
Abstract
Since their discovery more than 30 years ago, low-threshold T-type Ca2+ channels (T channels) have been suggested to play a key role in many EEG waves of non-REM sleep, which has remained exclusively linked to the ability of these channels to generate low-threshold Ca2+ potentials and associated high-frequency bursts of action potentials. Our present understanding of the biophysics and physiology of T channels, however, highlights a much more diverse and complex picture of the pivotal contributions that they make to different sleep rhythms. In particular, recent experimental evidence has conclusively demonstrated the essential contribution of thalamic T channels to the expression of slow waves of natural sleep and the key role played by Ca2+ entry through these channels in the activation or modulation of other voltage-dependent channels that are important for the generation of both slow waves and sleep spindles. However, the precise contribution to sleep rhythms of T channels in cortical neurons and other sleep-controlling neuronal networks remains unknown, and a full understanding of the cellular and network mechanisms of sleep delta waves is still lacking.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As this review is part of a special issue on T-type Ca2+ channels (T channels), it feels appropriate to firstly provide the reader with an overview of the stereotyped sequences of electrical waves that are recorded in the EEG during natural non-REM sleep. Importantly, while the source(s) of the electrical waves observed in scalp EEG recordings are located in the upper layers of the neocortex, their generator(s) are the dynamical interactions between the different neuronal network activities that are expressed by various component neurons of the corticothalamic loop. In humans, the occurrence of theta waves (3–7 Hz) over a generally desynchronized EEG characterizes the first stage of non-REM sleep, while in stage 2 occasional K-complexes and slow waves start to appear. Sleep spindles are also present in stage 2, either in isolation or associated with a K-complex. The EEG in stage 3 sleep still presents spindle episodes but also shows clearly defined periods of delta waves (0.5–4 Hz) that together with slow waves (<1–2 Hz) become the predominant activity as sleep deepens into stage 4. This smooth and progressive transition from stage 1 to stage 4 non-REM sleep is invariably accompanied at the neuronal level by a reduction in the depolarizing tone exerted by cholinergic, monoaminergic, and histaminergic afferents from brainstem and mammillary body onto both cortical and thalamic neurons [60] (with some exceptions, see ref. [60]), leading to a progressive hyperpolarization of the majority of cortical and thalamic neurons [42, 78, 89].
The first insight into the role of T channels in sleep waves came from their discovery in thalamic neurons, and in particular the finding that activation of these channels, following a period of membrane hyperpolarization, leads to a voltage waveform known as the low-threshold Ca2+ potential (LTCP) or low-threshold spike [30, 32, 46, 47, 55]. Since then, the main and only widely recognized function of thalamic T channels in sleep waves has been that of providing the rhythmic LTCP-mediated sequences of high-frequency bursts of action potentials that characterize the cellular activity of these neurons during sleep spindles and delta waves as well as at the start of an up state of sleep slow waves [7, 8, 45, 53, 54, 61, 76, 81, 82, 84, 92]. However, the role of the T channels in sleep and non-REM EEG oscillations can no longer be restricted to the stereotypical LTCPs of thalamic neurons, since (1) it involves other physiological voltage waveforms that are dependent on the “window current” of these channels (i.e., I Twindow) [10, 43, 44, 91, 93], and (2) because non-thalamic neuronal populations, e.g. those in the neocortex and in sleep-controlling brain regions, show a marked expression of T channels [28, 35, 38, 40, 41, 48, 71, 87]. This short review will address these issues after presenting a brief overview on the biophysics of T channels, and in particular on I Twindow and its physiological consequences for neuronal excitability (for detailed descriptions of the molecular genetics, biophysics, and neuronal cell type distribution of T channels, see other contributions to this special issue). In addition, as few experiments have so far analyzed the role of T currents in naturally sleeping animals, we will also discuss how the current view of T channel function in sleep may be clouded by the speculative extrapolations of data obtained either in brain slices or in anesthetized preparations where EEG waves similar, though not identical, to those observed during the various stages of natural non-REM sleep can be recorded (see [21]). Finally, we will highlight the current difficulty in correctly identifying the cellular and network mechanisms of delta waves because of the partial overlap of their frequency band with that of slow waves.
Biophysics and physiological impact of the “window current” generated by T channels
Since their original characterization in primary sensory neurons [11, 13, 36, 65], two main types of native T type Ca2+ currents that display either “fast” or “slow” activation and inactivation kinetics were reported [45, 68]. Cloning of the three low-threshold Ca2+ channel genes (Cav3) further confirmed this crude categorization. Cav3.1 and Cav3.2 (α1G and α1H, respectively) generate low-threshold Ca2+ currents displaying fast activation and inactivation mechanisms [18, 69], while Cav3.3 (α1I) shows much slower kinetics [52]. Regardless of this difference in gating kinetics, all native and recombinant channels share the same basic voltage dependence with an activation threshold and a nearly complete steady-state inactivation around −60 mV [68].
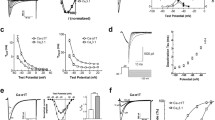
However, a closer look at the gating properties of the T channels reveals that the steady-state activation and inactivation curves overlap (Fig. 1(a)). Therefore, in this voltage region that corresponds to neuronal resting membrane potentials, a few T channels are not inactivated and their open probability is close but not equal to zero, hence a tonic T current (i.e., I Twindow) is generated. Since the activation and inactivation curves are obtained by fitting currents that in this voltage region are obviously very small, a precise estimation of I Twindow, particularly for native channels, is difficult to achieve, and thus great caution should be used in interpreting these data. Nevertheless, investigations on recombinant channels have suggested that Cav3.3 channels may generate a larger and more depolarized I Twindow (see Fig 7 in [68]; [15]) than that elicited by Cav3.1 and Cav3.2 channels. These differences in I Twindow should be carefully considered when assessing the precise role of this tonic current in the excitability of neurons that possess different complements (and different subcellular distributions) of the three isoforms of T channels. In particular, the glutamatergic thalamocortical (TC) neurons only express Cav3.1 channels while the GABAergic neurons of the nucleus reticularis thalami (NRT) possess Cav3.3 channels in addition to a small component of Cav3.2 channels [45, 87].

The window T current tightly controls thalamic neuron excitability. a Normalized activation and steady-state inactivation curves of T current recorded in a TC neuron from the rat ventrobasal thalamic nucleus. The activation curve was constructed by successive step depolarizations from −80 to −45 mV (2.5 mV increments) preceded by a 1-s hyperpolarizing pre-pulse to −100 mV. Inactivation of the T channels was induced using a 1-s pre-pulse of increasing potential (from −100 to −60 mV with 2.5 mV increments) and the resulting channel availability was estimated from the normalized current amplitude measured at −50 mV. Data were fitted by Boltzmann equations. Inset illustrates the voltage dependence of the steady-state channel activation (window current) estimated from the product of the Boltzmann fits of the normalized activation and inactivation curves. b The window T current evoked by a 10-s-long depolarizing voltage ramp from −100 to −40 mV preceded by a 1-s hyperpolarizing prepulse to −100 mV is fully blocked by the selective T channel blocker TTA-P2 (1 μM). B2 Voltage dependence of the window T current shown in B1. c In the continuous presence of trans-ACPD, recording from a cat TC neuron in an thalamic intralaminar nucleus reveals a slow oscillation (top trace) consisting of regularly recurring up and down states intermixed with much longer up states with continuous tonic action potentials firing. Each down state starts with a clear inflection point leading to a stereotypical large hyperpolarizing potential that, upon I h activation, slowly repolarizes the neuron up to the LTCP threshold (see also Fig. 2c). Following the block of I h with ZD 7288 (middle trace), the neuron exhibits two stable resting membrane potentials. Transitions between stable equilibrium potentials are evoked by short steps of positive or negative injected currents (I inj) that trigger an LTCP and switch off I Twindow, respectively. Upon application of TTA-P2 that progressively blocks the T channel population, the bistable behavior quickly disappears due to the decrease in I Twindow while enough T channels remain to evoke LTCPs (lower trace). B1–B2: reproduced with permission from [34]
Although I Twindow is an inherent biophysical property of all T channels, its amplitude in some neurons may be too small, thus precluding a significant physiological role in cellular excitability. However, both TC and NRT neurons express especially large T currents [6, 9, 31, 34] and a significant number of T channels are still de-inactivated around −60 mV. Thus, although the open probability of the channels is very low at these potentials, an I Twindow of about 30 pA can be measured in TC neurons using voltage ramps that are slow enough to achieve steady-state equilibrium between activation and inactivation of the T channels (Fig. 1(b)). Block of this tonic current with the specific T channel antagonist, TTA-P2, induces a 3-mV hyperpolarization of TC neurons held at −60 mV but has no effect when the neuron is held at a membrane potential outside the voltage range of I Twindow activation (see Fig. 3 in [34]). As predicted from the biophysics of recombinant Cav3.3 channels, an even larger hyperpolarization (5 mV) is observed upon application of TTA-P2 in NRT neurons [34], demonstrating that I Twindow play a crucial role in setting the resting membrane potential of both TC and NRT neurons. Moreover, when metabotropic glutamate or muscarinic receptors are activated, the interplay between the characteristic bell-shaped voltage dependence of I Twindow (see inset in Fig. 1(a)) and the leak current creates a marked (up to 20 mV) bistability of the resting membrane potential of TC and NRT neurons (Fig. 1(c), middle trace) [20, 44]. The shift between these two stable membrane potentials can occur spontaneously as an intrinsic mechanism (leading to the appearance of repetitive up and down states of sleep slow waves, see next section) (Fig. 1(c), top trace) [43] or can amplify small-amplitude subthreshold synaptic potentials leading to the generation of a rebound LTCP (see Fig. 6 in [93]).
Importantly, it is possible to block membrane bistability with the T channel blocker TTA-P2 while leaving the LTCP and its associate firing almost intact (Fig. 1(c), bottom trace). Indeed, up and down states quickly disappear upon a short period of TTA-P2 application that slightly reduces the functional T channel population whereas the full block of LTCPs requires much longer antagonist application (see Fig. 7 in [34]). These two experiments clearly demonstrated that the high density of T channels expressed in thalamic neurons far exceeds that required to generate a LTCP [9, 34]. Therefore, it is highly probable that such high density of T channels provides the significant number of de-inactivated channels at depolarized potentials that are required for the full repertoire of the physiological responses of thalamic neurons. Finally, it is important to point out that these T channels available at depolarized membrane potentials not only generate I Twindow in low-open probability conditions but are also recruited by synaptic activities and intrinsic noise that, by mediating a drastic increase in open probability, generate additional transient T currents that boost post-synaptic potentials (see Fig. 1 in [29]).
Thalamic and cortical T channel contribution to sleep slow waves
Sleep slow waves are one of the fundamental EEG wave of non-REM sleep (Fig. 2a). They are present in almost all non-REM sleep stages [1–3, 37, 58, 75, 77], underlie sleep K-complexes [2, 3], and group together periods of sleep spindles [3, 62] and delta waves [2, 77]. The cellular counterpart of the sleep slow rhythm recorded in the EEG is the regular recurrence of a depolarized (up) state and a hyperpolarized (down) state of the membrane potential, that occurs synchronously in all cortical [77, 85, 86] and thalamic neurons [3, 16, 37, 58, 62, 75, 77, 79].

Contribution of thalamic T channels to sleep slow waves. a EEG slow wave and corresponding wavelet scalograms recorded in a rat during natural sleep. Continuous thalamic microdialysis injection of a solution containing the T channel antagonist TTA-P2 (3 mM) reduces the frequency of slow waves compared to the injection of artificial cerebrospinal fluid (Control). b EEG slow wave recorded in a rat during ketamine/xylazine anesthesia. Continuous thalamic microdialysis injection of a solution containing 3 mM of TTA-P2 decreases the frequency of slow waves compared to the injection of artificial cerebrospinal fluid (Control). c Schematic diagram of two cycles of slow waves in a TC neuron shows the various contributions of T channels to this oscillation. a–c: reproduced with permission from [26] and [20]
In cortical neurones, sleep slow waves result from intense excitatory and inhibitory synaptic barrages that generate the up state and their absence that causes the down state [73, 85, 86, 89]. Although T currents are not considered to play a major role in this process, such powerful synaptic activity and the resulting changes in membrane potential do of course engage a variety of voltage-dependent channels, including T channels. Indeed clear examples of LTCPs can be seen in recordings from cortical neurons during slow waves in anesthetized animals [16], as one would expect from the presence of (1) all three T channel isoforms in the neocortex [87] and (2) LTCPs in layers V–VI pyramidal neurons [28, 38, 41], as well as in somatostatin [48] and VIP [71] interneurons recorded in slices. It is surprising, therefore, that no study has directly investigated so far the role of T channels in the activity of different cortical neurons during slow waves of non-REM sleep.
Because thalamic lesions do not suppress slow waves in anesthetized cats [85] and up and down states can be recorded in neocortical slices [17, 73] and in an isolated cortical gyrus in vivo during anesthesia [88], these EEG slow waves were originally viewed as a cortically generated rhythm [12, 14, 88]. however, up and down states and slow waves similar to those observed in vivo can be recorded in thalamic slices [10, 21, 43], and a recent study has conclusively shown that selective block of thalamic firing by tetrodotoxin markedly reduces the frequency of EEG slow waves both in anesthetized and naturally sleeping rats [26]. Thus, a dynamic interplay between the synaptically driven neocortical oscillator and the thalamic oscillators of slow waves is necessary for the full expression of these waves of natural non-REM sleep.
As far as the thalamic oscillators are concerned, many studies have clearly demonstrated that the T channels of TC and NRT neurons contribute to the expression of sleep slow waves in three ways. Firstly, by evoking the LTCP (with high-frequency burst of action potentials) that almost invariably marks the start of every up state (Fig. 2c). Secondly (as mentioned in the previous section), by providing the membrane potential bistability that underlies the up and down state transitions, i.e., up state = I Twindow “on” and down state = I Twindow “off” (Fig. 2c) [19, 22]. Thirdly, by providing the selective Ca2+ entry that is required to activate (1) the Ca2+-activated, non-selective cation current (I CAN), which tightly controls the durations of the up states [43] (Fig. 2c), and (2) the Ca2+-activated, K+ channels (SK type) that contribute to the hyperpolarization that follows an LTCP [23, 24] (for a comprehensive biophysical description of these mechanisms and a list of the other currents contributing to sleep slow waves in thalamic neurons, see ref [22]).
On the basis of all these data, one would expect slow waves of natural sleep to be compromised in the absence of thalamic T channels. Surprisingly, the original study in mice with global knockout of the Cav3.1 isoform of the T channels reported no change in EEG slow wave power [50]. Similar results were observed in mice with a “putative thalamic-selective” knockout of the same T channel isoform, though recombination was also present in some cortical regions and hypothalamic nuclei [4]. However, the negative results of these two studies cannot be simply interpreted as indicating a lack of involvement of the Cav3.1 isoform in slow waves since (1) compensation by other T channel isoforms or other voltage- and transmitter-gated channels might have occurred in the thalamus of these two types of KO mice, and(2) Cav3.1 T channels that are strongly expressed in brain areas other than the thalamus (see section below) were definitively knocked-out in these mice, with unpredictable consequences on slow waves and other sleep rhythms. Confirmation of an essential role for thalamic T channels in sleep slow waves has finally been provided by experiments where optogenetics and neuronal ensemble recordings were combined with localized thalamic microdialysis injections of the selective T channel antagonist TTA-P2 [26]. Thalamic dialysis concentration of TTA-P2 that fully blocks T channel mediated burst firing produces a consistent reduction in the frequency of slow waves during anesthesia and natural non-REM sleep (Fig. 2a, b). In addition, block of thalamic T channels suppresses the ability of selective optogenetic activation of TC neurons to entrain EEG slow waves [26]. These data, therefore, provide conclusive evidence on the essential role played by thalamic T channels in slow wave of non-REM sleep.
Thalamic and cortical T channel contribution to sleep spindles
A typical sleep spindle is a waxing and waning wave that lasts for a few seconds, has a frequency of 12–15 Hz in humans (but 8–12 Hz in rodents), and can occur in isolation from other sleep waves though it is mostly observed in close association with a K-complex (Fig. 3(A1, A2)) [3, 27, 62]. LTCPs are present at almost every cycle of the spindle wave in NRT neurons, and occasionally in TC neurones (Fig. 3(A3, A4)). The firing associated with the LTCPs generated by NRT neurons evokes GABAA IPSPs in TC neurons, some of which provide enough time- and voltage-dependent removal of T channel inactivation so that an LTCP (with or without the associated high-frequency burst) can then be generated. In turn, the LTCP-evoked firing of TC neurons elicits EPSPs in NRT neurons which help to trigger LTCPs at spindle frequency (Fig. 3(A3, A4)).

Contribution of thalamic T channels to sleep spindles and comparison of delta waves recorded in vivo and in vitro. A1 Filtered (5–15 Hz) (bottom trace) and unfiltered (upper trace) examples of a spindle recorded in a rat during natural sleep (graph shows the corresponding wavelet scalogram). Note the occurrence of the spindle immediately after a down state. A2 Spindles recorded in a cat during natural light sleep and anesthesia (top and bottom set of traces, respectively). Note the association with a K-complex in the boxed spindles. A3 Top trace: intracellular recording from a cat NRT neuron under barbiturate anesthesia shows rhythmic LTCPs superimposed on a depolarizing envelope during a spindle wave. Bottom trace: in contrast, in vitro intracellular activity from a ferret neuron during spindle-like oscillations recorded in a slice preparation revealed LTCPs of increasing amplitude followed by a progressively larger afterhyperpolarization. A4 In vivo intracellular recording from a cat TC neurone under barbiturate anesthesia shows that the NRT-evoked IPSPs occasionally give rise to LTCPs. The resulting increase in intracellular Ca2+ concentration can progressively activate I h, producing an increasing depolarizing drive that contributes to spindle wave termination. B1 EEG trace showing delta waves at 2.5–3.5 Hz in a naturally sleeping rat and corresponding wavelet scalogram. B2 In vitro recording of a TC neuron in an adult cat slice displays continuous oscillations at delta frequency upon hyperpolarizing DC current injection (top trace). In contrast, in vivo recordings in a cat TC neuron under ketamine/xylazine anesthesia clearly showed that delta waves occur as transient episodes during the down state of slow waves (bottom trace). A1–B2: reproduced with permission from [92], [70], [79], [26], [80]
In addition to the LTCPs, another key contribution of T channels to the electrical activity of TC neurons during sleep spindles is to provide the Ca2+ entry that regulates the cAMP-mediated upregulation of I h [56, 57]. It has been postulated that the potentiation of this inward current leads to a progressively larger depolarization of TC neurones during a spindle wave and ultimately to their inability to generate LTCPs, thus contributing to the spindle wave termination. A similar mechanism may occur in NRT neurons where the presence of HCN isoforms [63, 64] and I h [10, 72] has now been demonstrated. Other roles of T channels in NRT neurons during sleep spindles include the Ca2+ entry necessary to activate SK channels [24] and potentially I CAN [10].
This well-accepted mechanism of spindle wave generation based on the recruitment of both TC and NRT T channels has emerged from intracellular recordings in anesthetized animals (often after systemic injection of barbiturates to increase the occurrence of spindle waves) and in in vitro slice preparations. Therefore, caution should be used when interpreting these results obtained in such different experimental conditions and extrapolating this mechanism to natural sleep spindles. Indeed, spindle waves tend not to occur in association with a K-complex under barbiturate anesthesia, contrary to what is observed during ketamine/xylazine anesthesia and natural sleep. In addition, the LTCPs of NRT neurons in vivo during spindle waves emerge from a depolarizing envelope, whereas spindle-like activity in vitro consists of LTCPs of increasing amplitude, each followed by a progressively larger afterhyperpolarization (Fig. 3(A3)).
Unfortunately, data obtained so far in naturally sleeping T channel KO mice do not help to clarify the role of the different T channel isoforms in the sleep spindle. Thus, genetic knockout of Cav3.1 channels (which abolishes T current in all TC neurons) was originally reported to significantly decrease EEG power in the 8–10 Hz frequency band [50], although some remaining spindles showing a reduced amplitude were still present, whereas a later study by the same group reported no effect when spindle events were filtered at 6–15 Hz [51]. In Cav3.3 KO mice, no difference in the 10–12 Hz EEG power was observed when compared to wild-type animals [6]. Only when the analysis was restricted to periods of transitions from non-NREM to REM sleep (i.e., when sleep spindles are more prominent), a reduction of the EEG power in the 10–12-Hz frequency band measured about 30 s before REM sleep onset was observed [6]. Since in NRT neurons, LTCPs are observed at every cycle of spindle waves both in vivo [30, 39] and in vitro [92] (see Fig. 3(A3)), the absence of a clear effect of genetically deleting Cav3.3 channels (the main isoform present in NRT neurons) on this sleep rhythm is highly surprising. These contradictory results may once again be in part explained by compensatory mechanisms that occur in these T channel isoform KO mice but may also result from difficulties in clearly identifying spindle episodes in the EEG of naturally sleeping mice. Indeed, although local field potential recordings in deep cortical areas reveal a comparable profile of spindles in humans and mice, spindles are not clearly apparent in EEG traces from mice and their identification require sophisticated analysis to assess EEG spectral changes at the level of individual sleep stage transitions [5, 94]. Notwithstanding these contrasting results in transgenic mice, recent experiments in rats, using microdialysis of TTA-P2 in the somatosensory thalamus and NRT, provide conclusive evidence of a drastic decrease of spindle waves both during anesthesia and natural sleep [26], confirming the key role of thalamic T channels in the generation of this sleep rhythm.
Finally, since many cortical neurones possess a vast repertoire of T channels (see previous section) and the waveform of cortical spindle waves spans the voltage region of T channel activation/inactivation, a contribution of these cortical channels to the fine tuning of EEG sleep spindles would be expected, although to the best of our knowledge, this has not so far been rigorously investigated.
Thalamic and cortical T channel contribution to delta waves
In TC neurons, membrane potential oscillations at delta frequency (0.5–4 Hz) that consist of rhythmically occurring LTCPs were the first T channel-dependent activity whose mechanism was fully elucidated in vitro [53, 54, 61, 76]. This work, together with in vivo studies in anesthetized animals [33, 66, 67, 83], strongly suggested that delta oscillations in TC neurones are fully determined in a pacemaker fashion by the time and voltage dependencies of the h, T, and K+ channels, in both TC and NRT neurons [10, 54, 61] stressing the key role of thalamic T channels in the generation of these waves.
One important issue that has often been overlooked, however, is that the majority of EEG delta waves of natural sleep, and its thalamic counterpart the delta oscillation, do not occur in very long periods, as a somewhat inaccurate interpretation of the initial in vitro studies (Fig. 3(B2), top trace) might led to conclude. Indeed, thalamic delta oscillations appear to occur mostly in discrete groups during the down state of slow waves in both TC and NRT neurons (Fig. 3(B2), bottom trace) [43, 79, 90]. We are unaware of any evidence supporting the presence of long period of delta oscillations in TC neurons in vivo during natural sleep, raising the question of whether these long sequences of repetitive LTCPs are only observed in thalamic slices. An additional point of concern when interpreting EEG data is the ambiguity in the definition of delta waves of natural sleep and their potential overlap with slow waves. In other words, it is possible that EEG delta waves at the lower end of their frequency range (i.e., 0.5–2 Hz) may correspond to a thalamic (and cortical) cellular activity characterized by up and down states (i.e., slow waves) and not by repetitive LTCPs (compare Figs. 2(a) and 3(B1)).
Together with compensatory mechanisms (as highlighted in previous section), the above two issues might explain the contradictory results on delta waves obtained in mice with genetic ablation of T channels. Thus, total KO of the Cav3.1 isoform was shown to induce a marked decrease in the power density of delta frequency band (selected as 2–6.5 Hz) [50] whereas in the “putative thalamic-selective” Cav3.1 KO mouse, there is a moderate increase in EEG spectral power within the delta frequency range (selected as 1–4 Hz) [4]. Moreover, no change in delta frequency power was observed in global Cav3.3 KO mice [6], while the impact of deleting the Cav3.2 isoform on sleep rhythms has not yet been analyzed. To complicate this picture further, systemic injection of selective antagonists of all T channel isoforms has been shown to dose-dependently increase delta waves and reduce slow waves in wild-type mice and rats, respectively [26, 49].
As far as cortical T channels are concerned, the firing input at delta frequency from TC to cortical neurones might clearly play an important contribution to the expression of delta waves of natural sleep in the EEG. However, membrane potential oscillations at delta frequency have been observed in cortical neurones [77] and the full mechanisms of the delta waves (i.e., the relative contribution of synaptic and intrinsic conductances, including a precise role for T channels) in cortical neurons remains to be fully elucidated.
Contribution of T channels in other brain regions to the expression of sleep waves
The difficulty in the interpretation of sleep studies using global, genetic, or pharmacological T channel block that we have outlined in the previous sections clearly stems from our lack of knowledge of the contribution of T channels not only in cortical neurons but also in other brain networks that control and/or modulate natural sleep. In fact, neurons expressing LTCPs are present in the ventrolateral preoptic nucleus [40] and in the lateral hypothalamus (i.e., hypocretin-orexin-expressing neurons) [35], two strongly interacting regions that belong, respectively, to the sleep-promoting and ascending arousal pathways [74] (Fig. 4). The potential presence of T channels mediated electrical events (e.g. LTCPs, I Twindow, T channel-dependent Ca2+ modulation or activation of various voltage-gated channels, etc.) in neurons of these pathways should therefore be investigated to gain a full understanding of the role of T channels in sleep. In this respect, it is clear that manipulating T channels not only has an impact on various sleep rhythms but also on sleep architecture and the transitions between wake and sleep as well as between non-REM and REM sleep. Both global and “putative thalamic-selective” Cav3.1 KO mice (but not cortical KO mice) show an increased number of frequent brief awakenings that interrupt non-REM sleep and a delayed sleep onset [4, 50]. Moreover, systemic injection of TTA-A2/P2 in wild-type mice acutely reduces the mean time spent in active wake [49] and induces dose-dependent behavioral and EEG changes indicative of sedation/sleep in rats [59].

Interconnected brain areas involved in sleep control and EEG sleep waves generation that present T-type dependent activity. Example of LTCPs evoked from hyperpolarized membrane potentials in response to depolarizing current injections in layer V/VI cortical neuron (1), TC (2) and NRT (3) neurons, ventrolateral preoptic neuron (VLPO, 4) and hypocretin-orexin expressing neurons (hcrt/orx, 5) of the lateral hypothalamus. Both tonic firing and LTCP are presented in 1, 4, and 5. Traces reproduce with permission from [28], [34], [40], [25]
Conclusions
After 30 years since the discovery of T channels in thalamic neurons, we have a very comprehensive view of their precise contribution to slow waves and spindles of natural sleep, including the role of LTCPs and I Twindow. A similar understanding of delta waves is still missing, in part because of lack of appropriate studies and uncertainties on an appropriate classification of these waves. Moreover, we still know very little about the contribution of T channels in neurons of the neocortex and other neuronal networks involved in sleep. Undoubtedly, the development of selective blockers for different T channel isoforms and of conditional and area-selective KO animals will contribute to unravel the full role played by these widely expressed voltage-gated channels in sleep waves and architecture.
References
Achermann P, Borbely AA (1997) Low-frequency (< 1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience 81:213–222
Amzica F, Steriade M (1997) The K-complex: its slow (<1-Hz) rhythmicity and relation to delta waves. Neurology 49:952–959
Amzica F, Steriade M (2002) The functional significance of K-complexes. Sleep Med Rev 6:139–149
Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S (2005) Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A 102:1743–1748
Astori S, Wimmer RD, Luthi A (2013) Manipulating sleep spindles—expanding views on sleep, memory, and disease. Trends Neurosci 36:738–748
Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Luthi A (2011) The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A 108:13823–13828
Bal T, von Krosigk M, McCormick DA (1995) Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol Lond 483:665–685
Bal T, von Krosigk M, McCormick DA (1995) Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J Physiol Lond 483:641–663
Bessaih T, Leresche N, Lambert RC (2008) T current potentiation increases the occurrence and temporal fidelity of synaptically evoked burst firing in sensory thalamic neurons. Proc Natl Acad Sci U S A 105:11376–11381
Blethyn KL, Hughes SW, Tóth TI, Cope DW, Crunelli V (2006) Neuronal basis of the slow (<1Hz) oscillation in neurons of the nucleus reticularis thalami in vitro. J Neurosci 26:2474–2486
Bossu JL, Feltz A (1986) Inactivation of the low-threshold transient calcium current in rat sensory neurones: evidence for a dual process. J Physiol 376:341–357
Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW (2012) Control of sleep and wakefulness. Physiol Rev 92:1087–1187
Carbone E, Lux HD (1984) A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310:501–502
Chauvette S, Crochet S, Volgushev M, Timofeev I (2011) Properties of slow oscillation during slow-wave sleep and anesthesia in cats. J Neurosci Off J Soc Neurosci 31:14998–15008
Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P (2002) Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol 540:3–14
Contreras D, Steriade M (1995) Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci 15:604–622
Cossart R, Aronov D, Yuste R (2003) Attractor dynamics of network UP states in the neocortex. Nature 423:283–288
Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, Perez-Reyes E (1998) Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res 83:103–109
Crunelli V, Blethyn KL, Cope DW, Hughes SW, Parri HR, Turner JP, Toth TI, Williams SR (2002) Novel neuronal and astrocytic mechanisms in thalamocortical loop dynamics. Philos Trans R Soc London B-Biol Sci 357:1675–1693
Crunelli V, Cope DW, Hughes SW (2006) Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium 40:175–190
Crunelli V, Hughes SW (2010) The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci 13:9–17
Crunelli V, Toth TI, Cope DW, Blethyn K, Hughes SW (2005) The 'window' T-type calcium current in brain dynamics of different behavioural states. J Physiol 562:121–129
Cueni L, Canepari M, Adelman JP, Luthi A (2009) Ca(2+) signaling by T-type Ca(2+) channels in neurons. Arch Eur J Physiol 457:1161–1172
Cueni L, Canepari M, Lujan R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Luthi A (2008) T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci 11:683–692
Cvetkovic-Lopes V, Eggermann E, Uschakov A, Grivel J, Bayer L, Jones BE, Serafin M, Muhlethaler M (2010) Rat hypocretin/orexin neurons are maintained in a depolarized state by TRPC channels. PLoS ONE 5:e15673
David F, Schmiedt JT, Taylor HL, Orban G, Di Giovanni G, Uebele VN, Renger JJ, Lambert RC, Leresche N, Crunelli V (2013) Essential thalamic contribution to slow waves of natural sleep. J Neurosci Off J Soc Neurosci 33:19599–19610
De Gennaro L, Ferrara M (2003) Sleep spindles: an overview. Sleep Med Rev 7:423–440
de la Pena E, Geijo-Barrientos E (1996) Laminar localization, morphology, and physiological properties of pyramidal neurons that have the low-threshold calcium current in the guinea-pig medial frontal cortex. J Neurosci Off J Soc Neurosci 16:5301–5311
Deleuze C, David F, Behuret S, Sadoc G, Shin HS, Uebele VN, Renger JJ, Lambert RC, Leresche N, Bal T (2012) T-type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci Off J Soc Neurosci 32:12228–12236
Deschenes M, Paradis M, Roy JP, Steriade M (1984) Electrophysiology of neurons of lateral thalamic nuclei in cat: resting properties and burst discharges. J Neurophysiol 51:1196–1219
Destexhe A, Neubig M, Ulrich D, Huguenard J (1998) Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci 18:3574–3588
Domich L, Oakson G, Steriade M (1986) Thalamic burst patterns in the naturally sleeping cat: a comparison between cortically projecting and reticularis neurones. J Physiol Lond 379:429–449
Dossi RC, Nunez A, Steriade M (1992) Electrophysiology of a slow (0.5-4 Hz) intrinsic oscillation of cat thalamocortical neurones in vivo. J Physiol Lond 447:215–234
Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N (2010) Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J Neurosci Off J Soc Neurosci 30:99–109
Eggermann E, Bayer L, Serafin M, Saint-Mleux B, Bernheim L, Machard D, Jones BE, Muhlethaler M (2003) The wake-promoting hypocretin-orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci Off J Soc Neurosci 23:1557–1562
Fedulova SA, Kostyuk PG, Veselovsky NS (1985) Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones. J Physiol 359:431–446
Ferri R, Rundo F, Bruni O, Terzano MG, Stam CJ (2005) Dynamics of the EEG slow-wave synchronization during sleep. Clin Neurophysiol Off J Int Fed Clin Neurophysiol 116:2783–2795
Friedman A, Gutnick MJ (1987) Low-threshold calcium electrogenesis in neocortical neurons. Neurosci Lett 81:117–122
Fuentealba P, Timofeev I, Steriade M (2004) Prolonged hyperpolarizing potentials precede spindle oscillations in the thalamic reticular nucleus. Proc Natl Acad Sci U S A 101:9816–9821
Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, Audinat E, Muhlethaler M, Serafin M (2000) Identification of sleep-promoting neurons in vitro. Nature 404:992–995
Hamill OP, Huguenard JR, Prince DA (1991) Patch-clamp studies of voltage-gated currents in identified neurons of the rat cerebral cortex. Cereb Cortex 1:48–61
Hirsch JC, Fourment A, Marc ME (1983) Sleep-related variations of membrane potential in the lateral geniculate body relay neurons of the cat. Brain Res 259:308–312
Hughes SW, Cope DW, Blethyn KL, Crunelli V (2002) Cellular mechanisms of the slow (1 Hz) oscillation in thalamocortical neurons in vitro. Neuron 33:947–958
Hughes SW, Cope DW, Toth TI, Williams SR, Crunelli V (1999) All thalamocortical neurones possess a T-type Ca2+ 'window' current that enables the expression of bistability-mediated activities. J Physiol Lond 517:805–815
Huguenard JR, Prince DA (1992) A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci 12:3804–17
Jahnsen H, Llinas R (1984) Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol 349:205–26
Jahnsen H, Llinas R (1984) Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol 349:227–47
Kawaguchi Y, Kubota Y (1993) Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol 70:387–396
Kraus RL, Li Y, Gregan Y, Gotter AL, Uebele VN, Fox SV, Doran SM, Barrow JC, Yang ZQ, Reger TS, Koblan KS, Renger JJ (2010) In vitro characterization of T-type calcium channel antagonist TTA-A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther 335:409–417
Lee J, Kim D, Shin HS (2004) Lack of delta waves and sleep disturbances during non-rapid eye movement sleep in mice lacking alpha1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A 101:18195–18199
Lee J, Song K, Lee K, Hong J, Lee H, Chae S, Cheong E, Shin HS (2013) Sleep spindles are generated in the absence of T-type calcium channel-mediated low-threshold burst firing of thalamocortical neurons. Proc Natl Acad Sci U S A 110:20266–20271
Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E (1999) Cloning and expression of a novel member of the low voltage-activated T- type calcium channel family. J Neurosci 19:1912–1921
Leresche N, Jassik-Gerschenfeld D, Haby M, Soltesz I, Crunelli V (1990) Pacemaker-like and other types of spontaneous membrane potential oscillations of thalamocortical cells. Neurosci Lett 113:72–77
Leresche N, Lightowler S, Soltesz I, Jassik-Gerschenfeld D, Crunelli V (1991) Low-frequency oscillatory activities intrinsic to rat and cat thalamocortical cells. J Physiol Lond 441:155–174
Llinas R, Jahnsen H (1982) Electrophysiology of mammalian thalamic neurones in vitro. Nature 297:406–408
Lüthi A, McCormick DA (1998) Periodicity of thalamic synchronized oscillations: the role of Ca2+- mediated upregulation of Ih. Neuron 20:553–563
Lüthi A, McCormick DA (1999) Modulation of a pacemaker current through Ca(2+)-induced stimulation of cAMP production. Nat Neurosci 2:634–641
Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G (2004) The sleep slow oscillation as a traveling wave. J Neurosci Off J Soc Neurosci 24:6862–6870
McCafferty C, David F, Venzi M, Di Giovanni G, Orban G, Uebele VN, Renger JJ, Lambert RC, Leresche N, V C (2012) T-type calcium channels of cortical and thalamocortical neurons are necessary for absence seizures.Society for Neuroscience. New Orleans USA
McCormick DA (1992) Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol 39:337–388
McCormick DA, Pape HC (1990) Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol Lond 431:291–318
Molle M, Marshall L, Gais S, Born J (2002) Grouping of spindle activity during slow oscillations in human non-rapid eye movement sleep. J Neurosci Off J Soc Neurosci 22:10941–10947
Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ (2000) Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res 81:129–139
Notomi T, Shigemoto R (2004) Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol 471:241–276
Nowycky MC, Fox AP, Tsien RW (1985) Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316:440–443
Nunez A, Amzica F, Steriade M (1992) Intrinsic and synaptically generated delta (1-4 Hz) rhythms in dorsal lateral geniculate neurons and their modulation by light-induced fast (30-70 Hz) events. Neuroscience 51:269–284
Nunez A, Curro Dossi R, Contreras D, Steriade M (1992) Intracellular evidence for incompatibility between spindle and delta oscillations in thalamocortical neurons of cat. Neuroscience 48:75–85
Perez-Reyes E (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83:117–161
Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH (1998) Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 391:896–900
Pirchio M, Turner JP, Williams SR, Asprodini E, Crunelli V (1997) Postnatal development of membrane properties and delta oscillations in thalamocortical neurons of the cat dorsal lateral geniculate nucleus. J Neurosci 17:5428–5444
Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E (1999) Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci Off J Soc Neurosci 19:5228–5235
Rateau Y, Ropert N (2006) Expression of a functional hyperpolarization-activated current (Ih) in the mouse nucleus reticularis thalami. J Neurophysiol 95:3073–3085
Sanchez-Vives MV, McCormick DA (2000) Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci 3:1027–1034
Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010) Sleep state switching. Neuron 68:1023–1042
Simon NR, Manshanden I, da Silva FH L (2000) A MEG study of sleep. Brain Res 860:64–76
Soltesz I, Lightowler S, Leresche N, Jassik-Gerschenfeld D, Pollard CE, Crunelli V (1991) Two inward currents and the transformation of low-frequency oscillations of rat and cat thalamocortical cells. J Physiol Lond 441:175–197
Steriade M (2003) The corticothalamic system in sleep. Front Biosci J Virtual Libr 8:d878–d899
Steriade M, Amzica F, Nunez A (1993) Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. J Neurophysiol 70:1385–1400
Steriade M, Contreras D, Curro Dossi R, Nunez A (1993) The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci 13:3284–3299
Steriade M, Deschênes M (1998) Cellular thalamic mechanisms. Elsevier, Amsterdam
Steriade M, Deschenes M, Domich L, Mulle C (1985) Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol 54:1473–1497
Steriade M, Domich L, Oakson G, Deschenes M (1987) The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol 57:260–273
Steriade M, Dossi RC, Nunez A (1991) Network modulation of a slow intrinsic oscillation of cat thalamocortical neurons implicated in sleep delta waves: cortically induced synchronization and brainstem cholinergic suppression. J Neurosci 11:3200–3217
Steriade M, McCormick DA, Sejnowski TJ (1993) Thalamocortical oscillations in the sleeping and aroused brain. Science 262:679–685
Steriade M, Nunez A, Amzica F (1993) Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci 13:3266–3283
Steriade M, Nunez A, Amzica F (1993) A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci 13:3252–3265
Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA (1999) Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19:1895–1911
Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M (2000) Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex 10:1185–1199
Timofeev I, Grenier F, Steriade M (2001) Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc Natl Acad Sci U S A 98:1924–1929
Timofeev I, Steriade M (1996) Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J Neurophysiol 76:4152–4168
Toth TI, Hughes SW, Crunelli V (1998) Analysis and biophysical interpretation of bistable behaviour in thalamocortical neurons. Neuroscience 87:519–523
von Krosigk M, Bal T, McCormick DA (1993) Cellular mechanisms of a synchronized oscillation in the thalamus. Science 261:361–364
Williams SR, Toth TI, Turner JP, Hughes SW, Crunelli V (1997) The 'window' component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol Lond 505:689–705
Wimmer RD, Astori S, Bond CT, Rovo Z, Chatton JY, Adelman JP, Franken P, Luthi A (2012) Sustaining sleep spindles through enhanced SK2-channel activity consolidates sleep and elevates arousal threshold. J Neurosci Off J Soc Neurosci 32:13917–13928
Acknowledgments
VC work in this field is supported by The Wellcome Trust (grant 91882); NL and RCL work by Agence Nationale de la Recherche (grant MNMP-2009) and CNRS (LEA 528).
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is published as part of the special issue on T-type channels.
Rights and permissions
About this article
Cite this article
Crunelli, V., David, F., Leresche, N. et al. Role for T-type Ca2+ channels in sleep waves. Pflugers Arch - Eur J Physiol 466, 735–745 (2014). https://doi.org/10.1007/s00424-014-1477-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-014-1477-3