
Abstract
Mutations in the KCNK18 gene that encodes the TRESK K2P potassium channel have previously been linked with typical familial migraine with aura. Recently, an atypical clinical case has been reported in which a male individual carrying the p.Trp101Arg (W101R) missense mutation in the KCNK18 gene was diagnosed with intellectual disability and migraine with brainstem aura. Here we report the functional characterization of this new missense variant. This mutation is located in a highly conserved residue close to the selectivity filter, and our results show although these mutant channels retain their K+ selectivity and calcineurin-dependent regulation, the variant causes an overall dramatic loss of TRESK channel function as well as an initial dominant-negative effect when co-expressed with wild-type channels in Xenopus laevis oocytes. The dramatic functional consequences of this mutation thereby support a potentially pathogenic role for this variant and provide further insight into the relationship between the structure and function of this ion channel.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The KCNK18 gene codes for the TWIK-related spinal cord K+ channel (TRESK). The relevant protein (K2P18.1) belongs to the two-pore domain (K2P) family of K+ channels involved in the control of cellular electrical excitability [7, 8, 24]. The regulation of TRESK activity by the calcium-dependent phosphatase calcineurin [3, 4], as well as its expression in dorsal root [27] and trigeminal ganglia [2] led to a proposed role for this channel in a variety of pain pathways [26]. In particular, a frameshift mutation (F139Wfsx24) in TRESK was identified in a multigenerational pedigree where it co-segregated with typical migraine with aura [18]. Furthermore, functional analysis revealed that this mutation caused a dominant-negative loss of TRESK function and that the truncated subunit was also capable of down regulating wild-type channel function. This therefore highlighted a potential role for KCNK18 in the pathogenesis of familial migraine with visual aura [18]. Interestingly, a very recent study [23] has shed further light on the unusual molecular mechanism by which this particular frameshift mutation works by introducing an alternative translation initiation site to produce a truncated protein that not only down regulates wild-type TRESK activity, but also that of several unrelated K2P channels in trigeminal ganglion neurons. However, the relationship of other missense variants in TRESK to migraine and/or any other disease states remains unclear.
Recently, an atypical clinical case has been reported in which a male individual was diagnosed with intellectual disability (IQ 65) at the age of 10. It was reported that the patient presented with an acute confused state, agitation, speech impairment, disorientation, and loss of sense of place and time at the age of 12. It was also reported that approximately 1 h before this change occurred, he had experienced severe head pain and dizziness accompanied by vomiting and nausea, aggravated by movement. There was a familial history of migraine, with both his mother and elder sister having a history of migraine attacks with visual impairment and nausea, and mild intellectual disability. The formulated diagnosis for the proband was intellectual disability associated with migraine with brainstem aura. Targeted next generation sequencing and Sanger sequencing revealed that the proband harbored a heterozygous missense mutation (c.301 T > C; p.Trp101Arg; W101R) in KCNK18 that was maternally inherited [11]. However, at the time, the potential effects of this mutation on TRESK channel function were not assessed and its pathogenic relevance not corroborated. In this study, we have investigated the functional effect of the W101R variant to probe the potential association between TRESK channel dysfunction, migraine with brainstem aura and intellectual disability. Our results demonstrate a significant functional impairment of TRESK channel activity associated with this mutation.
Materials and methods
Molecular biology
Human TRESK was subcloned between the 5′ and 3′ UTR of the Xenopus β-globin gene in the oocyte expression vector, pFAW. The W101R mutant was introduced by site directed mutagenesis and confirmed by automated sequencing. mRNA for wild-type and mutant channels was synthesized using the T7 mMESSAGE mMACHINE kit (Ambion, Life Technologies, Carlsbad, CA, USA) and mRNA concentrations were quantified by spectrophotometric analysis prior to injection.
Electrophysiological recordings
Unless otherwise stated, equal quantities of either wild-type or mutant mRNAs were microinjected into defolliculated Xenopus laevis oocytes according to standard protocols and in accordance with international standards of animal care, the Maltese Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Whole-cell currents were recorded 1 to 6 days after injection, at room temperature (20–22 °C), using the two-electrode voltage clamp method (Axoclamp-2B, Axon DIGIDATA 1550B, Axon Instruments, Foster City, CA, Axon). Oocytes were held in a small recording chamber and were continuously perfused with control low K+ extracellular solution, (in mM 95.4 NaCl, 2 KCl, 1.8 CaCl2, 5 HEPES pH 7.5 with NaOH). Recording electrodes were back-filled with 3 M KCl and their resistances varied from 0.3 to 1 MΩ. Currents were filtered at 100 Hz and digitized at 1 kHz for analysis. Oocytes were held at −80 mV, voltage commands were applied, and currents were recorded using Clampex 10.7 software (Axon Instruments, Foster City, CA, USA). TRESK currents were usually measured in low K+ solution, at the end of 1 s long voltage steps from a holding potential of − 80 mV delivered in 20 mV increments from − 120 mV to + 60 mV. For the experiments with ionomycin, TRESK currents were measured in high K+ solution (in mM 17.4 NaCl, 80 KCl, 1.8 CaCl2, 5 HEPES, pH 7.5 with NaOH) at the end of 300 ms-long voltage steps from 0 mV to −100 mV. Ionomycin (free acid form), was made as a stock solution of 1 mM in DMSO and diluted in the high K+ solution to the appropriate test concentration (0.5 μM) on the day of the experiment. Data were analyzed with Clampfit 10.7 (Axon instruments, Foster City, CA, USA), Igor and Kaleida Graph programs.
Statistics
Data are given as mean values ± standard error of the mean (SEM), where n represents the number of oocytes. Results were reproducible in at least 2–3 different batches of oocytes. Statistical significance was determined using a student’s t test. When error bars are not shown they are smaller than the size of the symbol.
Results
Location of the missense variant in the TRESK channel
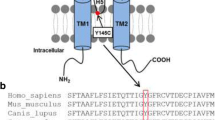
Using a previously generated homology model of human TRESK [1], the predicted location of the W101R missense variant within the TRESK channel is shown in Fig. 1c. This tryptophan (W101) is located within the first extracellular loop, upstream of the selectivity filter and within the first pore domain (P1) (Fig. 1a, b). Interestingly, this residue also resides within a very highly conserved region and is found at this position in all 15 members of the human K2P family (Fig. 1a). Furthermore, the structure reveals W101 to be in close proximity to the first pore-helix and adjacent to the selectivity filter which has been implicated in the control of K2P channel gating [22]. The highly conserved nature of this residue and its predicted location therefore suggest that it may play a critical role in the structure and/or function of the TRESK channel.

Localization of the W101 residue. a Alignment of human K2P channel sequences in the region of the W101R missense variant. This indicates that this highly conserved residue (in bold) is located adjacent to Pore-Helix 1 and the selectivity filter of the first pore-domain (P1). b Schematic topology of the human K2P TRESK subunit showing the position of the W101R variant. c Homology model of the TRESK channel in this region shows the predicted location of this highly-conserved tryptophan, just before the first pore-helix
Loss of TRESK function in the W101R variant
We next examined whether the W101R variant affects the functional properties of human TRESK channels. Figure 2a shows representative whole-cell basal currents recorded from Xenopus oocytes injected with mRNA encoding either wild-type (WT) or mutant W101R TRESK channel. While the WT TRESK channels exhibited large outwardly-rectifying K+ currents similar to those previously reported [17, 24], the W101R mutant expressed markedly reduced current amplitudes (Fig. 2a, b). Although the W101R currents were very small compared to WT, residual current could still be detected for this variant (Fig. 2) and when greater quantities of mRNA (5 ng) were injected, we found that larger whole-cell currents could be recorded indicating that the channel is not completely non-functional.

W101R mutation reduces the basal current and abolishes the ability of TRESK channels to control the cell resting potential. a Representative families of current traces for the WT (left) and W101R (right) channels. Note that unlike the WT channel, W101R produces little current. The inset on the right shows an enlargement of the W101R currents. Recordings were done in K+ 2 mM. Cells membrane potential was held at -80 mV. Each family of currents was evoked by 1 s long voltage steps from −120 mV to 60 mV, with 20 mV increments. The recordings were performed 48 h after the injection of 1 ng of mRNA for each channel type. b Average of plateau current as a function of voltage (IV relationships) calculated from experiments as in A for WT (circles) and W101R (squares). The data points are mean ± standard error (n = 12). The averaged current amplitudes for W101R channels are statistically different from those of WT from − 40 to + 60 mV (p < 0.001). c Resting membrane potentials from individual oocytes uninjected (red dots), injected with 1 ng of WT or W101R mRNA and recorded 24 (gray dots) or 48 (blue dots) hours after injection. Note that while the expression of the WT channels shifts the resting potential towards K+ reversal potential, the W101R has little effect even at 48 h post-injection of the relevant mRNA. The dots in c represent single cell recordings. The data are mean ± standard deviation. At 24 and 48 h the statistical differences are as follow: WT vs W101R, p < 0.001; WT vs uninjected, p < 0.001; W101R vs uninjected, n.s.
TRESK channels typically produce a leak K+ current that stabilizes the resting membrane potential and we observed that the expression of WT TRESK shifted the cell resting membrane potential of the oocytes towards the K+ equilibrium potential (EK) (Fig. 2c). Assuming an intracellular K+ concentration of ~ 140 mM in the oocyte, the predicted value for EK is −107 mV. This hyperpolarizing effect was observed at both 24 and 48 h after injection of WT RNA. On the other hand, the resting membrane potential of oocytes expressing the mutant channel were only marginally more negative than uninjected oocytes at both time periods (Fig. 2c).
Given the location of this mutation close to the selectivity filter which forms an important part of the gating mechanism in K2P channels, we next examined whether the residual W101R mutant currents were still capable of responding to calcineurin activation. In WT TRESK channels expressed in Xenopus oocytes, an increase of intracellular Ca2+ produced by the calcium ionophore ionomycin, produces a calcineurin-dependent activation of TRESK [3, 4]. We therefore determined the effect of ionomycin on this TRESK variant. As expected, 0.5 μM ionomycin induced a large, robust and reversible activation of WT TRESK (Fig. 3a). However, we also found that ionomycin activated the W101R variant, although the absolute values of Ca2+-activated whole-cell currents were significantly smaller than that for the WT channel (Fig. 3a–d). The response of un-injected control oocytes to ionomycin was insignificant as previously reported [28] (Fig. 3).

Ionomycin activates both WT and W101R TRESK channels. Representative data points showing ionomycin activation of TRESK WT and W101R currents as well as endogenous currents from uninjected oocytes (a, b). Currents were evoked from oocytes injected with 0.13 or 1 ng of mRNA of the corresponding and indicated channel type and by 300 ms long voltage steps from 0 mV to − 100 mV. The sampled data represent the average of 50 ms long period of the steady-state currents recorded at − 100 mV. The steady-state basal current amplitude (IBasal) and the ionomycin activated current amplitude (IIono) is indicated in panel b. Ionomycin activates both WT and W101R channels but have little effect on uninjected oocytes. Note that the current decay upon reapplication of a solution containing 2 mM K+ in the recording chamber is due to the reduced K+ concentration rather than ionomycin washout. Bar graphs (c, d) showing the mean steady-state basal currents (IBasal) and the ionomycin activated currents (IIono) both recorded in K+ 80 mM. The data are mean ± standard error (*p < 0.05; **p < 0.01). Normalized ionomycin activation (IIono/IBasal) for the WT, W101R TRESK (injected with 0.13 ng or 1 ng) and uninjected oocytes (e). Each data point represents the steady state ionomycin activated current divided by the steady-state basal current (IIono/IBasal) recorded in the presence of 80 mM extracellular K+. The statistical differences are as follow: WT vs W101R (0.13 ng of mRNA) n.s.; WT vs W101R (1 ng of mRNA) p < 0.001 (mean ± sd)
Interestingly, we found that extent of activation (IIono/IBasal ratios) for both WT and W101R TRESK appeared similar when a lower amount of mRNA (0.13 ng) was injected (Fig. 3e). But, when larger amounts of mRNA (1 ng) were injected the IIono/IBasal ratios for W101R were far more variable and possibly greater than that observed for WT TRESK (Fig. 3e). However, due to the relatively small size of the unstimulated IBasal currents for the W101R mutant these values may be overestimated. Nevertheless, these results suggest that the W101R mutation reduces the basal activity of the TRESK channels, but does not markedly impair their ability to respond to calcineurin activation.
The location of the W101R mutation near the selectivity filter of the channel (Fig. 1) might also alter the K+ selectivity of the channel. To address this, the extracellular K+ concentration was gradually increased by replacing Na+ with K+ while measuring the reversal potential of the currents. We found no difference between the WT and W101R reversal potential plots, indicating the mutant channel retains normal K+ selectivity (Fig. 4) and oocytes expressing these mutant W101R channels remained viable for many days, indicating a lack of cellular toxicity that is sometimes associated with altered ionic selectivity [25].

The W101R mutation does not alter the ion selectivity of TRESK channel. The reversal potentials for the WT and W101R TRESK channels are plotted vs the corresponding extracellular K+ concentration and fitted with the Nernst’s equation. The slope of the fit is 49 and 48 for WT and W101R TRESK channels, respectively (each data point for WT and W101R represents the mean ± SEM and are not statistically different; n = 8)
K2P channels assemble as dimers and the reported proband was heterozygous for the W101R variant. Previous studies that examined loss-of-function mutations in the TRESK channel also report a reduction in WT channel activity when mutant and WT TRESK are co-expressed in a 1:1 ratio, i.e. a situation that mimics the heterozygous state [18]. We therefore examined whether the W101R variant had a similar effect. Co-injection of WT and W101R mutant mRNAs in a 1:1 ratio reduced the whole-cell currents nearly 70% at day 1 post mRNA injection (Fig. 5a). Similar reductions were also observed when the WT:W101R mRNA ratio was increased to 1:5 and the time course of whole cell current expression was also altered (Fig. 5b). Overall, our results suggest that the W101R variant has the potential to produce a dominant negative effect by co-assembly with WT TRESK channels.

Expression time course for WT TRESK, W101R and WT:W101R channels in Xenopus oocytes. Steady-state whole-cell current amplitudes were recorded at +60 mV and plotted as a function of days after the injections of the indicated mRNAs at 1:1 (a) and 1:5 (b) ratios into oocytes drawn from the same frog (data points are mean ± SEM; n = 10, significance: *p < 0.05, **p < 0.01, ***p < 0.001 vs WT values)
Discussion
The majority of intellectual disability in patients remain undiagnosed, and this can have considerable adverse effects on the proband and family members, such as failure to identify proper management, as well as failure to provide anticipatory support and neurological prognosis. In this study, we have functionally characterized a newly identified missense variant in the KCNK18 gene which may provide important insights into the proposed association between this gene and the reported phenotype of the described patient. This W101R variant was identified in a male proband first diagnosed with a disability characterized by limited intellectual function associated with increasing neurological dysfunction including migraine with brainstem aura. The mutation was maternally inherited, and the mother also presented with mild intellectual disability as well as migraine attacks with visual impairment and nausea. The results presented here, therefore expand the phenotypic spectrum associated with TRESK channel dysfunction, a phenomenon not uncommon in K+ channelopathies (e.g., Episodic Ataxia Type 1, EA1) where in addition to the core symptoms of a disease, unexpected atypical symptoms and/or co-morbidities can be present [5, 6].
The reduction in whole cell current associated with this W101R variant is consistent with the location of this mutation adjacent to the selectivity filter where substitution of a large highly conserved aromatic side-chain with a positively charged arginine is presumably disruptive to channel structure and/or function, especially since the selectivity filter has been implicated in the control of K2P channel gating [19]. However, the mutated channels retained their K+ selectivity indicating that the structure of the filter is not dramatically compromised. Interestingly, despite its low basal currents, the W101R mutation still retained its ability to respond to calcineurin-dependent activation, and this may be significant in differentiating variants that have a complete loss-of-function with those that retain not only some activity, but also the ability to respond to Ca2+-dependent signaling pathways. Whether the mutation leads to an intrinsic loss of function and/or problems with channel structure/assembly that impair its trafficking to the cell surface remains to be determined, though such questions are best be addressed in more native cell types. Nevertheless, our results clearly indicate compromised TRESK channel activity associated with this variant that reduces the channel’s ability to perform its physiological role, especially in the control of the resting membrane potential.
The ability of the W101R variant to functionally interact with, and reduce the activity of, the co-expressed WT TRESK, as would be observed in the heterozygous state, could also further reduce overall TRESK channel activity through heteromeric co-assembly. Often in K+ channel diseases the mutant subunits further reduce channel activity through ‘dominant-negative’ co-assembly. For tetrameric K+ channels this can sometimes result in a > 90% suppression of channel activity [12,13,14,15]. For dimeric K2P channels, however, this effect is not predicted to be as severe because at least 25% homomeric WT channels exist. The W101R variant initially exerts a strong dominant negative effect on the WT subunit, although this effect appears to gradually reduce over time. The reasons for this are unclear and further investigation of the underlying cell biology and trafficking of this mutant channel in a more relevant line will be required to fully understand this effect. Nevertheless, our results show that the mutant subunit clearly has the potential to exert a dominant negative effect when initially coexpressed with WT subunits. The phenotypic effects of a dominant negative mutation are also predicted to be different to that of a complete gene deletion which would be unable to co-assemble. Notably, complete loss of TRESK function by means of genetic ablation of Kcnk18 in mice causes mechanical and thermal hyperalgesia or exaggerated nocifensive behaviors during an inflammatory headache model [10, 21].
TRESK channels are expressed in a number of neuronal populations in different regions of the central and peripheral nervous systems in addition to the spinal cord and dorsal root ganglia [16, 17, 19] and can be regulated by Gαq-signaling pathways [4, 8, 20]. This strongly suggests that TRESK may contribute broadly to the maintenance and regulation of the resting membrane potential in these cell types; channel dysfunction during development of the CNS may therefore also need to be considered because K+-dependent alterations of neuronal membrane potential and excitability are known to cause serious neurodevelopmental disorders with intellectual disability [9]. Establishing a causative role of this TRESK channel variant in neuronal dysfunction clearly requires further investigation, nevertheless the severely perturbed functional properties associated with this variant provides important information towards this goal.
References
Andres-Enguix I, Shang L, Stansfeld P et al (2012) Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci Rep 2:237. https://doi.org/10.1038/srep00237
Bautista DM, Sigal YM, Milstein AD, Garrison JL, Zorn JA, Tsuruda PR, Nicoll RA, Julius D (2008) Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat Neurosci 11(7):772–779. https://doi.org/10.1038/nn.2143
Czirjak G, Enyedi P (2006) Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J Biol Chem 281:14677–14682
Czirjak G, Toth ZE, Enyedi P (2004) The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J Biol Chem 279:18550–18558
D'Adamo MC, Gallenmüller C, Servettini I et al (2015) Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol 5:525. https://doi.org/10.3389/fphys.2014.00525
D'Adamo MC, Hasan S, Guglielmi L et al (2015) New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci 9:317. https://doi.org/10.3389/fncel.2015.00317
Enyedi P, Czirják G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90:559–605. https://doi.org/10.1152/physrev.00029.2009
Enyedi P, Czirják G (2015) Properties, regulation, pharmacology, and functions of the K2p channel, TRESK. Pflugers Arch 467(5):945–958. https://doi.org/10.1007/s00424-014-1634-8
Guglielmi L, Servettini I, Caramia M, Catacuzzeno L, Franciolini F, D’Adamo MC, Pessia M (2015) Update on the implication of potassium channels in autism: K+ channelautism spectrum disorder. Front. Cell Neurosci 9:34. https://doi.org/10.3389/fncel.2015.00034
Guo Z, Qiu CS, Jiang X, Zhang J, Li F, Liu Q et al (2019) TRESK K+ channel activity regulates trigeminal nociception and headache. eNeuro 6(4):ENEURO.0236-19.2019. https://doi.org/10.1523/ENEURO.0236-19.2019
Han JY, Jang JH, Park J, Lee IG (2018) Targeted next-generation sequencing of Korean patients with developmental delay and/or intellectual disability. Front Pediatr 6:391. https://doi.org/10.3389/fped.2018.00391
Imbrici P, Cusimano A, D'Adamo MC, De Curtis A, Pessia M (2003) Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels. Pflugers Arch 446(3):373–379. https://doi.org/10.1007/s00424-002-0962-2
Imbrici P, D'Adamo MC, Cusimano A, Pessia M (2007) Episodic ataxia type 1 mutation F184C alters Zn2+−induced modulation of the human K+ channel Kv1.4 Kv1.1/Kvbeta1.1. Am J Physiol Cell Physiol 292(2):C778–C787. https://doi.org/10.1152/ajpcell.00259.2006
Imbrici P, Gualandi F, D'Adamo MC, Masieri MT, Cudia P, de Grandis D, Mannucci R, Nicoletti I, Tucker SJ, Ferlini A, Pessia M (2008) A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 157(3):577–587. https://doi.org/10.1016/j.neuroscience.2008.09.022
Imbrici P, D'Adamo MC, Grottesi A, Biscarini A, Pessia M (2011) Episodic ataxia type 1 mutations affect fast inactivation of K+ channels by a reduction in either subunit surface expression or affinity for inactivation domain. Am J Physiol Cell Physiol 300(6):C1314–C1322. https://doi.org/10.1152/ajpcell.00456.2010
Kang D, Kim D (2006) TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am J Physiol Cell Physiol 291(1):C138–C146. https://doi.org/10.1152/ajpcell.00629.2005
Kang D, Mariash E, Kim D (2004) Functional expression of TRESK-2, a new member of the tandem-pore K+ channel family. J Biol Chem 279:28063–28070
Lafrenière RG, Cader MZ, Poulin JF, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafrenière F, McLaughlan S, Dubé MP, Marcinkiewicz MM, Ramagopalan S, Ansorge O, Brais B, Sequeiros J, Pereira-Monteiro JM, Griffiths LR, Tucker SJ, Ebers G, Rouleau GA (2010) A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16(10):1157–1160. https://doi.org/10.1038/nm.2216
Mathie A, Al-Moubarak E, Veale EL (2010) Gating of two pore domain potassium channels. J Physiol 588:3149–3156
Pergel E, Lengyel M, Enyedi P, Czirják G (2019) TRESK (K2P18.1) background Potassium Channel is activated by novel-type protein kinase C via Dephosphorylation. Mol Pharmacol 95(6):661–672. https://doi.org/10.1124/mol.119.116269
Pettingill P, Weir GA, Wei T, Wu Y, Flower G, Lalic T, Handel A, Duggal G, Chintawar S, Cheung J, Arunasalam K, Couper E, Haupt LM, Griffiths LR, Bassett A, Cowley SA, Cader MZ (2019) A causal role for TRESK loss of function in migraine mechanisms. Brain 142(12):3852–3867. https://doi.org/10.1093/brain/awz342
Piechotta PL, Rapedius M, Stansfeld et al (2011) The pore structure and gating mechanism of K2P channels. EMBO J 30(17):3607–3619. https://doi.org/10.1038/emboj.2011.268
Royal P, Andres-Bilbe A, Avalos Prado P et al (2019) Migraine-associated TRESK mutations increase neuronal excitability through alternative translation initiation and inhibition of TREK. Neuron 101:232–45 e6
Sano Y, Inamura K, Miyake A, Mochizuki S, Kitada C, Yokoi H, Nozawa K, Okada H, Matsushime H, Furuichi K (2003) A novel two-pore domain K+ channel, TRESK, is localized in the spinal cord. J Biol Chem 278:27406–27412. https://doi.org/10.1074/jbc.M206810200
Tucker SJ, Pessia M, Moorhouse AJ, Gribble F, Ashcroft FM, Maylie J, Adelman JP (1996) Heteromeric channel formation and Ca2+-free media reduce the toxic effect of the weaver Kir 3.2 allele. FEBS Lett 390(3):253–257. https://doi.org/10.1016/0014-5793(96)00635-7
Weir GA, Pettingill P, Wu Y, Duggal G, Ilie AS, Akerman CJ, Cader MZ (2019) The role of TRESK in discrete sensory neuron populations and somatosensory processing. Front Mol Neurosci 12:170. https://doi.org/10.3389/fnmol.2019.00170
Yoo S, Liu J, Sabbadini M, Au P, Xie GX, Yost CS (2009) Regional expression of the anesthetic-activated potassium channel TRESK in the rat nervous system. Neurosci Lett 465(1):79–84. https://doi.org/10.1016/j.neulet.2009.08.062
Yoshida S, Plant S (1992) Mechanism of release of Ca2+ from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. J Physiol 458:307–318
Acknowledgments
We gratefully acknowledge the financial support of the University of Malta Research, Innovation and Development Trust (RIDT), the Biotechnology and Biological Sciences Research Council (BBSRC), and United Arab Emirates University (Grants n. 31M452 and 31M468).
Author information
Authors and Affiliations
Contributions
PI designed the study, analyzed data, and wrote the paper; ENA performed electrophysiological experiments and analyzed data; SH critically discussed the results and reviewed the literature; SJT and MP critically revised the manuscript and supervised the work; MCD designed the study, supervised the work and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose. No competing financial interests exist.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Channelopathies: from mutation to diseases in Pflügers Archiv—European Journal of Physiology
Rights and permissions
About this article

Cite this article
Imbrici, P., Nematian-Ardestani, E., Hasan, S. et al. Altered functional properties of a missense variant in the TRESK K+ channel (KCNK18) associated with migraine and intellectual disability. Pflugers Arch - Eur J Physiol 472, 923–930 (2020). https://doi.org/10.1007/s00424-020-02382-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-020-02382-5