer NT, Ryman BE, Whelan WJ (1976) The action pattern of amylomaltase from Escherichia coli. Eur J Biochem 69:105–115
Peat S, Whelan WJ, Rees WR (1956) The enzymic synthesis and degradation of starch (part XX). The disproportionating enzyme (D-enzyme) of the potato. J Chem Soc 1956:44–53
Peavey DG, Steup M, Gibbs M (1977) Characterization of starch breakdown in the intact spinach chloroplast. Plant Physiol 60:305–308
Preiss J, Okita TW, Greenberg E (1980) Characterization of the spinach leaf phosphorylases. Plant Physiol 66:864–869
Pugsley AP, Dubrevil C (1988) Molecular characterization of malQ, the structural gene for the Escherichia coli enzyme amylomaltase. Mol Microbiol 2:473–479
Ritte G, Raschke K (2003) Metabolite export of isolated guard cell chloroplasts of Vicia faba. New Phytol 159:195–202
Schneider A, Häusler RE, Kolukisaoglu Ü, Kunze R, van der Graaff E, Schwacke R, Catoni E, Desimone M, Flügge U-I (2002) An Arabidopsis thaliana knock-out mutant of the chloroplast triose phosphate/phosphate translocator is severely compromised only when starch synthesis, but not starch mobilization is abolished. Plant J 32:685–699
Servaites JC, Geiger DR (2002) Kinetic characteristics of chloroplast glucose transport. J Exp Bot 53:1–11
Shirokane Y, Ichikawa K, Suzuki M (2000) A novel enzymic determination of maltose. Carbohydr Res 329:699–702
Steup M, Latzko E (1979) Intracellular localization of phosphorylases in spinach and pea leaves. Planta 145:69–75
Steup M, Schächtele C, Latzko E (1980) Purification of a non-chloroplastic α-glucan phosphorylase from spinach leaves. Planta 148:168–173
Stitt M, ap Rees T (1980) Carbohydrate breakdown by chloroplasts of Pisum sativum. Biochim Biophys Acta 627:131–143
Stitt M, Heldt HW (1981) Physiological rates of starch breakdown in isolated intact spinach chloroplasts. Plant Physiol 68:755–761
Stitt M, Quick WP (1989) Photosynthetic carbon partitioning: Its regulation and possibilities for manipulation. Physiol Plant 77:633–641
Szmelcman S, Schwartz M, Silhavy TJ, Boos W (1976) Maltose transport in Escherichia coli K12. A comparison of transport kinetics in wild-type and λ–resistant mutants with the dissociation constants of the maltose binding protein as measured by fluorescence quenching. Eur J Biochem 65:13–19
Takaha T, Smith SM (1999) The functions of 4-α-glucanotransferases and their use for the production of cyclic glucans. Biotech Gen Eng Rev 16:257–280
Takaha T, Yanase M, Okada S, Smith SM (1993) Disproportionating enzyme (4-α-glucanotransferase — EC 2.4.1.25) of potato — purification, molecular cloning, and potential role in starch metabolism. J Biol Chem 268:1391–1396
Takaha T, Critchley J, Okada S, Smith SM (1998) Normal starch content and composition in tubers of antisense potato plants lacking D-enzyme (4-α-glucanotransferase). Planta 205:445–451
Terada Y, Fujii K, Takaha T, Okada S (1999) Thermus aquaticus ATCC 33923 amylomaltase gene cloning and expression and enzyme characterization: production of cycloamylose. Appl Environ Microbiol 65:910–915
Trethewey RN, Smith AM (2000) Starch metabolism in leaves. In: Leegood RC, Sharkey TD, von Caemmerer S (eds) Photosynthesis: physiology and metabolism. Kluwer, Dordrecht, pp 205–231
Weigel D, Ahn JH, Blázquez MA, Borevitz JO, Christensen SK, Fankhauser C, Ferrándiz C, Kardailsky I, Malancharuvil EJ, Neff MM, Nguyen JT, Sato S, Wang Z-Y, Xia Y, Dixon RA, Harrison MJ, Lamb CJ, Yanofsky MF, Chory J (2000) Activation tagging in Arabidopsis. Plant Physiol 122:1003–1013
Weise SE, Weber APM, Sharkey TD (2003) Maltose is the major form of carbon exported from the chloroplast at night. Planta (in press)
Wiese A, Gröner F, Sonnewald U, Deppner H, Lerchl J, Hebbeker U, Flügge U-I, Weber A (1999) Spinach hexokinase I is located in the outer envelope membrane of plastids. FEBS Lett 461:13–18
Wintermans JFGM, DeMots A (1965) Spectrophotometric characteristics of chlorophylls a and b and their pheophytins in ethanol. Biochim Biophys Acta 109:448–453
Yang Y, Steup M (1990) Polysaccharide fraction from higher plants which strongly interacts with the cytosolic phosphorylase isozyme I. Isolation and characterization. Plant Physiol 94:960–969
Acknowledgements
This research was supported by the US Department of Energy under grant DE-FG02-99ER 20345. We thank the Biotech Center knockout facility of UW-Madison for access to T-DNA tagged plants. We also thank Donna E. Fernandez, Andreas P.M. Weber, and Sean E. Weise for their advice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu, Y., Sharkey, T.D. The role of amylomaltase in maltose metabolism in the cytosol of photosynthetic cells. Planta 218, 466–473 (2004). https://doi.org/10.1007/s00425-003-1127-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-003-1127-z