
Abstract
Obesity and insulin resistance are considered the main causes of nonalcoholic fatty liver disease (NAFLD), and oxidative stress accelerates the progression of NAFLD. Free fatty acids, which are elevated in the liver by obesity or insulin resistance, lead to incomplete oxidation in the mitochondria, peroxisomes, and microsomes, leading to the production of reactive oxygen species (ROS). Among the ROS generated, H2O2 is mainly produced in peroxisomes and decomposed by catalase. However, when the H2O2 concentration increases because of decreased expression or activity of catalase, it migrates to cytosol and other organelles, causing cell injury and participating in the Fenton reaction, resulting in serious oxidative stress. To date, numerous studies have been shown to inhibit the pathogenesis of NAFLD, but treatment for this disease mainly depends on weight loss and exercise. Various molecules such as vitamin E, metformin, liraglutide, and resveratrol have been proposed as therapeutic agents, but further verification of the dose setting, clinical application, and side effects is needed. Reducing oxidative stress may be a fundamental method for improving not only the progression of NAFLD but also obesity and insulin resistance. However, the relationship between NAFLD progression and antioxidants, particularly catalase, which is most commonly expressed in the liver, remains unclear. Therefore, this review summarizes the role of catalase, focusing on its potential therapeutic effects in NAFLD progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined as the accumulation of excessive fat in the liver, with at least 5% of hepatocytes containing triglyceride (TG) or steatosis occurring in at least 5% of the liver volume or weight of patients, who consume less than 30 and 20 g of alcohol per day in men and women, respectively [2]. NAFLD is now the most common liver disease in all age groups, with 14–30% of patients developing the disease because of high obesity and overweight, and has emerged as a serious clinical problem [2].
NAFLD includes a variety of stages from hepatocellular steatosis to inflammatory nonalcoholic steatohepatitis, fibrosis, and cirrhosis [2], and in this review, NAFLD is used as a comprehensive word for these diseases (Fig. 1). The pathogenesis of NAFLD is described in detail below using the “two-hit” model. The first stage of NAFLD is steatosis, which is a simple fatty liver. Steatosis occurs when the cytoplasm of hepatocytes contains more than 5% TG (Fig. 1) [157]. If hepatocyte injury as hepatocyte ballooning and cell death, inflammatory infiltration, and/or fibrosis occurs, nonalcoholic steatohepatitis (NASH) results (Fig. 1) [40]. Cirrhosis is a condition in which hepatocytes are replaced by scar tissue, and 10–29% of patients with NASH develop into cirrhosis within 10 years (Fig. 1) [8].

The diagram of progression of nonalcoholic fatty liver disease. TG, triglyceride
Most patients with NAFLD exhibit obesity, diabetes, or dyslipidemia, and thus, metabolic syndrome is recognized as the greatest risk factors for NAFLD development [28]. Oxidative stress is also thought to contribute to the progression of simple steatosis to steatohepatitis, fibrosis, and cirrhosis [43], such as by increasing lipid peroxide levels [140]. Antioxidants are abundant in the liver [86], and thus, decreased antioxidant defense is a major factor promoting oxidative stress in patients with NAFLD. Decreases in antioxidant factors including coenzyme Q10, Cu/Zn-superoxide dismutase, catalase, glutathione, and glutathione S-transferase correlate with the severity of NAFLD [86, 151, 170]. Machado et al. [97] suggested that expression of antioxidant enzymes was reduced in the liver of NASH mice compared to that of obese steatosis mice, resulting in higher oxidative stress than in obese steatosis mice. The livers of NASH mice showed more fibrosis and inflammation than those of steatosis mice, and indicators of cell death were significantly greater in NASH mice than in steatosis mice. Thus, oxidative stress is an attractive therapeutic target for therapy in patients with fatty liver disease.
The most important intracellular antioxidants in the human body are superoxide dismutase (SOD), glutathione peroxidase (GPX), and catalase [153]. Catalase is a common enzyme found in all organisms. Catalase is present in peroxisomes, where it decomposes two hydrogen peroxide (H2O2) molecules into two H2O molecules and O2 (2H2O2 → 2H2O + O2). Both catalase and GPX degrade H2O2, but GPX has a higher affinity for H2O2 compared to catalase [70]. Thus, H2O2 is typically decomposed by GPX under normal conditions. However, as the concentration of H2O2 increases, catalase shows a greater contribution to H2O2 degradation [169]. Additionally, H2O2 is relatively stable among reactive oxygen species, and thus, it can easily move away from its production site to show a concentration gradient [133]. Catalase expression in the cardiac mitochondria is elevated by a high-fat diet, which elevates catalase expression to remove excess H2O2 produced by increased lipid metabolism [137]. In another report, cytochrome C oxidase and GPX were not significantly involved in the rate of H2O2 consumption of highly purified rat liver mitochondria, while H2O2 consumption was significantly inhibited by the catalase inhibitor KCN or aminotriazole [142]. Thus, catalase contributes to mitochondrial protection against endogenous or exogenous H2O2, and mitochondrial catalase in the liver may be a new therapeutic strategy for liver disease. Most studies on cytosolic catalase have been performed using erythrocytes. Cytosolic catalase in erythrocytes mainly protects erythrocytes from highly exogenous H2O2; when catalase was inhibited, GPX did not prevent H2O2-induced oxidative stress [139]. Few studies have examined cytosolic catalase in the liver. In 1984, it was reported that purified rat liver cytosolic catalase showed enzymatic activity reaching the levels of purified peroxisome catalase [144]. In 1992, a study of the changes in peroxisomal and cytosolic catalase activity over time indicated that cytosolic catalase can be incorporated into peroxisomes [49]. This means that catalase is mainly present in peroxisomes, but also contributes to decomposition of H2O2 generated at other sites in the cell, such as the mitochondria and cytosol, and is essential for overcoming intracellular oxidative stress.
Therefore, we focus our attention on the relationship between catalase and NAFLD, as the role of catalase in fatty liver development is often overlooked. This review discusses the findings of studies on the activity or expression of catalase in NAFLD models.
NAFLD pathogenesis and oxidative stress
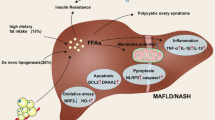
NAFLD exhibits excessive accumulation of TG because of excessive influx of free fatty acids (FFAs) and/or increased de novo lipogenesis without significant alcohol consumption [26]. Although the pathogenesis of NAFLD remains unclear, the “two-hit” model is considered as the most probable cause of NAFLD (Fig. 2) [43]. FFAs in hepatocytes are transmitted from adipose tissue or produced by de novo synthesis [46]. Hepatic FFAs are mainly used for β-oxidation or synthesized into TG, and NAFLD occurs when β-oxidation is reduced and TG synthesis is increased (Fig. 2). Insulin resistance enhances lipolysis in adipose tissue and increases the inflow of FFAs into the liver. Excess FFAs in liver are mostly synthesized as TG because of β-oxidation overload. The mechanisms of excess fat accumulation and insulin resistance in the liver are the “first hit,” while the “second hit” are oxidative stress, lipid peroxidation, proinflammatory cytokines, and adipocytokines derived from adipose tissue. When β-oxidation is overloaded in hepatocytes, reactive oxygen species (ROS) are produced, initiating the “second hit” and leading to the development of NASH from simple fatty liver (Fig. 2) [47].

Mechanisms during the progression to nonalcoholic steatohepatitis by obesity and insulin resistance. The development of NASH is primarily initiated by risk factors including rich food and lack of exercise that cause obesity and insulin resistance. Obesity leads to hyperglycemia and hyperlipidemia and further contributes to insulin resistance in adipose tissue and muscle. Insulin resistance promotes lipolysis in adipose tissue and inhibits uptake of glucose in muscle, further increasing circulating FA and glucose. FA and glucose, which are excessively infused into the liver, undergo oxidation overload and are mainly used for TG synthesis. On the other hand, ROS produced by overload of β-oxidation induces lipid peroxidation, inflammation, and fibrosis and develops steatosis to NASH. FA, fatty acid; TG, triglyceride; ROS, reactive oxygen species
NAFLD pathogenesis is related to hepatic and adipose tissue insulin resistance and underlying metabolic syndrome, which is a combination of conditions such as diabetes, obesity, and dyslipidemia [25, 64]. For example, people who meet the criteria for metabolic syndrome are at least twofold more likely to develop NAFLD compared to normal people, and more than 90% of patients with NAFLD have metabolic syndrome [91, 100]. Obesity is the most important risk factor for NAFLD. In one study, 74% of obese subjects (30–50% overweight, 534 male) developed NAFLD, which was 4.6-fold higher than the rate in normal people (4079 male) [116]. Among patients who are pathologically obese and underwent bariatric surgery for weight loss, 84–96% have steatosis and 2–12% have severe fibrosis or cirrhosis [18, 41, 45, 57]. The prevalence of NAFLD is estimated to be at least twofold more common among those who meet the criteria for metabolic syndrome [91]. Among patients with NAFLD, more than 90% of cases are characterized by metabolic syndrome [100]. Diabetes was reported in 33–50% of patients with NAFLD, while insulin resistance affected as many as 75% of patients [104].
Oxidative stress may trigger the development stage of NAFLD (Fig. 2). Increased oxidative stress, decreased hepatic ATP, and inflammation impair mitochondrial bioenergetics, function, and morphology [140]. Metabolic diseases such as obesity, diabetes, and dyslipidemia and oxidative stress are related and interact. The serum levels of H2O2 and malondialdehyde, which are oxidative stress markers, are significantly higher in patients diagnosed with metabolic syndrome and NAFLD than those with only metabolic syndrome [7]. Additionally, the inflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-6 were also significantly higher in patients diagnosed with both diseases than in patients diagnosed with metabolic syndrome alone [7]. Excessive FFA flow into the liver leads to saturation of the oxidative pathway and an incomplete β-oxidation pathway. Incomplete β-oxidation in the mitochondria of liver tissue increases mitochondrial ROS, including superoxide, H2O2, and hydroxyl radicals, resulting in DNA mutations and lipid peroxidation [125]. Pessayre et al. [125] also suggested that ROS and lipid peroxidation products contribute to mitochondrial dysfunction and hepatic mitochondrial dysfunction contributes to the genesis of NASH lesions. In NAFLD, increased expression and activity of cytochrome P450 2E1 (CYP2E1) is an important source of ROS, which is the starting point of oxidative stress and continuously deteriorates the function of hepatic mitochondria [14]. Additionally, elevated microsomal CYP4A enzymes promoted ROS production in steatosis and ablation of the CYP4A gene in an animal model of steatohepatitis decreased hepatic inflammation and fibrosis [87, 184].
NAFLD pathogenesis and insulin/IGF-1 signaling
Insulin-like growth factor-1 (IGF-1) is mainly secreted in hepatocytes by growth hormone (GH) [147]. The main function of IGF-1 via IGF receptors (IGF1R, IGF2R) in the liver is organ development, growth, and regeneration, and it regulates the cell cycle progression, proliferation, and differentiation of hepatocytes [76]. In general, IGF-1 protects liver function and is known to play a very important role in hepatic hormonal regulation and metabolism [36, 37, 130].
Among the various physiological functions of IGF-1, it plays a role in chronic liver disease process from simple fatty liver to cirrhosis, hepatitis, and hepatocellular carcinoma [1, 112]. Abundant data suggest the role of IGF-1 in the development of chronic liver disease and that IGF-1 administration can reduce fibrosis and improve overall liver function. Efstratiadis et al. [48] showed that IGF-1 levels were low in patients with metabolic syndrome and that IGF-1 is an independent prognostic factor for hepatic steatosis. The results of Völzke et al. [171] and Galiano et al. [56] showed that liver steatosis is associated with low serum IGF-1, and IGF-1 levels were further decreased during the progression to NASH. Interestingly, in the case of adult GH deficiency, GH replacement therapy significantly reversed NASH and reduced inflammation and oxidative stress markers [158], indirectly indicating that IGF-1 has an NAFLD-improving effect in humans.
Additionally, IGF-1 concentration may be important in the pathogenesis of T2DM by regulating the insulin sensitivity or maintenance of beta cell mass [147]. Sandhu et al. [143] showed that elevated serum IGF-1 levels are associated with a decreased risk of developing T2DM. It is unclear whether insulin resistance causes fatty liver or if fatty liver causes hepatic and peripheral (muscle, adipose tissue) insulin resistance. However, there is a close relationship between NAFLD and hepatic insulin resistance and decreased systemic insulin sensitivity. Insulin resistance induces excessive accumulation of TG in hepatocytes by increasing the release of FFAs from adipose tissue. Particularly, it has been shown that hepatic insulin resistance was found to be more important in NAFLD pathogenesis than muscle or adipose tissue insulin resistance using a tissue-specific insulin-resistant mouse model [19]. Hepatic insulin resistance is associated with impaired glycogenesis and increased gluconeogenesis and glycogenolysis [24, 35]. Marchesini et al. [98] reported that the glucose disposal rate, a measure of insulin sensitivity, was reduced by 45–50% in NAFLD compared to that in normal subjects. Additionally, insulin resistance promotes the progression from simple steatosis to NASH and becomes more severe as it progresses to NASH [58, 100]. Hyperinsulinemia directly induces oxidative stress and promotes the secretion of components of the extracellular matrix associated with hepatic stellate cell proliferation and fibrosis progression [121, 154].
In summary, IGF-1 improves insulin resistance, reduces ROS, enhances mitochondrial function, and reduces TG accumulation in hepatocytes. However, little is known about the direct association between IGF-1 and catalase activity. However, IGF-1-deficient mice treated with IGF-1 have been reported to have increased hepatic catalase activity, while other antioxidant enzymes were unchanged [118]. Thus, GH and IGF-1 treatments have shown the potential for liver and mitochondrial protection and antioxidant effects, suggesting the clinical applicability of these hormones. Further studies are needed to clarify the exact mechanism of GH or IGF-1 in regulating catalase activity.
When lipid peroxidation is increased by excessive ROS in hepatocytes, inflammatory cytokines, which promotes apoptosis and inflammation, such as TNF-α, IL-6, and IL-1, are induced in Kupffer cells [13, 140, 167]. ROS, together with the products of lipid peroxidation, increases the secretion of TNF-α, which plays an important role in cell death, inflammation, and fibrosis [13]. TNF-α increases the lipid peroxidation of mitochondrial membranes, exacerbating and further inducing oxidative stress [156]. In vitro studies showed that IL-6 promotes insulin resistance through a variety of mechanisms [73, 78, 148], and human studies have also demonstrated the role of IL-6 in the pathogenesis of type 2 diabetes mellitus (T2DM) [82, 128, 132, 165]. The role of IL-6 in NAFLD pathogenesis is unclear, as the results of studies on the relationship between IL-6 levels and NAFLD are controversial [4, 56, 66, 88, 163, 172]. However, because insulin resistance is included in the “first hit” of NAFLD pathogenesis, the effect of IL-6 on NAFLD is relatively predictable. IL-1α and IL-1β play crucial roles in the conversion from steatosis to steatohepatitis [72].
Serum IL-1 levels are significantly higher in patients with NAFLD with increasing histological grades and severity of fibrosis [83]. The role of various cytokines including TNF-α, IL-6, IL-8, IL-1, and IL-18 in the development of insulin resistance and NAFLD has been widely studied [69, 174, 178]. Recent studies also suggested that inflammation caused by oxidative stress associated with cytokine activation is a cause of NASH development [115]. In conclusion, oxidative stress also increases inflammation and develops NAFLD in the liver tissue through systemic circulation.
Mechanisms regulating the activity and expression of catalase
Mammalian catalase expressed in humans, rats, and mice is a 240-kDa heme-containing protein of tetramers. It is encoded by a single structural gene that is highly evolutionarily conserved [15, 111, 134, 136], approximately 33 kb in length, and comprises 13 exons and 12 introns [111, 134, 136]. Catalase expression in mammals is regulated in a tissue-specific manner [38, 103]. Catalase expression levels between tissues, with the highest levels found in the liver, kidney, and blood and lowest levels found in the connective tissue and brain [5, 123, 141]. Catalase is known to be expressed or activated by multiple mechanisms such as genetic, epigenetic, and posttranscriptional processes as well as transcription factors [61]. During the past decade, peroxisome proliferator-activated receptor gamma (PPARγ), organic cation transporter 1 (Oct-1), nuclear respiratory factor (Nrf), and CCAAT-enhancer-binding protein beta (C/EBPβ) have been identified as catalase transcription factors [61].
First, PPARγ is the most well-known activator of transcription of catalase gene and is involved in catalase expression in humans, rats, and mice [61]. The PPAR-response element was detected in the rodent catalase promoters [59, 117] and catalase expression was increased by PPARγ agonists (e.g., 15-deoxy delta prostaglandin J2, pioglitazone, rosiglitazone) in rat oligodendrocytes, cardiomyocytes, fibroblasts, and astrocytes [17, 32, 77, 180]. In addition, the expression of catalase was reduced in the neurons of PPARγ mutant mice, and expression of the prosurvival gene was impaired, leading to further damage resulting from oxidative stress [185]. In human melanocytes, catalase expression was also decreased when PPARγ was inhibited, whereas catalase levels were increased when 2,4,6-octatrienoic acid, a PPARγ activator, was administered [54].
Oct-1 and Nrf are potential activators for catalase expression. The POU (Pit-1, Oct, and Unc)-domain transcription factor Oct1 (POU2F1) regulates target genes, such as peroxiredoxin 2, interferon-activated gene 202B, and tissue inhibitor of metalloproteinase 3, and acts as a sensor of oxidative and metabolic stress [162]. In experiments using human hepatocellular carcinoma cell lines, POU2F1 bound to the catalase promoter at the octamer consensus sequence ATTAAATA and increased the expression of catalase [135]. Hypermethylation of the Oct1 promoter also reduced the expression of Oct1 and subsequently reduced the expression of catalase protein in hepatocarcinoma cells exposed to H2O2 [135]. Nrf2, a pleiotropic transcription factor involved in cell defense against oxidative stress, was reported to increase the expression of several antioxidant enzymes including catalase [168]. The expression of catalase in cells such as cardiac fibroblasts, macrophages, and cardiomyocytes isolated from Nrf2−/− mice was significantly lower than that in wild-type (WT) mice [187,188,189].
Taniguchi et al. [161] showed that C/EBPβ is involved in catalase gene activity by demonstrating that C/EBPβ binds to multiple initiation sites (CCAAT boxes and GT boxes) in the rat reuber hepatoma cell line and regulates gene transcription in the catalase promoter.
Finally, in addition to the various transcription factors identified, recent studies have shown that Akt/protein kinase B in the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) signaling pathway is important in the expression of catalase by modulating the activity of forkhead box O3a (FoxO3a). The FoxO3a transcription factor mainly regulates the expression of antioxidant enzymes such as catalase and mitochondrial SOD in rodents. Suppression of FoxO3a in cardiomyocytes isolated from rat reduced catalase mRNA and protein expression [159] and decreased catalase protein levels in rat vascular smooth muscle cells [90]. Inhibition of the PI3K/Akt pathway inhibited FoxO3a phosphorylation and blocked translocation outside the nucleus. mRNA and protein expression of catalase was increased when LY294002, a PI3K inhibitor, was used to treat rat vascular smooth muscle cells and human MCF-7 cancer cells [60, 90]. FoxO3a regulates catalase expression in cooperation with the transcriptional coactivators PPARγ coactivator 1 (PGC1α) and NAD-dependent deacetylase sirtuin1 (Sirt1). PGC1α is a regulatory factor for mitochondrial function and oxidative metabolism and is involved in regulating gene transcription involved in ROS detoxification. In double-knockout FoxO3a and PGC1α mouse embryonic fibroblast cells, catalase, SOD, and peroxiredoxins were decreased, while there was no effect on catalase expression in these cells with PGC1α single KO and FoxO3a overexpression. Thus, FoxO3a requires PGC1α to induce catalase expression [119]. Sirt1 is also involved in FoxO transcriptional activity and catalase expression. FoxO3a and PGC1α are activated by Sirt1-mediated deacetylation to increase FoxO3a/ PGC1α complex formation and regulate catalase expression [113, 120]. Upon exercise-stimulated Sirt1 activity in the cardiac and adipose tissues of rats, FoxO3a and catalase were increased [53] and Sirt1 inhibition of human proximal tubular cell lines decreased catalase expression, whereas overexpression of Sirt1 increased catalase expression [65].
Although various transcription factors that regulate catalase expression have been identified, further studies of the role of transcriptional regulatory elements in catalase gene expression by oxidative stress such as H2O2 are needed.
Catalase and oxidative stress in liver
As described above, not all cases of simple fatty liver progress to steatohepatitis. To produce inflammation and fibrosis in the simple fatty liver, a major stimulant known as “oxidative stress” is needed [129]. The production of ROS in hepatocytes occurs in the mitochondria [63], microsomes [172, 173], and peroxisomes; particularly, H2O2 is produced in the peroxisomal β-oxidation process when mitochondrial β-oxidation is saturated by fatty acid excess or damaged [166]. H2O2 is not a radical because it has no unpaired electrons. Therefore, it is relatively less reactive than ROS, but when combined with iron, severe damage can occur. Hydroxyl radical (•OH) is the most toxic and dangerous form of ROS. The hydroxyl radical is among the most powerful oxidizing agents and can react unselectively and rapidly with the surrounding chemicals as soon as it is produced. This hydroxyl radical is generated by the Fenton reaction, and H2O2 is the substrate for the Fenton reaction. In the Fenton reaction, iron(II) (Fe2+) is oxidized by hydrogen peroxide into iron(III) (Fe3+) to form a hydroxyl radical and hydroxide ion (OH−). In 1999, Bonkovsky et al. [20] found that hemochromatosis gene mutations were increased in patients with NASH. Hemochromatosis is a stimulating factor that accumulates iron in hepatocytes and causes oxidative damage by activating the Fenton reaction [152]. Thus, if H2O2 is not removed by catalase, hydroxyl radical is generated, resulting in additional oxidative stress.
The role of catalase in protecting cells and tissues against oxidative stress has been extensively studied. As expected, overexpression of catalase in murine fibroblasts, human bronchial epithelial cells, and human umbilical vein epithelial cells was shown to be resistant to toxicity of H2O2 and oxidant-mediated injury from exposure to hyperoxia [50, 95, 182]. Additionally, transgenic mice overexpressing catalase were protected following myocardial injury caused by administration of adriamycin and hypertension because of norepinephrine or angiotensin treatment [74, 179]. Thus, catalase plays a role in cellular antioxidant defense mechanisms by suppressing the accumulation of H2O2 [68]. According to Ho et al. [68], the degree of antioxidant activity of catalase depends on the type of tissue, and catalase showed an excellent ability to remove H2O2 from the liver. This is consistent with a report showing catalase is very highly expressed in the liver [39].
Therefore, catalase is thought to play a very important role in NAFLD and NASH. Abundant experimental results obtained using cells, animals, and humans support this prediction.
Catalase and NAFLD: in vivo and in vitro studies
According to the results of Spitz et al. [150], catalase activity was increased by 20-fold with increase in H2O2 concentrations in Chinese hamster fibroblasts. Overexpression of catalase (up to 80%) in Drosophila melanogaster did not affect the lifespan of the fly but conferred strong resistance to H2O2 [175]. As described above, this supports that catalase is closely related to oxidative stress resistance caused by H2O2 (Table 1).
HepG2 cells treated with various concentrations of H2O2 (0, 0.25, 0.5, and 1 mM) for 24 h increased the mRNA expression of catalase in a dose-dependent manner [108]. After overexpression of catalase in the cytosol or mitochondria of HepG2 cells, Bai et al. [11] measured intracellular H2O2 levels induced by exogenously added H2O2 or antimycin A, which led to cytotoxicity and apoptosis. As a result, both cytosolic catalase and mitochondrial catalase were shown to equally decrease H2O2 and protect cells from cytotoxicity or apoptosis induced by oxidative stress.
In animal experiments that led directly to the induction of fatty liver using H2O2, catalase activity in the liver was significantly increased, while GSH and GPX were significantly lower than in the control group [122]. In this experiment, catalase plays a major role in H2O2 decomposition compared to GSH or GPX. Low molecular weight fucoidan extracted from the brown seaweed Laminaria japonica Areschoug is known to contribute to the prevention of metabolic syndrome such as diabetes, obesity, hyperlipidemia, and fatty liver. The experimental results of Zheng et al. [186] showed that catalase activity was increased in db/db mice treated with fucoidan at 80 mg/kg/day by gavage for 7 weeks, resulting in decreased lipotoxicity-related oxidative stress, thus preventing NAFLD. Thus, obesity/diabetes-induced NAFLD can be prevented by inhibiting oxidative stress via catalase activation. Brady et al. [21] observed that the hepatic mitochondrial and peroxisomal oxidative capacities of obese mice were increased and catalase activity was significantly elevated compared to those of lean mice. The oxidative capacities indicate mitochondrial oxygen consumption (ng-atoms of O consumed/min per mg of protein) and peroxisomal palmitoyl-CoA oxidation (nmol/min per mg of peroxisomal protein). In agreement with the results of Brady et al. [21], Murphy et al. [109] reported that catalase activity and β-oxidation in the peroxisomal fraction of liver from obese mice were increased compared to those in lean mice.
Because oxidative stress contributes to NAFLD (NASH, fatty liver), researchers have examined whether the loss of catalase increases the susceptibility to the disease. Despite feeding of normal diet, catalase KO (CKO) mice showed higher fatty liver compared to the WT mice as age increased [61], and the high-fat diet caused liver injury in only 2 weeks in CKO mice [127]. In a comparison of young (7 weeks) WT mice and old (16 months) WT mice, hepatic H2O2 increased and hepatic steatosis, hepatocyte apoptosis, and hepatic fibrosis were reported as hepatic aging parameters [3]. To confirm whether catalase affects the fat accumulation in the liver, first, we administrated a high-fat diet to WT and CKO mice for 4 weeks; fat accumulation was increased in the CKO mice but not in WT mice (Fig. 3a). Next, we analyzed liver extracted from 5-, 10-, and 37-week-old WT and CKO mice fed normal chow diet. As a result, there were no morphological differences in the liver between WT and CKO mice aged 5 and 10 weeks, but we observed fatty liver in CKO mice aged 37 weeks (Fig. 3b). Consistent with previous studies [67, 127], these findings suggest that CKO mice were more vulnerable to nonalcoholic fatty liver, which can be caused by a high-fat diet or aging, compared to WT mice.

Histopathological difference in liver of wild-type (WT) and catalase KO (CKO) mice. a Hematoxylin and eosin-stained liver sections isolated from mice fed normal diet (ND) or high-fat diet (HFD) (× 200). b Liver was extracted from 5-, 10-, and 37-week-old WT and CKO mice fed ND and H&E stained (× 200)
Catalase and NAFLD: human studies
Clinical studies examining the expression or activity of catalase in fatty liver occurrence showed conflicting results (Table 1).
Several studies reported that antioxidant enzyme activity is decreased as fatty liver worsens and the defense mechanism against oxidative stress in the cytosol and mitochondria is damaged. Screekumar et al. [151] collected liver samples from patients with NASH in the absence of other (viral, drug/toxin, autoimmune, or metabolic) causes of steatosis, cirrhosis by hepatitis C virus (HCV), cirrhosis by primary biliary cirrhosis (PBC), and normal subjects through biopsy and measured hepatic gene expression by high-density synthetic oligonucleotide microarray analysis. In the analysis of hepatic gene expression, the mRNA expression of catalase was significantly downregulated in patients with NASH compared to that in all other groups. Videla et al. [170] analyzed the parameters associated with oxidative stress in the liver for 31 patients with NAFLD. Catalase activities in patients with NASH were significantly reduced by 42 and 48% compared with patients with simple fatty liver and normal subjects, respectively, while changes in hepatic GPX activities were not observed. These changes promote CYP2E1 activity, which increases the production of ROS, free radicals, and reactive mediates, and increased CYP2E1 activity in the liver inactivates SOD and catalase as the disease progresses [87, 93], making the liver more susceptible to oxidative stress. In other in vitro experiments, the enhancement of CYP2E1 activity was shown to cause catalase inactivation and disease progression [51, 81]. Additionally, antioxidative capacity in the blood of patients with NASH was reduced, supporting further lowering of the ferric-reducing ability of plasma (FRAP) values, compared to that of patients with simple fatty liver [170]. FRAP is a simple method for determining the antioxidant capacity by measuring the degree of reduction of ferric to ferrous at low pH using colorimetric assay. In another experiment using blood from 35 patients with NAFLD, catalase activity was reduced by approximately 18.5% compared to that from normal subjects [42]. Because type IV collagen, a marker of fibrosis, was increased in these patients, patients with NAFLD were considered as NASH models rather than a model of simple fatty liver. Additionally, erythrocytic catalase activity was significantly reduced in patients with various stages (17 mild, 7 moderate, and 2 severe) of fatty liver compared to that in normal subjects [146], and 51 patients with NASH also showed significantly decreased systemic catalase activity compared to healthy subjects [181]. Das et al. [42] found that blood is resistant to oxidative stress because it contains various antioxidants such as catalase, SOD, GPX, glutathione S-transferase, and glutathione reductase (GSR), but excessive production of superoxide radical can inactivate catalase.
In contrast to the above reports, studies have shown that as the fatty liver is aggravated, catalase activity increases. RT-qPCR analysis of pediatric nonalcoholic steatohepatitis livers revealed a 12.7-fold increase in mRNA expression of catalase compared to that in controls, with no change in GPX and GSR [44]. In 2005, catalase activity in the livers of NAFLD patients was increased by approximately 30% compared to those in normal livers, but there was no difference between the erythrocytes of patients with NAFLD and those of normal subjects [124]. Perlemuter et al. [124] explained that these results indicate that circulating antioxidant defenses did not reflect hepatic peroxidation and that oxidative stress occurs in the liver of NAFLD patients. Baker et al. [12] and Moya et al. [108] reported that catalase activity was increased in the livers of 6 and 36 patients with NASH, respectively, compared to that in normal livers. Kohjima et al. [79] analyzed fatty acid metabolism and antioxidant-related gene expression using liver tissue samples from 26 patients with NAFLD (including 4 patients with NASH). As a result, genes involved in fatty acid oxidation and catalase were overexpressed in patients with NAFLD, which is considered to neutralize ROS produced by fatty acid oxidation. Strikingly, the mRNA expression of catalase was elevated by tenfold in patients with NAFLD compared to that in normal subjects, whereas glutathione synthetase (GSS) was not changed [79]. These results are similar to the results of Desai et al. [44].
The expression levels of catalase in normal, steatosis, and NASH were 0.21, 1.21, and 1.08 (mean of mRNA level of gene compared to that of β-actin), respectively, and significantly enhanced in patients with NAFLD compared to those in normal subjects [9]. These results suggest that catalase expression is altered by the fatty liver stage, although there was no statistical difference between steatosis and NASH. Increased catalase activity in the livers of NASH may be related to increased peroxides, which are produced in the peroxisomes of the fatty liver by increased fatty acid oxidation [20]. This suggests that excessive fatty acids provided by consumption of a high-fat diet increase fatty acid oxidation and thus catalase activity.
Catalase and NAFLD: role of catalase in the development of NAFLD
Based on the above results, the inhibitory effect of catalase on the progression of NASH is shown in Fig. 4. When the concentration of H2O2 is increased in hepatocytes, it can diffuse to other cell organelles because of the concentration difference. H2O2 induces oxidative stress through the Fenton reaction and leads to lipid peroxidation, inflammation, fibrosis, and cell injury. Additionally, steatosis develops into NASH through a combination of oxidative stress and TG pools, which is increased by excessive FFAs (Fig. 4). Catalase can disrupt this sequence of procedures. Particularly, catalase, which shows increased activity when the H2O2 concentration is high, can prevent NAFLD from becoming more severe.

Mechanisms of catalase function to suppress development of NAFLD. If excessive FFAs in the liver persist, the β-oxidative function of mitochondria becomes overloaded and oxidations of FFAs are activated in peroxisomes and microsomes compensatively. Thus, H2O2 is generated in peroxisomes and microsomes, and at the same time, catalase is activated in peroxisomes to decompose H2O2. Catalase suppresses the development of steatosis into NASH by blocking Fenton reaction, lipid peroxidation, inflammation, fibrosis, and cell injury by H2O2. FA, fatty acid; TG, triglyceride; CAT, catalase
In most cell and animal experiments, the activity or expression of catalase was measured during H2O2 treatment or steatosis induced by obesity. Because oxidative stress is initiated during an early stage of steatosis, consistently high catalase activity is observed. In a clinical study, catalase expression or activity in patients with NAFLD was not consistent because the process was analyzed at different stages of NAFLD. Thus, catalase has different functions in NAFLD and NASH. Importantly, our research group recently demonstrated that elimination of catalase easily causes steatosis in mice by promoting excessive lipid accumulation. This suggests that catalase plays a protective role as an antioxidant in the livers of NASH, given that oxidative stress is an important therapeutic or preventative target for patients with NASH.
During catalase overexpression in liver disease, the exact mechanism of the main upstream signaling is unclear. Only phenomenological results (catalase activity or mRNA levels up- or downregulated) have been reported for the expression of catalase associated with NAFLD. Catalase activity or mRNA expression increases with H2O2 treatment (by ROS stimulation) [108, 122, 150]. A study of human subjects showed that catalase expression differs according to the stage of NAFLD, which is mainly increased in the early stage of NAFLD and decreased in the terminal stage of NASH. Although this may be because many cells are destroyed and become nonfunctional, the exact mechanism of catalase in regulating NAFLD requires further analysis.
NAFLD has been associated with hepatocellular carcinoma (HCC) because of its synergistic interactions with other risk factors of HCC such as chronic HCV infection. HCC is the most common primary liver cancer type, accounting for 85% of liver cancers [101]. HCV and hepatitis B virus (HBV) infection account for approximately 54.9 and 9.5% of HCC cases, respectively, and are a major cause of HCC [183]. Wnt, mitogen-activated protein kinase cascade, and Ras are major molecular biomarkers of HCC pathogenesis, and ROS has been implicated in HCC [101]. The pathogenesis mechanism of HCC known to date is that the oxidative stress is increased in the pre-HCC stage in which hepatic steatosis is present and further exacerbated by the viral core protein [94, 106, 107]. These events ultimately compromise the mitochondrial and cell signaling pathways. At the same time, the viral core protein induces cell proliferation through a mitogen-activated protein kinase pathway containing c-Jun N terminal kinase, p38, and extracellular signal-regulated kinase [80, 101]. Eventually, excessive oxidative stress and abnormal cell proliferation cause HCC. Thus, it has been suggested that oxidative stress contributes to the progression and worsening of HCC and viral hepatitis [110, 155].
In fact, the precise mechanism of antioxidant enzyme disruption in HCC is unknown, but a unique pathological character of HCC is the dramatic downregulation of antioxidant enzymes that constitute the most important free radical scavenger systems such as catalase, SOD, and GPX [27, 92, 114], and HBV or HCV infection reduces antioxidative defense in patients. Kumar et al. [84] measured antioxidant parameters in the blood of 25 patients with chronic viral hepatitis including HCV and HBV. As a result, the catalase activity in the chronic viral hepatitis group was significantly lower than that in the healthy group, but GPX activity was significantly increased and SOD activity was not different. In another study using blood from HCC patients, there was no difference in catalase activity in patients with HBV, while GPX activity was significantly decreased; in patients with HCV, there was no difference in GPX activity, while catalase activity was significantly decreased [94]. In 13 HCC tissue samples infected with HBV, catalase and GPX activities were significantly decreased compared to those in normal tissues. Additionally, catalase activity in the liver tissue of patients with HCC was lower in both tumor tissue and tumor-free tissue compared to that in healthy liver tissues [16].
Changes in the antioxidant systems of cancer cells during tumor growth result in various outcomes, but the antioxidant system is clearly compromised. Thus, a variety of natural and synthetic antioxidants have been used to treat HCC, although it is not known whether these agents can suppress the abnormal regulation of cellular redox by HCC and reduce viral replication [96]. Therefore, improving the antioxidant system in the body by increasing catalase activity may help inhibit tumor worsening.
Chen et al. produced the first transgenic mouse model [Tg(CAT)] in which expression of catalase was increased in all tissues using an 80-kb genomic DNA fragment containing 5′ and 3′ flanking regions as well as the human catalase gene [29]. This [Tg(CAT)] mouse showed catalase activity that was up to fourfold higher than in the WT littermates with similar characteristics such as growth rate, body weight, body composition, and fertility [30]. The levels of other major antioxidant enzymes such as SOD and GPX did not differ from those of the WT [30]. Hepatocytes and skin fibroblasts isolated from [Tg(CAT)] mice showed strong resistance to H2O2 but were sensitive to γ-irradiation [30]. Overexpression of catalase in the heart inhibited oxidative injury by doxorubicin, ischemia-reperfusion, and hypoxia-reoxygenation in vivo [74, 89]. Overexpression of catalase in pancreatic islets of mice inhibited STZ-induced diabetes and islets showed resistance to H2O2 [177]. Kidney-specific catalase overexpression inhibited ROS generation and apoptosis in the kidneys of STZ-induced diabetic mice [22]. Overexpression of catalase in the heart of mice prolonged the lifespan and inhibited cardiac protein damage and contractile defects due to aging [176]. Overexpression of catalase has been reported to be more susceptible to radiation, but has more advantages such as antioxidant, antidiabetic, and extended life span. However, a liver-specific catalase overexpression model has not been reported, and further experiments are needed to determine the mechanism of catalase overexpression in the liver.
Summary and future perspective
Because there are no effective therapeutic drugs, the current therapy for NAFLD relies on weight loss and exercise. However, a variety of insulin-sensitive substances, antioxidants, and drugs have been shown to be beneficial. One of the most popular agents is vitamin E. Recent randomized clinical trials comparing vitamin E to placebo showed that vitamin E can be used as a primary drug in adults with fatty liver [85, 145]. However, it is unknown whether vitamin E is effective in diabetic patients with hepatitis, simple hepatitis, or liver cirrhosis due to hepatitis. There are also no data regarding the safety of long-term treatment with high-dose antioxidants [71]. Metformin is an antidiabetic agent that lowers blood glucose and insulin resistance. Nondiabetic patients with NAFLD were tested to determine the effects of metformin compared to vitamin E, resulting in a marked weight loss effect in the metformin group [23]. However, metformin with rosiglitazone did not show any improvement in patients with NASH compared to rosiglitazone alone [164], and metformin did not improve histological features in patients with pediatric NASH [85]. Additionally, Marchesini et al. [99] found that metformin was not superior to dietary intervention and lifestyle changes. Glucagon-like peptide-1 receptor (GLP-1R) agonists are a relatively new class of drugs for treating type 2 diabetes, and typically include liraglutide. Liraglutide has been reported to be not only beneficial as an anti-diabetes agent but also effective in patients with NAFLD by causing weight loss and inducing antioxidant and anti-inflammatory effects [55, 102, 105, 138]. However, while liraglutide administration has been reported to be effective for weight loss and glycemic control, it has no effect on fatty liver contents [126, 160] and has not been shown to affect hepatic fibrosis [149]. Previous studies suggested that liraglutide can improve NAFLD indirectly through obesity and diabetic improvement, but it may not be suitable for directly treating NAFLD. A dietary phenolic compound, resveratrol, is known to have a wide range of positive health effects, including antioxidant, anti-inflammatory, anti-cancer, anti-obesity, anti-diabetes, and anti-aging effects. Resveratrol was shown to prevent metabolic diseases by upregulating fatty acid oxidation and insulin sensitivity, uncoupling protein 2, and inhibiting lipogenic genes and oxidative stress through various experiments using animals and cells [6, 10, 31, 33, 75, 131]. However, it is difficult to determine the dose-dependency of the effects of resveratrol on NAFLD in mouse [34] and rat [62] experiments, and thus, the appropriate dose of resveratrol to treat NAFLD cannot be evaluated. Additionally, the results of Foruzan et al. [52] showed that when patients with NAFLD were administered 500 mg per day of resveratrol for 12 weeks, the levels of inflammation markers and hepatocyte apoptosis were reduced when lifestyle modifications were also made [52]. Thus, resveratrol alone is not an effective therapeutic material, and additional studies are needed to evaluate its clinical application. There is no effective drug for the wide variety of patients with different characteristics.
Because catalase is an antioxidant enzyme that orchestrates adaptation to intracellular redox perturbation, it may be effective for reducing oxidative stress caused by not only NAFLD but also obesity and diabetes. To date, however, few studies have examined the development of NAFLD, focusing on catalase function. It is essential to understand the oxidative stress and antioxidant system according to the detailed stages of NAFLD and to establish a method for effective expression of antioxidants. Increasing the activity of catalase in combination with weight loss and exercise therapy may be a natural method for overcoming the limitations of monotherapy.
References
Abbas Z, Shazi L (2015) Pattern and profile of chronic liver disease in acute on chronic liver failure. Hepatol Int 9(3):366–372
Abd El-Kader SM, El-Den Ashmawy EM (2015) Non-alcoholic fatty liver disease: the diagnosis and management. World J Hepatol 7(6):846–858
Abdelmegeed MA, Choi Y, Ha SK, Song BJ (2016) Cytochrome P450-2E1 promotes aging-related hepatic steatosis, apoptosis and fibrosis through increased nitroxidative stress. Free Radic Biol Med 91:188–202
Abiru S, Migita K, Maeda Y, Daikoku M, Ito M, Ohata K, Nagaoka S, Matsumoto T, Takii Y, Kusumoto K, Nakamura M, Komori A, Yano K, Yatsuhashi H, Eguchi K, Ishibashi H (2006) Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int 26(1):39–45
Aebi H, Suter H (1971) Acatalasemia. Adv Hum Genet 2:143–199
Ahn J, Cho I, Kim S, Kwon D, Ha T (2008) Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J Hepatol 49(6):1019–1028
Al-Gayyar MM, Shams ME, Barakat EA (2012) Fish oil improves lipid metabolism and ameliorates inflammation in patients with metabolic syndrome: impact of nonalcoholic fatty liver disease. Pharm Biol 50(3):297–303
Argo CK, Caldwell SH (2009) Epidemiology and natural history of non-alcoholic steatohepatitis. Clin Liver Dis 13(4):511–531
Ashla AA, Hoshikawa Y, Tsuchiya H, Hashiguchi K, Enjoji M, Nakamuta M, Taketomi A, Maehara Y, Shomori K, Kurimasa A, Hisatome I, Ito H, Shiota G (2010) Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease. Hepatol Res 40(6):594–604
Bagul PK, Middela H, Matapally S, Padiya R, Bastia T, Madhusudana K, Reddy BR, Chakravarty S, Banerjee SK (2012) Attenuation of insulin resistance, metabolic syndrome and hepatic oxidative stress by resveratrol in fructose-fed rats. Pharmacol Res 66(3):260–268
Bai J, Rodriguez AM, Melendez JA, Cederbaum AI (1999) Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J Biol Chem 274(37):26217–26224
Baker SS, Baker RD, Liu W, Nowak NJ, Zhu L (2010) Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS One 5(3):e9570. https://doi.org/10.1371/journal.pone.0009570
Begriche K, Igoudjil A, Pessayre D, Fromenty B (2006) Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion 6(1):1–28
Begriche K, Knockaert L, Robin MA, Fromenty B (2011) Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin Res Hepatol Gastroenterol 35(10):630–637
Bell GI, Najarian RC, Mullenbach GT, Hallewell RA (1986) cDNA sequence coding for human kidney catalase. Nucleic Acids Res 14(13):5561–5562
Bellisola G, Casaril M, Gabrielli GB, Caraffi M, Corrocher R (1987) Catalase activity in human hepatocellular carcinoma (HCC). Clin Biochem 20(6):415–417
Bernardo A, Bianchi D, Magnaghi V, Minghetti L (2009) Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J Neuropathol Exp Neurol 68(7):797–808
Beymer C, Kowdley KV, Larson A, Edmonson P, Dellinger EP, Flum DR (2003) Prevalence and predictors of asymptomatic liver disease in patients undergoing gastric bypass surgery. Arch Surg 138(11):1240–1244
Biddinger SB, Kahn CR (2006) From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 68:123–158
Bonkovsky HL, Jawaid Q, Tortorelli K, LeClair P, Cobb J, Lambrecht RW, Banner BF (1999) Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol 31(3):421–429
Brady LJ, Brady PS, Romsos DR, Hoppel CL (1985) Elevated hepatic mitochondrial and peroxisomal oxidative capacities in fed and starved adult obese (ob/ob) mice. Biochem J 231(2):439–444
Brezniceanu ML, Liu F, Wei CC, Tran S, Sachetelli S, Zhang SL, Guo DF, Filep JG, Ingelfinger JR, Chan JS (2007) Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int 71(9):912–923
Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, David E, Rizzetto M, Marchesini G (2005) A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol 100(5):1082–1090
Bugianesi E, McCullough AJ, Marchesini G (2005) Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42(5):987–1000
Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferrannini E, Rizzetto M (2005) Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 48(4):634–642
Cao SS, Kaufman RJ (2014) Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21(3):396–413
Casaril M, Corso F, Bassi A, Capra F, Gabrielli GB, Stanzial AM, Nicoli N, Corrocher R (1994) Decreased activity of scavenger enzymes in human hepatocellular carcinoma, but not in liver metastases. Int J Clin Lab Res 24(2):94–97
Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ (2012) The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55(6):2005–2023
Chen X, Mele J, Giese H, Van Remmen H, Dollé ME, Steinhelper M, Richardson A, Vijg J (2003) A strategy for the ubiquitous overexpression of human catalase and CuZn superoxide dismutase genes in transgenic mice. Mech Ageing Dev 124(2):219–227
Chen X, Liang H, Van Remmen H, Vijg J, Richardson A (2004) Catalase transgenic mice: characterization and sensitivity to oxidative stress. Arch Biochem Biophys 422(2):197–210
Chen S, Li J, Zhang Z, Li W, Sun Y, Zhang Q, Feng X, Zhu W (2012) Effects of resveratrol on the amelioration of insulin resistance in KKAy mice. Can J Physiol Pharmacol 90(2):237–242
Chen T, Jin X, Crawford BH, Cheng H, Saafir TB, Wagner MB, Yuan Z, Ding G (2012) Cardioprotection from oxidative stress in the newborn heart by activation of PPARγ is mediated by catalase. Free Radic Biol Med 53(2):208–215
Cho IJ, Ahn JY, Kim S, Choi MS, Ha TY (2008) Resveratrol attenuates the expression of HMG-CoA reductase mRNA in hamsters. Biochem Biophys Res Commun 367(1):190–194
Cho SJ, Jung UJ, Choi MS (2012) Differential effects of low-dose resveratrol on adiposity and hepatic steatosis in diet-induced obese mice. Br J Nutr 108(12):2166–2175
Choudhury J, Sanyal AJ (2004) Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis 8(3):575–594
Clemmons DR (2004) Role of insulin-like growth factor in maintaining normal glucose homeostasis. Horm Res 62(Suppl 1):77–82
Clemmons DR (2004) The relative roles of growth hormone and IGF-1 in controlling insulin sensitivity. J Clin Invest 113(1):25–27
Clerch LB (1995) A 3′ untranslated region of catalase mRNA composed of a stem-loop and dinucleotide repeat elements binds a 69-kDa redox-sensitive protein. Arch Biochem Biophys 317(1):267–274
Cohen G, Dembiec D, Marcus J (1970) Measurement of catalase activity in tissue extracts. Anal Biochem 34:30–38
Cohen JC, Horton JD, Hobbs HH (2011) Human fatty liver disease: old questions and new insights. Science 332(6037):1519–1523
Crespo J, Fernández-Gil P, Hernández-Guerra M, Cayón A, Mayorga M, Domínguez-Diez A, Fernández-Escalante JC, Pons-Romero F (2001) Are there predictive factors of severe liver fibrosis in morbidly obese patients with non-alcoholic steatohepatitis? Obes Surg 11(3):254–257
Das KS, Balakrishnan V, Mukherjee S, Vasudevan DM (2008) Evaluation of blood oxidative stress-related parameters in alcoholic liver disease and non-alcoholic fatty liver disease. Scand J Clin Lab Invest 68(4):323–334
Day CP, James OF (1998) Steatohepatitis: a tale of two “hits”? Gastroenterology 114(4):842–845
Desai S, Baker SS, Liu W, Moya DA, Browne RW, Mastrandrea L, Baker RD, Zhu L (2014) Paraoxonase 1 and oxidative stress in paediatric non-alcoholic steatohepatitis. Liver Int 34(1):110–117
Dixon JB, Bhathal PS, O'Brien PE (2001) Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 121(1):91–100
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115(5):1343–1351
Duvnjak M, Lerotić I, Barsić N, Tomasić V, Virović Jukić L, Velagić V (2007) Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol 13(34):4539–4550
Efstratiadis G, Tsiaousis G, Athyros VG, Karagianni D, Pavlitou-Tsiontsi A, Giannakou-Darda A, Manes C (2006) Total serum insulin-like growth factor-1 and C-reactive protein in metabolic syndrome with or without diabetes. Angiology 57(3):303–311
Eriksson AM, Lundgren B, Andersson K, DePierre JW (1992) Is the cytosolic catalase induced by peroxisome proliferators in mouse liver on its way to the peroxisomes? FEBS Lett 308(2):211–214
Erzurum SC, Lemarchand P, Rosenfeld MA, Yoo JH, Crystal RG (1993) Protection of human endothelial cells from oxidant injury by adenovirus-mediated transfer of the human catalase cDNA. Nucleic Acids Res 21(7):1607–1612
Escobar JA, Rubio MA, Lissi EA (1996) Sod and catalase inactivation by singlet oxygen and peroxyl radicals. Free Radic Biol Med 20(3):285–290
Faghihzadeh F, Adibi P, Rafiei R, Hekmatdoost A (2014) Resveratrol supplementation improves inflammatory biomarkers in patients with nonalcoholic fatty liver disease. Nutr Res 34(10):837–843
Ferrara N, Rinaldi B, Corbi G, Conti V, Stiuso P, Boccuti S, Rengo G, Rossi F, Filippelli A (2008) Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res 11(1):139–150
Flori E, Mastrofrancesco A, Kovacs D, Ramot Y, Briganti S, Bellei B, Paus R, Picardo M (2011) 2,4,6-Octatrienoic acid is a novel promoter of melanogenesis and antioxidant defence in normal human melanocytes via PPAR-γ activation. Pigment Cell Melanoma Res 24(4):618–630
Gao H, Zeng Z, Zhang H, Zhou X, Guan L, Deng W, Xu L (2015) The glucagon-like peptide-1 analogue Liraglutide inhibits oxidative stress and inflammatory response in the liver of rats with diet-induced non-alcoholic fatty liver disease. Biol Pharm Bull 38(5):694–702
García-Galiano D, Sánchez-Garrido MA, Espejo I, Montero JL, Costán G, Marchal T, Membrives A, Gallardo-Valverde JM, Muñoz-Castañeda JR, Arévalo E, De la Mata M, Muntané J (2007) IL-6 and IGF-1 are independent prognostic factors of liver steatosis and non-alcoholic steatohepatitis in morbidly obese patients. Obes Surg 17(4):493–503
Gholam PM, Kotler DP, Flancbaum LJ (2002) Liver pathology in morbidly obese patients undergoing Roux-en-Y gastric bypass surgery. Obes Surg 12(1):49–51
Gholam PM, Flancbaum L, Machan JT, Charney DA, Kotler DP (2007) Nonalcoholic fatty liver disease in severely obese subjects. Am J Gastroenterol 102(2):399–408
Girnun GD, Domann FE, Moore SA, Robbins ME (2002) Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol 16(12):2793–2801
Glorieux C, Auquier J, Dejeans N, Sid B, Demoulin JB, Bertrand L, Verrax J, Calderon PB (2014) Catalase expression in MCF-7 breast cancer cells is mainly controlled by PI3K/Akt/mTor signaling pathway. Biochem Pharmacol 89(2):217–223
Glorieux C, Zamocky M, Sandoval JM, Verrax J, Calderon PB (2015) Regulation of catalase expression in healthy and cancerous cells. Free Radic Biol Med 87:84–97
Gómez-Zorita S, Fernández-Quintela A, Macarulla MT, Aguirre L, Hijona E, Bujanda L, Milagro F, Martínez JA, Portillo MP (2012) Resveratrol attenuates steatosis in obese Zucker rats by decreasing fatty acid availability and reducing oxidative stress. Br J Nutr 107(2):202–210
Grattagliano I, Vendemiale G, Lauterburg BH (1999) Reperfusion injury of the liver: role of mitochondria and protection by glutathione ester. J Surg Res 86(1):2–8
Grundy SM (1999) Hypertriglyceridemia, insulin resistance, and the metabolic syndrome. Am J Cardiol 83(9B):25F–29F
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Washida N, Tokuyama H, Hayashi K, Itoh H (2008) Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372(1):51–56
Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K, Aukrust P (2006) Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol 44(6):1167–1174
Heit C, Marshall S, Singh S, Yu X, Charkoftaki G, Zhao H, Orlicky DJ, Fritz KS, Thompson DC, Vasiliou V (2017) Catalase deletion promotes prediabetic phenotype in mice. Free Radic Biol Med 103:48–56
Ho YS, Xiong Y, Ma W, Spector A, Ho DS (2004) Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem 279(31):32804–32812
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259(5091):87–91
Jones DP, Eklöw L, Thor H, Orrenius S (1981) Metabolism of hydrogen peroxide in isolated hepatocytes: relative contributions of catalase and glutathione peroxidase in decomposition of endogenously generated H2O2. Arch Biochem Biophys 210(2):505–516
Jun DW (2012) Practice guideline for the diagnosis and management of non-alcoholic fatty liver disease. Korean J Gastroenterol 60(1):64–66
Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, Dinarello CA, Apte RN, Harats D (2011) Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 55(5):1086–1094
Kanemaki T, Kitade H, Kaibori M, Sakitani K, Hiramatsu Y, Kamiyama Y, Ito S, Okumura T (1998) Interleukin 1beta and interleukin 6, but not tumor necrosis factor alpha, inhibit insulin-stimulated glycogen synthesis in rat hepatocytes. Hepatology 27(5):1296–1303
Kang YJ, Chen Y, Epstein PN (1996) Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J Biol Chem 271(21):12610–12616
Kang W, Hong HJ, Guan J, Kim DG, Yang EJ, Koh G, Park D, Han CH, Lee YJ, Lee DH (2012) Resveratrol improves insulin signaling in a tissue-specific manner under insulin-resistant conditions only: in vitro and in vivo experiments in rodents. Metabolism 61(3):424–433
Kasprzak A, Adamek A (2012) The insulin-like growth factor (IGF) signaling axis and hepatitis C virus-associated carcinogenesis (review). Int J Oncol 41(6):1919–1931
Khoo NK, Hebbar S, Zhao W, Moore SA, Domann FE, Robbins ME (2013) Differential activation of catalase expression and activity by PPAR agonists: implications for astrocyte protection in anti-glioma therapy. Redox Biol 1:70–79
Knudsen ST, Bek T, Poulsen PL, Hove MN, Rehling M, Mogensen CE (2002) Macular edema reflects generalized vascular hyperpermeability in type 2 diabetic patients with retinopathy. Diabetes Care 25(12):2328–2334
Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, Nakamuta M (2007) Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 20(3):351–358
Koike K (2007) Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol 22(Suppl 1):S108–S111
Kono Y, Fridovich I (1982) Superoxide radical inhibits catalase. J Biol Chem 257(10):5751–5754
Kopp HP, Kopp CW, Festa A, Krzyzanowska K, Kriwanek S, Minar E, Roka R, Schernthaner G (2003) Impact of weight loss on inflammatory proteins and their association with the insulin resistance syndrome in morbidly obese patients. Arterioscler Thromb Vasc Biol 23(6):1042–1047
Kumar R, Prakash S, Chhabra S, Singla V, Madan K, Gupta SD, Panda SK, Khanal S, Acharya SK (2012) Association of pro-inflammatory cytokines, adipokines & oxidative stress with insulin resistance & non-alcoholic fatty liver disease. Indian J Med Res 136(2):229–236
Kumar A, Sharma A, Duseja A, Das A, Dhiman RK, Chawla YK, Kohli KK, Bhansali A (2013) Patients with nonalcoholic fatty liver disease (NAFLD) have higher oxidative stress in comparison to chronic viral hepatitis. J Clin Exp Hepatol 3(1):12–18
Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, Abrams SH, Scheimann AO, Sanyal AJ, Chalasani N, Tonascia J, Ünalp A, Clark JM, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR (2011) Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 305(16):1659–1668
Leclercq IA (2004) Antioxidant defence mechanisms: new players in the pathogenesis of non-alcoholic steatohepatitis? Clin Sci (Lond) 106(3):235–237
Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR (2000) CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest 105(8):1067–1075
Leite NC, Salles GF, Cardoso CR, Villela-Nogueira CA (2013) Serum biomarkers in type 2 diabetic patients with non-alcoholic steatohepatitis and advanced fibrosis. Hepatol Res 43(5):508–515
Li G, Chen Y, Saari JT, Kang YJ (1997) Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am J Phys 273(3 Pt 2):H1090–H1095
Li M, Chiu JF, Gagne J, Fukagawa NK (2008) Age-related differences in insulin-like growth factor-1 receptor signaling regulates Akt/FOXO3a and ERK/Fos pathways in vascular smooth muscle cells. J Cell Physiol 217(2):377–387
Liangpunsakul S, Chalasani N (2005) Unexplained elevations in alanine aminotransferase in individuals with the metabolic syndrome: results from the third National Health and Nutrition Survey (NHANES III). Am J Med Sci 329(3):111–116
Liaw KY, Lee PH, Wu FC, Tsai JS, Lin-Shiau SY (1997) Zinc, copper, and superoxide dismutase in hepatocellular carcinoma. Am J Gastroenterol 92(12):2260–2263
Lieber CS (1997) Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev 77(2):517–544
Lin CC, Liu WH, Wang ZH, Yin MC (2011) Vitamins B status and antioxidative defense in patients with chronic hepatitis B or hepatitis C virus infection. Eur J Nutr 50(7):499–506
Lindau-Shepard BA, Shaffer JB (1993) Expression of human catalase in acatalasemic murine SV-B2 cells confers protection from oxidative damage. Free Radic Biol Med 15(6):581–588
Lozano-Sepulveda SA, Bryan-Marrugo OL, Cordova-Fletes C, Gutierrez-Ruiz MC, Rivas-Estilla AM (2015) Oxidative stress modulation in hepatitis C virus infected cells. World J Hepatol 7(29):2880–2889
Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, Guy CD, Diehl AM (2015) Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10(5):e0127991. https://doi.org/10.1371/journal.pone.0127991
Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N (2001) Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 50(8):1844–1850
Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N (2001) Metformin in non-alcoholic steatohepatitis. Lancet 358(9285):893–894
Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, Rizzetto M (2003) Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 37(4):917–923
Marra M, Sordelli IM, Lombardi A, Lamberti M, Tarantino L, Giudice A, Stiuso P, Abbruzzese A, Sperlongano R, Accardo M, Agresti M, Caraglia M, Sperlongano P (2011) Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: an overview. J Transl Med 9:171
Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB, Steering Committee LEADER, Trial Investigators LEADER (2016) Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 375(4):311–322
Masters C, Pegg M, Crane D (1986) On the multiplicity of the enzyme catalase in mammalian liver. Mol Cell Biochem 70(2):113–120
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ (1999) Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 116(6):1413–1419
McAdam-Marx C, Nguyen H, Schauerhamer MB, Singhal M, Unni S, Ye X, Cobden D (2016) Glycemic control and weight outcomes for exenatide once weekly versus liraglutide in patients with type 2 diabetes: a 1-year retrospective cohort analysis. Clin Ther 38(12):2642–2651
Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, Imai K, Todoroki T, Kimura S, Koike K (2001) Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res 61(11):4365–4370
Moriya K, Todoroki T, Tsutsumi T, Fujie H, Shintani Y, Miyoshi H, Ishibashi K, Takayama T, Makuuchi M, Watanabe K, Miyamura T, Kimura S, Koike K (2001) Increase in the concentration of carbon 18 monounsaturated fatty acids in the liver with hepatitis C: analysis in transgenic mice and humans. Biochem Biophys Res Commun 281(5):1207–1212
Moya D, Baker SS, Liu W, Garrick M, Kozielski R, Baker RD, Zhu L (2015) Novel pathway for iron deficiency in pediatric non-alcoholic steatohepatitis. Clin Nutr 34(3):549–556
Murphy PA, Krahling JB, Gee R, Kirk JR, Tolbert NE (1979) Enzyme activities of isolated hepatic peroxisomes from genetically lean and obese male mice. Arch Biochem Biophys 193(1):179–185
Nair J, Srivatanakul P, Haas C, Jedpiyawongse A, Khuhaprema T, Seitz HK, Bartsch H (2010) High urinary excretion of lipid peroxidation-derived DNA damage in patients with cancer-prone liver diseases. Mutat Res 683(1–2):23–28
Nakashima H, Yamamoto M, Goto K, Osumi T, Hashimoto T, Endo H (1989) Isolation and characterization of the rat catalase-encoding gene. Gene 79(2):279–288
Nalbantoglu IL, Brunt EM (2014) Role of liver biopsy in nonalcoholic fatty liver disease. World J Gastroenterol 20(27):9026–9037
Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 280(16):16456–16460
Ngoka LC (2008) Dramatic down-regulation of oxidoreductases in human hepatocellular carcinoma hepG2 cells: proteomics and gene ontology unveiling new frontiers in cancer enzymology. Proteome Sci 6:29
Nomura K, Yamanouchi T (2012) The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem 23(3):203–208
Nomura F, Ohnishi K, Satomura Y, Ohtsuki T, Fukunaga K, Honda M, Ema M, Tohyama T, Sugita S, Saito M et al (1986) Liver function in moderate obesity—study in 534 moderately obese subjects among 4613 male company employees. Int J Obes 10(5):349–354
Okuno Y, Matsuda M, Kobayashi H, Morita K, Suzuki E, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I (2008) Adipose expression of catalase is regulated via a novel remote PPARgamma-responsive region. Biochem Biophys Res Commun 366(3):698–704
Olleros Santos-Ruiz M, Sádaba MC, Martín-Estal I, Muñoz U, Sebal Neira C, Castilla-Cortázar I (2017) The single IGF-1 partial deficiency is responsible for mitochondrial dysfunction and is restored by IGF-1 replacement therapy. Growth Hormon IGF Res 35:21–32
Olmos Y, Valle I, Borniquel S, Tierrez A, Soria E, Lamas S, Monsalve M (2009) Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J Biol Chem 284(21):14476–14484
Olmos Y, Sánchez-Gómez FJ, Wild B, García-Quintans N, Cabezudo S, Lamas S, Monsalve M (2013) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid Redox Signal 19(13):1507–1521
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306(5695):457–461
Ozkaya A, Sahin Z, Gorgulu AO, Yuce A, Celik S (2016) Geraniol attenuates hydrogen peroxide-induced liver fatty acid alterations in male rats. J Intercult Ethnopharmacol 6(1):29–35
Percy ME (1984) Catalase: an old enzyme with a new role? Can J Biochem Cell Biol 62(10):1006–1014
Perlemuter G, Davit-Spraul A, Cosson C, Conti M, Bigorgne A, Paradis V, Corre MP, Prat L, Kuoch V, Basdevant A, Pelletier G, Oppert JM, Buffet C (2005) Increase in liver antioxidant enzyme activities in non-alcoholic fatty liver disease. Liver Int 25(5):946–953
Pessayre D, Fromenty B (2005) NASH: a mitochondrial disease. J Hepatol 42(6):928–940
Petit JM, Cercueil JP, Loffroy R, Denimal D, Bouillet B, Fourmont C, Chevallier O, Duvillard L, Vergès B (2017) Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-NAFLD Study. J Clin Endocrinol Metab 102(2):407–415
Piao L, Choi J, Kwon G, Ha H (2017) Endogenous catalase delays high-fat diet-induced liver injury in mice. Korean J Physiol Pharmacol 21(3):317–325
Pickup JC, Chusney GD, Thomas SM, Burt D (2000) Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci 67(3):291–300
Portincasa P, Grattagliano I, Palmieri VO, Palasciano G (2005) Nonalcoholic steatohepatitis: recent advances from experimental models to clinical management. Clin Biochem 38(3):203–217
Postic C, Dentin R, Girard J (2004) Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab 30(5):398–408
Poulsen MM, Larsen JØ, Hamilton-Dutoit S, Clasen BF, Jessen N, Paulsen SK, Kjær TN, Richelsen B, Pedersen SB (2012) Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet. Nutr Res 32(9):701–708
Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286(3):327–334
Preiser JC (2012) Oxidative stress. JPEN J Parenter Enteral Nutr 36(2):147–154
Quan F, Korneluk RG, Tropak MB, Gravel RA (1986) Isolation and characterization of the human catalase gene. Nucleic Acids Res 14(13):5321–5335
Quan X, Lim SO, Jung G (2011) Reactive oxygen species downregulate catalase expression via methylation of a CpG island in the Oct-1 promoter. FEBS Lett 585(21):3436–3441
Reimer DL, Bailley J, Singh SM (1994) Complete cDNA and 5′ genomic sequences and multilevel regulation of the mouse catalase gene. Genomics 21(2):325–336
Rindler PM, Plafker SM, Szweda LI, Kinter M (2013) High dietary fat selectively increases catalase expression within cardiac mitochondria. J Biol Chem 288(3):1979–1990
Rizvi AA, Patti AM, Giglio RV, Nikolic D, Amato A, Al-Busaidi N, Al-Rasadi K, Soresi M, Banach M, Montalto G, Rizzo M (2015) Liraglutide improves carotid intima-media thickness in patients with type 2 diabetes and non-alcoholic fatty liver disease: an 8-month prospective pilot study. Expert Opin Biol Ther 15(10):1391–1397
Rocha S, Gomes D, Lima M, Bronze-da-Rocha E, Santos-Silva A (2015) Peroxiredoxin 2, glutathione peroxidase, and catalase in the cytosol and membrane of erythrocytes under H2O2-induced oxidative stress. Free Radic Res 49(8):990–1003
Rolo AP, Teodoro JS, Palmeira CM (2012) Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52(1):59–69
Ruis H (1979) The biosynthesis of catalase. Can J Biochem 57(9):1122–1130
Salvi M, Battaglia V, Brunati AM, La Rocca N, Tibaldi E, Pietrangeli P, Marcocci L, Mondovì B, Rossi CA, Toninello A (2007) Catalase takes part in rat liver mitochondria oxidative stress defense. J Biol Chem 282(33):24407–24415
Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ (2002) Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet 359(9319):1740–1745
Sando T, Konno K, Takei N, Sakamoto T, Higashi T (1984) Purification and characterization of rat liver cytosol catalase. Cell Struct Funct 9(2):125–133
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR (2010) Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 362(18):1675–1685
Sariçam T, Kircali B, Köken T (2005) Assessment of lipid peroxidation and antioxidant capacity in non-alcoholic fatty liver disease. Turk J Gastroenterol 16(2):65–70
Schmid C (1995) Insulin-like growth factors. Cell Biol Int 19(5):445–457
Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA (2003) Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 278(16):13740–13746
Smits MM, Tonneijck L, Muskiet MH, Kramer MH, Pouwels PJ, Pieters-van den Bos IC, Hoekstra T, Diamant M, van Raalte DH, Cahen DL (2016) Twelve week liraglutide or sitagliptin does not affect hepatic fat in type 2 diabetes: a randomised placebo-controlled trial. Diabetologia 59(12):2588–2593
Spitz DR, Adams DT, Sherman CM, Roberts RJ (1992) Mechanisms of cellular resistance to hydrogen peroxide, hyperoxia, and 4-hydroxy-2-nonenal toxicity: the significance of increased catalase activity in H2O2-resistant fibroblasts. Arch Biochem Biophys 292(1):221–227
Sreekumar R, Rosado B, Rasmussen D, Charlton M (2003) Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology 38(1):244–251
Stål P, Glaumann H, Hultcrantz R (1990) Liver cell damage and lysosomal iron storage in patients with idiopathic hemochromatosis. A light and electron microscopic study. J Hepatol 11(2):172–180
Steinbrenner H, Sies H (2009) Protection against reactive oxygen species by selenoproteins. Biochim Biophys Acta 1790(11):1478–1485
Svegliati-Baroni G, Ridolfi F, Di Sario A, Casini A, Marucci L, Gaggiotti G, Orlandoni P, Macarri G, Perego L, Benedetti A, Folli F (1999) Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology 29(6):1743–1751
Swietek K, Juszczyk J (1997) Reduced glutathione concentration in erythrocytes of patients with acute and chronic viral hepatitis. J Viral Hepat 4(2):139–141
Syn WK, Choi SS, Diehl AM (2009) Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis 13(4):565–580
Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL (2005) Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 288(2):E462–E468
Takahashi Y, Iida K, Takahashi K, Yoshioka S, Fukuoka H, Takeno R, Imanaka M, Nishizawa H, Takahashi M, Seo Y, Hayashi Y, Kondo T, Okimura Y, Kaji H, Kitazawa R, Kitazawa S, Chihara K (2007) Growth hormone reverses nonalcoholic steatohepatitis in a patient with adult growth hormone deficiency. Gastroenterology 132(3):938–943
Tan WQ, Wang K, Lv DY, Li PF (2008) Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J Biol Chem 283(44):29730–29739
Tang A, Rabasa-Lhoret R, Castel H, Wartelle-Bladou C, Gilbert G, Massicotte-Tisluck K, Chartrand G, Olivié D, Julien AS, de Guise J, Soulez G, Chiasson JL (2015) Effects of insulin glargine and liraglutide therapy on liver fat as measured by magnetic resonance in patients with type 2 diabetes: a randomized trial. Diabetes Care 38(7):1339–1346
Taniguchi M, Hashimoto M, Hori N, Sato K (2005) CCAAT/enhancer binding protein-beta (C/EBP-beta), a pivotal regulator of the TATA-less promoter in the rat catalase gene. FEBS Lett 579(25):5785–5790
Tantin D, Schild-Poulter C, Wang V, Haché RJ, Sharp PA (2005) The octamer binding transcription factor Oct-1 is a stress sensor. Cancer Res 65(23):10750–10758
Tarantino G, Conca P, Pasanisi F, Ariello M, Mastrolia M, Arena A, Tarantino M, Scopacasa F, Vecchione R (2009) Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur J Gastroenterol Hepatol 21(5):504–511
Torres DM, Jones FJ, Shaw JC, Williams CD, Ward JA, Harrison SA (2011) Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: a 12-month randomized, prospective, open- label trial. Hepatology 54(5):1631–1639
Tsigos C, Papanicolaou DA, Kyrou I, Defensor R, Mitsiadis CS, Chrousos GP (1997) Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. J Clin Endocrinol Metab 82(12):4167–4170
Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM (1995) Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 96(1):620–630
Ucar F, Sezer S, Erdogan S, Akyol S, Armutcu F, Akyol O (2013) The relationship between oxidative stress and nonalcoholic fatty liver disease: its effects on the development of nonalcoholic steatohepatitis. Redox Rep 18(4):127–133
Venugopal R, Jaiswal AK (1998) Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17(24):3145–3156
Verkerk A, Jongkind JF (1992) Vascular cells under peroxide induced oxidative stress: a balance study on in vitro peroxide handling by vascular endothelial and smooth muscle cells. Free Radic Res Commun 17(2):121–132
Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, Varela N, Contreras J, Lazarte R, Csendes A, Rojas J, Maluenda F, Burdiles P, Diaz JC, Smok G, Thielemann L, Poniachik J (2004) Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond) 106(3):261–268
Völzke H, Nauck M, Rettig R, Dörr M, Higham C, Brabant G, Wallaschofski H (2009) Association between hepatic steatosis and serum IGF1 and IGFBP-3 levels in a population-based sample. Eur J Endocrinol 161(5):705–713
Weltman MD, Farrell GC, Liddle C (1996) Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111(6):1645–1653
Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C (1998) Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 27(1):128–133
Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE (2008) Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol 103(6):1372–1379
William CO, Rajindar SS (1992) The effects of catalase gene overexpression on life span and resistance to oxidative stress in transgenic Drosophila melanogaster. Arch Biochem Biophys 297(1):35–41
Wu S, Li Q, Du M, Li SY, Ren J (2007) Cardiac-specific overexpression of catalase prolongs lifespan and attenuates ageing-induced cardiomyocyte contractile dysfunction and protein damage. Clin Exp Pharmacol Physiol 34(1–2):81–87
Xu B, Moritz JT, Epstein PN (1999) Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic Biol Med 27(7–8):830–837
Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ (2003) The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 112(1):91–100
Yang H, Shi M, VanRemmen H, Chen X, Vijg J, Richardson A, Guo Z (2003) Reduction of pressor response to vasoconstrictor agents by overexpression of catalase in mice. Am J Hypertens 16(1):1–5
Yang W, Zhang J, Wang H, Shen W, Gao P, Singh M, Fang N (2011) Peroxisome proliferator-activated receptor γ regulates angiotensin II-induced catalase downregulation in adventitial fibroblasts of rats. FEBS Lett 585(5):761–766
Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, Sanisoglu SY, Erdil A, Ates Y, Aslan M, Musabak U, Erbil MK, Karaeren N, Dagalp K (2005) Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am J Gastroenterol 100(4):850–855
Yoo JH, Erzurum SC, Hay JG, Lemarchand P, Crystal RG (1994) Vulnerability of the human airway epithelium to hyperoxia. Constitutive expression of the catalase gene in human bronchial epithelial cells despite oxidant stress. J Clin Invest 93(1):297–302
Younossi ZM, Otgonsuren M, Henry L, Venkatesan C, Mishra A, Erario M, Hunt S (2015) Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 62(6):1723–1730
Zhang X, Li S, Zhou Y, Su W, Ruan X, Wang B, Zheng F, Warner M, Gustafsson JÅ, Guan Y (2017) Ablation of cytochrome P450 omega-hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proc Natl Acad Sci U S A 114(12):3181–3185
Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, Grotta JC, Aronowski J (2009) Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci 29(19):6186–6195
Zheng Y, Liu T, Wang Z, Xu Y, Zhang Q, Luo D (2018) Low molecular weight fucoidan attenuates liver injury via SIRT1/AMPK/PGC1α axis in db/db mice. Int J Biol Macromol 112:929–936
Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y (2005) Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett 579(14):3029–3036
Zhu H, Jia Z, Misra BR, Zhang L, Cao Z, Yamamoto M, Trush MA, Misra HP, Li Y (2008) Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol 8(2):71–85
Zhu H, Jia Z, Zhang L, Yamamoto M, Misra HP, Trush MA, Li Y (2008) Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp Biol Med (Maywood) 233(4):463–474
Funding
This study was supported by a grant from the National Research Foundation of Korea (NRF-2018R1A2B2004429) and a grant funded by the Korean Government (MSIP) (No. 2014R1A5A2010008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shin, SK., Cho, HW., Song, SE. et al. Catalase and nonalcoholic fatty liver disease. Pflugers Arch - Eur J Physiol 470, 1721–1737 (2018). https://doi.org/10.1007/s00424-018-2195-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2195-z