
Abstract
NCX proteins explore the electrochemical gradient of Na+ to mediate Ca2+-fluxes in exchange with Na+ either in the Ca2+-efflux (forward) or Ca2+-influx (reverse) mode, whereas the directionality depends on ionic concentrations and membrane potential. Mammalian NCX variants (NCX1-3) and their splice variants are expressed in a tissue-specific manner to modulate the heartbeat rate and contractile force, the brain’s long-term potentiation and learning, blood pressure, renal Ca2+ reabsorption, the immune response, neurotransmitter and insulin secretion, apoptosis and proliferation, mitochondrial bioenergetics, etc. Although the forward mode of NCX represents a major physiological module, a transient reversal of NCX may contribute to EC-coupling, vascular constriction, and synaptic transmission. Notably, the reverse mode of NCX becomes predominant in pathological settings. Since the expression levels of NCX variants are disease-related, the selective pharmacological targeting of tissue-specific NCX variants could be beneficial, thereby representing a challenge. Recent structural and biophysical studies revealed a common module for decoding the Ca2+-induced allosteric signal in eukaryotic NCX variants, although the phenotype variances in response to regulatory Ca2+ remain unclear. The breakthrough discovery of the archaebacterial NCX structure may serve as a template for eukaryotic NCX, although the turnover rates of the transport cycle may differ ∼103-fold among NCX variants to fulfill the physiological demands for the Ca2+ flux rates. Further elucidation of ion-transport and regulatory mechanisms may lead to selective pharmacological targeting of NCX variants under disease conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Typical hallmarks of SLC8 (NCX) transporters
Genes, alternative splicing, and tissue-specific expression of NCX variants
The Na+/Ca2+ exchanger proteins represent an antiporter system that utilizes the electrochemical gradient of Na+ to catalyze Ca2+-extrusion from the cytosol or organelle matrix [15, 78, 125]. The SLC8 (NCX) gene family is one of five families belonging to the CaCA (Ca2+/cation antiporter) superfamily [26, 94, 125]. The phylogenetic tree of the SLC8 gene family and its place in the CaCA superfamily has been extensively reviewed [94, 115, 116] and will not be covered here. Briefly, members of the CaCA superfamily share similar topology, comprising two clusters; each cluster contains five or six transmembrane helices (TM), and two clusters are joined by a cytoplasmic loop of varying lengths [91, 94, 110, 125, 151, 154]. The CaCA proteins possess a conserved sequence motif in each cluster (α1 and α2 segments), which are involved in the ion transport events [91, 94, 125, 151].
The recently discovered crystal structure of archaebacterial Methanococcus jannaschii (NCX_Mj) represents a long-wanted breakthrough [91] and offers new opportunities for systematically testing the specific molecular mechanisms underlying the alternating access in the antiport system. The molecular weight of eukaryotic NCX proteins (typically containing 930–970 amino acids) is more than three times higher than that of archaebacterial NCX_Mj, which contains “only” 301 amino acid residues [91]. Although the crystal structure of eukaryotic NCX is still unavailable, recent evidence suggests that the total number of helices as well as their packing in the membrane might be quite similar in eukaryotic and archaebacterial NCX [91, 132], meaning that the size differences among phylogenetically distant NCXs are due to regulatory domains associated with eukaryotic NCX proteins (Fig. 1).

NCX structure. Archaebacterial and eukaryotic NCX contain ten trans-membrane helices (TM1-10). The cytosolic regulatory f-loop (∼500 amino acids) contains two Ca2+-binding CBD domains, CBD1 and CBD2, which are connected with the TM-5 and TM-6, respectively, though the long linkers. The auto-inhibitory segment, XIP (20 amino acids), is connected to the C-terminal of TM-5. The interdomain Ca2+-sensing module at the interface of the two CBD domains involves two amino acids on CBD1 (Asp499 and Asp500) and CBD2 (Arg532 and Asp565). Occupation of Ca3-Ca4 sites by Ca2+ may shift a dynamic equilibrium of disorder-to-order transition (population shift) due to charge neutralization and coordination, thereby constraining conformational freedom of the two-domain CBD tandem, rigidifying the NCX1 f-loop, and triggering allosteric signal transmission to the membrane domains. The crystal structure of NCX_Mj reveals an ion-binding cluster with four ion-binding sites, one for Ca2+ (SCa), and three for Na+ (Sext, Smid, Sint), posed in a diamond-shaped pattern. Twelve amino acid residues contribute to ligation of ions, whereas Asp240, Glu213, and Glu54 play a critical role in coordinating and transport of ions
Mammals express the SLC8A1 (NCX1), SLC8A2 (NCX2), and SLC8A3 (NCX3) genes; NCX1 and NCX3 undergo alternative splicing [94, 107, 125]. Recent evidence indicates that NCLX protein (Slc8b1), previously assigned to the NCKX family (the SLC24 gene family mediating the Na+/Ca2+ exchange coupled with the K+ co-transport), is a long-wanted mitochondrial Na+/Ca2+ exchanger [122] owing to a unique feature that enables it to transport either the Na+ or Li+ ion (but not K+) in exchange for Ca2+. The properties of NCKX [140] and NCLX [24, 121] were recently summarized and will not be discussed here. Three mammalian genes (NCX1-3) and their splice variants are expressed in a tissue-specific manner to mediate Ca2+ fluxes in many cell types [93, 125]. Namely, NCX2 and NCX3 have been found in the brain and skeletal muscles, whereas NCX1 is universally disseminated. At least 17 NCX1 and 5 NCX3 proteins are produced through alternative splicing, whereas no splice variants of NCX2 have been yet identified. The splice variants arise from a combination of six small exons (A, B, C, D, E, and F), whereas a mutually exclusive exon (either A or B) appears in each given splice-variant [86, 89]. Interestingly, the cardiac and neuronal variants contain exon A, whereas the kidney, stomach, and skeletal muscle variants include exon B (Fig. 2). The structural organization of A-F exons has physiological relevance. The splice-variant segment is exclusively allocated within the CBD2 domain [57] and modifies the response kinetics, dynamic range, and affinity of allosteric sensors [43, 47], thereby exhibiting positive, negative, or no response to regulatory Ca2+ [18–22]. For example, Ca2+ activates the brain (AD), cardiac (ACDEF), and kidney (BD) variants, although the Ca2+-induced relieve of Na+-dependent inactivation is observed only in the cardiac and brain variants [38, 39]. In contrast, the regulatory Ca2+ inhibits Drosophila NCX (CALX1.1), but it has no effect on the CALX1.2 variant [66, 114].

Splice variants of NCX. The splice segment is exclusively located on CBD2. Tissue-specific splice variants arise from a combination of six small exons A, B, C, D, E, and F, whereas a mutually exclusive exon (either A or B) shows up in every splice variant. The NMR structures of CBD1 (PDB 2FWS, orange), CBD2-AD (PDB 2FWU, red), and CBD2-BD (PDB code 2KLT, green) structures were superimposed on the template of the CBD12-E454K crystal structure (PBD 3US9) to show the position of the splice segment. Residues encoded by exons A and B are shown in blue and residues encoded by exon D are cyan
Ion-transport mechanisms and alternating access
NCX proteins represent a carrier-type “exchange-only” mechanism (otherwise known as antiporter or counter transport system) [79, 82, 85, 143], where a transmembrane electrochemical gradient of Na+ is utilized to couple stoichiometric and electrogenic ion exchange (3Na+:Ca2+) for each transport cycle [15, 130]. According to the unifying concept of an alternating access mechanism [41, 143], NCX might undergo a major conformational change involving an alternative exposure of transport sites to extracellular and intracellular sides of the membrane, where the ion binding to the transport-site pocket must promote a major conformational change, associated with alternative access [41, 78, 143]. The NCX proteins can mediate the partial reactions and the Na+/Na+ and Ca2+/Ca2+ exchanges as a part of the Na+/Ca2+ exchange cycle, although the physiological relevance of these partial reactions is unclear [15, 78, 79].
Kinetic studies reveal that the Na+/Ca2+ exchange cycle can be described as separate movements of 3Na+ and Ca2+ ions through the exchanger [80, 109], whereas the Na+-translocation accounts for a voltage-sensitive step of the exchange cycle [63, 81–83, 85]. Consistent with this, the recently derived crystal structure of archaebacterial NCX_Mj revealed three binding sites for Na+ and one binding site for Ca2+, where the simultaneous occupation of all four binding sites by 3Na+ and Ca2+ ions is thermodynamically forbidden [91]. Since the ion-transport pathways of archaebacterial and eukaryotic NCX proteins show striking similarities [75, 91, 132], phylogenetically distant NCX orthologs might share common mechanisms to drive alternative access. Electrostatic interactions of 3Na+ and Ca2+ ions with the ion-binding pocket of NCX might play a crucial role in generating critical transitions along the transport cycle [41, 88, 91]. Kinetic observations are consistent with the notion that the unloaded ion-binding domain bears two negative charges (z = −2), meaning that the ground-state E.Na3 species are positively charged (and voltage-sensitive) and the E.Ca species are electroneutral [81–85]. This working hypothesis is very attractive to test in light of the newly derived X-ray structure of NCX_Mj [91].
Turnover rates of the ion-transport cycle in phylogenetically distant NCX variants
It is widely accepted that mammalian NCX variants represent a “high-capacity/low-affinity” system capable of rapidly extruding large amounts of cytosolic Ca2+ in a relatively short time in order to maintain the proper dynamic balance between the Ca2+ entry and exit modes (e.g., the size of the Ca2+ transient amplitude and its decay kinetics in cardiomyocytes) [8, 9]. Mammalian NCX proteins are low-abundant proteins (<0.1 % of the total plasma membrane protein) owing to the high rates (2,500–5,000 s−1) of single-cycle turnover either in intact cardiomyocytes [109], excised patches of cardiomyocytes [63], or isolated sarcolemma vesicles [6]. These high rates of ion transport are compatible with the site density of 250–2,500 exchangers/μm2 in cardiomyocytes, corresponding to ∼106–107 copies of NCX per cell [6, 63, 106, 109]. Interestingly, there are at least 50-fold differences in the turnover rates among NCKX proteins [33, 136, 140]. Moreover, taking into account the protein expression levels and the ion flux activities of NCX_Mj, the calculated turnover rate of the transport cycle might be at least 103 times slower in archaebacterial NCX_Mj than in mammalian NCX. More extensive experimentation is required for quantifying the turnover rate of a single transport cycle in NCX_Mj and the relevant kinetic mechanisms governing its catalytic capacity. This issue is especially interesting in light of the striking structural similarities in ion transport pathways owing to the mammalian and archaebacterial NCX variants [75, 91, 132]. Again, these differences in the catalytic capacity may be physiologically relevant for mammalian NCX proteins since these proteins are “committed” to rapidly extrude the cytosolic Ca2+ from the cell within a limited time slot in order to fulfill the physiological demands of functionally diverse cell types.
The directionality of NCX-mediated Ca2+ flux
The Na+/Ca2+ exchange can occur either in the forward (Ca2+-efflux) or reverse (Ca2+-influx) mode. At constant RT/F (∼25 mV), the directionality of net Ca2+ flux depends on cytosolic and extracellular [Na+] and [Ca2+] and on the membrane potential [8–10, 15]. At fixed levels of extracellular [Na+]o and [Ca2+]o and resting membrane potential (E m = −80 mV), the electrochemical driving force for electrogenic (3Na+/Ca2+) exchange is E NCX = −32 mV.
Thus, at resting E m, the Ca2+-efflux is a thermodynamically favored mode (E NCX > E m), where the Na+/Ca2+ exchange changes the directionality when E NCX and E m become equal. The dynamic swings in [Na+]i, [Ca2+]i, and in membrane potential (e.g., in cardiomyocytes) modify the thermodynamic driving force, which actually governs the directionality of the net Ca2+ movements [8, 15]. Therefore, 10–20-fold changes in cytosolic [Ca2+]i and varying the membrane potential (from −90 mV to +50 mV) in ventricular cadiomyocytes modify the thermodynamic driving force (E NCX) and thus, the directionality of the Na+/Ca2+ exchange along the action potential [8, 9]. Moreover, at fixed extracellular [Na+] levels, even twofold changes in cytosolic [Na+]i can significantly affect the E NCX profile (and thus, the directionality) along the action potential since, according to the equation (see above), the changes in [Na]i are powered in the third degree. Although the Ca2+-efflux is a predominant mode of NCX under default conditions, the reverse mode (Ca2+-influx) may become functionally important under both physiological and pathophysiological conditions [10, 15, 79]. For example, in cardiac disease, a prolonged APD (action potential duration) and elevated [Na+]i promote Ca2+-entry through the reverse mode of NCX, thereby mediating life-threatening arrhythmogenic currents [8–10, 15].
NCX interaction with specific ion-transport systems
The coupling of the reverse mode NCX with specific ion-transport systems may mediate the net fluxes of Na+ and/or Ca2+, which may effectively integrate and regulate important physiological events on the cellular and systemic levels [105, 126, 137, 155]. Notably, the physiological relevance of the reverse mode NCX has been controversial for many years, but the contribution of NCX-mediated Ca2+-influx in EC-coupling, vascular constriction, and synaptic transmission has been underscored in recent publications [120, 126, 137]. According to these reports, the Ca2+-influx through the NCX protein is coupled with the Na+-efflux either through the voltage-sensitive or store-operated Na+ conducting channels, where a rapid increase in [Na+]i within the restricted space of a putative “Na+ microdomain” drives the NCX-mediated Ca2+-entry into the cell. More specifically, in smooth muscle cells, the Ca2+-influx through NCX1 can be coupled with Na+ transport via store-operated channels, TRPC6 and Orai1, where TRPC6, Orai1, and NCX1 are colocalized with α-2 Na+/K+ ATPases in the plasma membrane clusters near junctional SR [16, 126, 155–158]. The open question is: How do microdomains govern the net fluxes of Na+ and/or Ca2+ from a channel or exchanger (the source) to its target transducer or pump (the sink), while preventing “loss” of ions to the bulk cytosol? [126, 155]. To address this question, one needs detailed information about the architecture of the plasma membrane–SR junction including the position and site-density of relevant ion-transport systems as well as knowledge of the physical factors (e.g., viscosity) limiting the ion diffusion nearby the membrane space.
“Secondary” (kinetic) modulation of NCX
Kinetic modulation of NCX is mediated by interaction of cytosolic Ca2+, Na+, and H+ ions with sites that are not directly involved in Ca2+ and Na+ transport (translocation), thereby representing “secondary” or allosteric regulation of NCX [35–38, 59–63, 100]. Owing to hefty and speedy swings in cytosolic [Ca2+] during the action potential, the allosteric activation of NCX by Ca2+ is especially important in excitable tissues [8–10, 131]. For example, in cardiomyocytes, NCX-mediated ion-currents are elevated ∼25-fold when the cytosolic [Ca2+]i level rises from 0.1 to 2 μM, showing that the level is highly dependent on [Ca2+]i with an unusually high degree of cooperativity [21, 49]. Rapid removal of cytosolic Ca2+ in patch-clamp experiments results in slow inactivation (I2 state) of NCX, thereby exhibiting hallmark kinetic differences among tissue-specific NCX variants, which perhaps have physiological relevance [65, 100, 131]. Recent studies demonstrated that high-affinity Ca3–Ca4 sites on the CBD1 domain represent a primary Ca2+ sensor [27, 28, 118, 119], whereas the specific interdomain interactions between the two CBD domains result in slow dissociation of “occluded” Ca2+ from Ca3–Ca3 sites [44–48]. These interdomain interactions are “secondarily” modified by a tissue-specific splice segment located on CBD2 [43, 44], which may represent tissue-specific differences in slow inactivation (I2) of matching NCX variants [38, 39].
In patch-clamp experiments, a rise in cytosolic [Na+] rapidly stimulates eukaryotic NCX (due to Na+ interaction to transport sites), followed by hallmark slow inactivation of the exchanger, which levels off at certain steady-state levels of ion current (I1 inactivation state) [59–62]. The amplitude and kinetics of I1 inactivation characteristically differ among NCX variants [38, 39], whereas the elevation of cytosolic [Ca2+] relieves Na+-dependent inactivation [59–62, 65]. The Na+-dependent inactivation of NCX cannot be explained by direct interaction of Na+ with Ca2+ binding sites on CBD domains, because Na+ has no effect on Ca2+ binding to these regulatory sites in the isolated preparations of CBD1, CBD2, or CBD12 [21, 23, 44, 47]. It was suggested that the “regulatory Na+ site” may be located on the “catenin-like domain” at the terminals of the cytosolic f-loop [57], but there are no data that support this. Most probably, the Na+-dependent inactivation involves the ion-transport sites and the XIP region, as originally suggested by Hilgemann and collaborators [59–62, 65, 131], although the underlying structure–activity relationships remain unclear. Anyway, Ca2+ binding to the CaI site of CBD2 relieves Na+-dependent inactivation [27, 28, 118, 119], although it is presently unclear how the allosteric signal is transmitted from CBD2 to transport sites.
Eukaryotic NCX is extremely sensitive to mild cytosolic acidification (a pH decrease from 7.2 to 6.9 results in nearly 90 % inactivation of NCX), thereby emphasizing the physiological relevance of NCX “proton block” under acidosis and ischemia conditions [35–37]. In general, protons may interact with transport and/or regulatory domains, although there is no evidence that within the physiological range of pH the protons affect the ion binding affinity and/or transport rates. According to Doering and Lederer, there are two components that contribute to proton inhibition of NCX in patch-clamp experiments, one of which is fast but the other is slow and requires cytoplasmic Na+ [36, 37]. In isolated preparations of CBDs, Na+ has no effect on the competition of Ca2+ and H+ for Ca2+-binding to CBD12 [21, 23, 44], which is consistent with Na-independent proton block of NCX. The mechanism underlying Na-dependent proton-inhibition of NCX remains unclear. Most probably, the protonation of CBD sites prevents the Ca2+-dependent displacement of inhibitory Na+ from the transport sites, since protons can effectively compete for Ca2+ binding to CBDs, thereby causing a dramatic shift in [Ca]i-dependent activation of NCX in cardiomyocytes [21]. Thus, CBDs may function as a dual Ca2+/pH sensor whereas, under acidosis/ischemia conditions, the interaction of protons with CBDs decreases Ca2+ affinity for allosteric activation and thus shuts down the NCX activation to prevent NCX-mediated arrhythmogenic ion-currents [21].
Structure–function relationships
NCX_Mj as a template structure for the CaCA superfamily
This structure describes the outward-facing (extracellular) conformation of NCX_Mj with ten trans-membrane helices, TM1-10 (Fig. 1). The ion-binding pocket contains four ion-binding sites: one for Ca2+ (SCa), and three for Na+ (Sext, Smid, Sint), arranged in a diamond-shaped configuration. Two apparent passageways allow separate access for Na+ and Ca2+ ions to a central ion-binding pocket, where 12 residues contribute to Na+ and Ca2+ ligation (four in TM2 and TM7, and two in TM3 and TM8). Similar passageways for ion access and ion-pockets might exist in NCX_Mj [91], mammalian NCX [75, 132], and NCKX [2, 140] proteins, although the specific structural differences might be responsible for the differences in the ion-binding selectivity and turnover rates among the related proteins.
In contrast to previous argumentations, recent experimental approaches imply a ten-helix structure with a similar packing of TMs and ion-transport pathways in the archaebacterial and eukaryotic NCX variants [75, 132]. Therefore, the basic mechanisms underlying ion transport might be similar in phylogenetically distant NCX variants, but the catalytic capacity or regulation can widely differ among NCX orthologs. This tentative conclusion seems to be encouraging, since a better understanding of the basic ion transport mechanisms in NCX_Mj might help in elucidating physiologically more relevant features of mammalian NCX proteins such as the dynamic features of regulation and pharmacological targeting of tissue-specific NCX variants under altered conditions.
In sharp contrast with eukaryotic NCX, the cytosolic loop between TM5 and TM6 is extremely short in NCX_Mj, meaning that this loop cannot be a prototype for a large cytosolic f-loop (∼500 amino acids) of eukaryotic NCX bearing the regulatory CBD domains (Fig. 1). Most importantly, eight helices of NCX_Mj (TM2-5 and TM7-10) generate a tightly packed hub (which is perpendicularly inserted into the membrane), whereas two long and tilted helices (TM1 and TM6) are limply packed in front of a rigid eight-helix core [91]. This structure originally predicted the sliding of TM1 and TM6 toward the rigid core helices, which is a hallmark feature representing a major conformational change associated with alternating access. However, it remains unclear how partial occupation of four sites by Ca2+ or 3 Na+ ions drives the sliding of TM1/TM6 to initiate alternative access.
In 2013, the crystal structures of inward (cytosol)-facing conformations were resolved for three members of the Ca2+/H+ (CAX) exchanger (which also belongs to the CaCA superfamily), showing astonishing structural similarities to outward-facing NCX_Mj [91, 110, 151, 154]. This breakthrough provided valuable information on the trajectory of helix movements associated with alternative access in NCX and CAX proteins, although the dynamic features of conformational changes underlying the helix sliding remain unclear. An interesting outcome of these new structures is that, in both NCX and CAX, the gap between TM2 and TM7 might hasten the sliding of the gating bundles (TM1 and TM6) in the inward-facing apo state (in order to prevent ion-leakage). According to this scenario, ion binding to a yet undefined site may result in the gap’s closure, thereby allowing the sliding motion of the gating bundle. Thus, the open question is how the ion-protein interactions drive the alternative exposure of the ion-binding pocket.
One possibility is that the ion-selectivity and electrostatic properties of a four-site ion-binding cluster (Sext, Smid, Sint, and SCa sites arranged in a diamond-shaped configuration) dictate the conformational transitions associated with alternative access. More specifically, based on crystal data, it is reasonable to assume that the Sext and Sint sites have high selectivity to the Na+ ion, whereas the Smid and SCa sites are less selective to Na+ and thus, these sites can be occupied either by the Ca2+ or Na+ ions [91]. If so, the flickering of the Ca2+ or Na+ ions between the “vacant sites” (Smid and SCa) may involve specific interactions of charged amino acids (E54, E213, and D240) with ligated ions, which thermodynamically favor the gap closure between TM2 and TM7 to promote alternative access. The challenge is to identify the relevant ion-protein interactions that result in alternating exposure of the ion-binding pocket.
Strikingly, the NCX, NCKX, and CAX families contain structurally related hallmark segments, α1 (on TM2 and TM3) and α2 (on TM7 and TM8) [91, 110, 140, 151, 154], thereby suggesting that the principal mechanism underlying ion-transport and alternating access might be similar among CaCA proteins. In addition, it is unclear to what extent the fine nuances of the α1 and α2 sequence contribute to differences in the ion-transport rates owing to NCX variants. Notably, all eukaryotic NCX variants (including CALX) contain a huge cytosolic f-loop between TM5 and TM6, bearing a number of regulatory domains [27, 28, 57, 107, 125], which are completely absent in NCX_Mj [91]. At the N terminus of the f-loop is located a positively charged auto-inhibitory XIP sequence (20 amino acids), exhibiting an α-helix structure [90, 101], where this region is involved in both Na+ and PIP2 regulation [65, 90]. The cytosolic loop-f contains Ca2+ binding regulatory domains, CBD1 and CBD2, which are connected in a head-to-tail fashion to form a CBD12 tandem [12, 13, 57, 58]. CBD1 contains a primary allosteric Ca2+ sensor (Ca3-Ca4 sites) [27, 28, 119], the affinity and kinetics of which are modulated by a spliced segment located on CBD2 [12, 43, 58].
Structure-dynamic features of regulatory CBD domains
High-resolution X-ray and nuclear magnetic resonance (NMR) studies of isolated CBD1 and CBD2 domains revealed an immunoglobulin-like β-sandwich structure with seven antiparallel β-strands containing four Ca2+ binding sites (Ca1–Ca4) on CBD1 and two Ca2+ sites (CaI–CaII) on CBD2 [12, 57, 108]. In the cardiac, brain, and kidney variants, the Ca3 and Ca4 sites of CBD1 have high affinity for Ca2+ binding (K d = 0.05–0.2 μM), whereas the remaining two sites of CBD1, namely, Ca1 and Ca2, exhibit low affinity (K d > 20 μM) for Ca2+ binding [23, 43, 44, 47, 57]. In the cardiac and brain variants, the CaI site of CBD2 binds Ca2+ with a K d value of 2–10 μM, whereas the CaII site of CBD2 exhibits lower affinity (K d > 20 μM) for Ca2+ binding [13, 44, 47, 58]. Mutant analysis of full-size NCX revealed that, in the cellular system, only three of the six Ca2+ sites (Ca3 and Ca4 on CBD1 and CaI on CBD2) contribute to [Ca2+]-dependent regulation of NCX [27, 28, 118, 119]. Namely, in the cellular system, the Ca3–Ca4 sites govern the “affinity” of the primary allosteric sensor (K 0.5 ∼ 0.3 μM), whereas the CaI site is involved in [Ca2+]-dependent alleviation of Na+-dependent inactivation, showing a K 0.5 value of ∼10 μM [28, 118, 119]. Most probably, the low-affinity sites (Ca1, Ca2, and CaII) are Mg2+ rather than Ca2+ sites, which are constitutively occupied by Mg2+ under physiologically relevant ionic conditions [23, 25, 46]. Moreover, occupation of Ca1–Ca2 sites by Mg2+ decreases the affinity of the primary sensor (Ca3–Ca4 sites), whereas the occupation of the CaII site by Mg2+ increases the affinity of the CaI site [23, 46, 47]. The rationale behind this is that this occupation maintains the properties of regulatory Ca2+ sensors within a physiologically relevant range, thereby exhibiting K d values of 0.2–10 μM and Ca2+ off-rates of 0.02–150 s−1 [47].
There is an increasing body of evidence for synergistic interactions between CBDs, either in the isolated CBD12 or intact NCX [23, 58, 76, 139], where the short interdomain linker (501-HAGIFT-506) encodes unique information governing the flexibility of CBD movements and Ca2+-driven coupling for decoding and transmission of allosteric signals [23, 45]. For example, in order to couple Ca2+-driven conformational transitions in CBDs, it is obligatory to have a glycine residue at position 503 not only in isolated CBD12 [45] but also in intact NCX [99]. Recently derived crystal structures of an isolated two-domain tandem (CBD12) revealed a unique interface between the CBD domains, whereas the occupation of the Ca3–Ca4 sites by Ca2+ generates interdomain salt-bridges in which R532 (located in CBD2) tethers D565 from CBD2 with D499 and D500 from CBD1 [48, 153]. Interestingly, these bifurcated salt-bridges are obligatory for slow Ca2+ dissociation and for Ca2+-induced restriction of CBD movements (Fig. 1), thereby representing a hallmark feature for regulatory coupling of CBDs [23, 45–47]. Namely, the sequential dissociation of two Ca2+ ions from Ca3–Ca4 sites involves a rapid dissociation of the first Ca2+ ion, followed by a slow dissociation of the second (occluded) Ca2+ ion (the off-rate of which is 20–50 times slower than for the first Ca2+ ion) [23, 45, 46]. The slow off-rates of occluded Ca2+ (measured by stopped-flow) differ among the cardiac, kidney, and brain variants (k s = 0.02–0.5 s−1) [43, 47] and correlate with the slow inactivation kinetics (I2), observed for matched NCX variants in patch-clamp experiments [38, 39].
A fundamental question is how is the information about Ca2+ binding to the primary allosteric sensor on CBD1 (Ca3−Ca4 sites) decoded, diversified, and propagated to ion-transport domains, whereas the positive, negative, or no sustained response to regulatory Ca2+ is realized in diverse NCX variants. The SAXS [57, 58], FRET [76], and NMR [139] were explored to resolve the dynamic mechanisms underlying the Ca2+-induced conformational transitions in the isolated preparations of the two-domain CBD12 tandem. Advanced ensemble optimization method (EOM) SAXS analyses revealed that, as a result of Ca2+ binding to the Ca3–Ca4 sites, more constrained conformational states become highly populated at a dynamic equilibrium in the absence of global conformational transitions in the CBDs’ alignment [46]. This conclusion is consistent with NMR analyses describing a similarly extended shape for both the apo and Ca2+-bound states of CBD12, where Ca2+ binding results in rigidified motions of CBDs [139]. Moreover, EOM-SAXS data can rationalize crystallographic data, showing that the interdomain angles between CBDs in Ca2+-bound form are nearly identical in NCX1.4 [48] and CALX1.1 [153] owing to positive and negative responses to regulatory Ca2+, respectively. Since the structural organization of the CBD interface is highly homologous (if not identical) among NCX and CALX orthologs (showing a positive, negative, or no sustained response to regulatory Ca2+), it is reasonable to conclude that a primary mechanism for decoding the allosteric signal is very common among diverse NCX phenotypes [48]. The question is how the allosteric signal from Ca2+ binding regulatory domains (CBD1 and CBD2) to ion-transport sites of NCX (located 80-90 Å away) is propagated in specific cases when the negative, positive, or no response to regulatory Ca2+ is instigated. This issue is especially interesting in light of principle mechanisms proposed for transmission of allosteric signals in multi-domain proteins [96, 148].
Up- and downstream signaling cascades
Metabolic regulation of NCX holds particular promise due to its pathophysiological relevance and potential outlook for devising selective blockers and activators possessing clinical relevance. NCX is regulated by small cytosolic effectors such as PIP2, phosphoarginine, ATP, and putative endogenous inhibitors [4, 7, 22, 35, 50]. Since these issues were extensively reviewed in the past, only the recent contributions will be covered here.
NCX, MAPK-cascade, and related regulatory pathways
To evaluate the MAPK-dependent regulation of NCX, recent work examined the expression levels and activity of specific NCX1-3 isoforms in PC12 cells under conditions in which the ERK1/2, JNK, and p38 MAPKs were shut down, pharmacologically blocked, or activated with NGF [141]. These studies demonstrated isoform-dependent regulation of NCX in a MAPK-cascade-specific manner. Namely, upon NGF stimulation, both ERK1/2 and p38 upregulated NCX1 and NCX3, whereas only p38 had the capacity to downregulate NCX2. Notably, in NGF-untreated cells (where NCX1 and NCX3 were controlled by JNK and ERK1/2, respectively), NCX2 was totally MAPK-independent. Moreover, p38 does not regulate the basal expression levels of any NCX1-3 isoform in PC12 cells, whereas in cardiomyocytes p38 upregulates NCX1 [141]. These findings may be useful for selective pharmacological targeting of predefined NCX isoforms. This issue is especially interesting in conjunction with brain injuries (e.g., brain ischemia and stroke), where the specific expression profiles of NCX isoforms are observed under pathophysiological conditions [17–19].
The involvement of NCX in NO-induced cellular toxicity in neurobrastoma, astrocytes, and microgila cells has been recently documented [145, 146]. Interestingly, in neurobrastoma and astrocyte cells, NO stimulates reverse mode NCX presumably through a cGMP/protein kinase G (PKG)-dependent mechanism, accompanied by elevated [Ca2+]i levels, ROS production, and phosphorylation of ERK, JNK, and p38 MAPK, which finally ends in apoptosis. In contrast, in microglia, NO stimulates forward mode NCX, which results in ER Ca2+ depletion and ER stress associated with apoptotic cell death [117, 145, 146]. The relevant mechanisms could be of special interest in view of the role of NO in neurodegenerative disorders (including Alzheimer’s and Parkinson’s diseases), because the Ca2+ and NO signaling pathways may synergistically interact with each other to promote neurotoxicity. An open-ended problem concerning the mechanisms underlying NO-mediated NCX activation is that the actual phosphorylation of NCX by PKG has not yet been demonstrated at any level of an experimental system (in vitro, in situ, or in vivo). It is possible that NO-mediated NCX activation involves phosphorylation–dephosphorylation of yet an unidentified player (e.g., phospholemman), which may directly interact with NCX to modulate its activity. Further resolution of the underlying molecular mechanisms may help in devising selective blockers for specific neuronal NCX variants, which could be beneficial for suppressing neurodegenerative progression.
A recent report reveals that reverse mode NCX (Ca2+ entry) activates the plasma membrane PKCα of endothelial cells to promote VEGF-induced ERK1/2 phosphorylation and angiogenesis [3]. Besides the endothelial cells, the reverse-mode NCX was reported to be compulsory for ERK1/2 activation in neuroblastoma cells [144, 145]. The significance of these findings is that NCX may activate ERK1/2 downstream of thrombin and angiopoietin, two pathways that are associated with tumor angiogenesis and evasion of anti-angiogenic therapy. This research area becomes especially interesting in light of the fact that the relevant regulatory pathways may represent major therapeutic targets in anti-angiogenic cancer and chiefly contribute to drug resistance in VEGF and related signaling pathways. Therefore, it is challenging to look for and identify new drug candidates that selectively inhibit tissue-specific NCX isoforms, with the goal of hampering drug-resistant cancer forms.
NCX and phospholemman
Phospholemman (PLM) is a 72-amino acid phosphoprotein with a single transmembrane domain, which is highly expressed in cardiomyocytes to regulate Na+/K+-ATPase [124], Na+/Ca2+ exchange [30, 31], and L-type Ca2+ channel [52] activities; thus, it can affect the heart rate and/or the contractility force. Phosphorylation of Ser68 by PKA is an important regulatory mode, resulting in NCX inhibition and activation of Na+/K+-ATPase in cardiomyocytes [31, 32, 124]. Notably, the cytoplasmic tail of PLM interacts with two short segments on NCX located on the f-loop, whereas the PLM-dependent regulation is independent of Ca2+ interaction with CBDs [31].
Under catecholamine stress conditions (when PLM is phosphorylated at Ser68 by PKA), inhibition of Na+/K+-ATPase by PLM is relieved, whereas NCX activity is suppressed [30–32, 124]. Therefore, under stress conditions, phosphorylated PLM may decrease arrhythmia risks (by suppressing Ca2+ and Na+ overload through Na+/K+-ATPase activation), and phosphorylated PLM may preserve the inotropic capacity of muscle contractility (by decelerating Ca2+ efflux through NCX inhibition). Interestingly, PLM-S68E mutations in adult cardiomyocytes lead to NCX inhibition, which therefore represents the potential capacity for enhancing [Ca2+]i-transients and contractility [31–33]. Thus, this approach may be used for specific inhibition of NCX under disease-related conditions, whereas the targeting of PLM-Ser68 could be beneficial for improving cardiac performance under stress.
Physiological functions
Although NCX variants are associated with diverse regulatory machinery to fit the tissue-specific physiological demands of Ca2+ homeostasis, the partial contributions of NCX variants to specific cells and systemic functions remain largely unclear. The present article focuses only on selected topics covering the recent advances.
Knockout mouse models of NCX
Early studies with the NCX knockout models provide information on NCX contributions to physiological functions on a systemic level [67, 128, 134]. Recent studies with organ-specific knock-out mouse models revealed more specific contributions of NCX variants to heart contractility, arrhythmia, and ischemic damage [50, 113, 120, 157], to smooth muscle vasoconstriction and blood pressure regulation [155, 156, 158], and to brain activities (hippocampal long-term potentiation in learning, cerebral reperfusion damage, stroke, preconditioning, etc.) [17–19, 73, 74, 141]. Interestingly, the total deletion of the Slc8a1 gene results in NCX1-null embryos that cannot perform a spontaneous heart beat and consequently die at early stages of development, whereas those mice with cardiac-specific knockout of NCX1.1 can live to adulthood [50, 67, 128]. The accumulating data suggest that NCX variants contribute to specific systemic functions and organ protection. For example, ablation of NCX1 protects against heart ischemia–reperfusion injury [113], whereas mice lacking the brain NCX2 (the major isoform of the brain) exhibit an enhanced capacity for learning and memory [74].
Recent studies with ventricular-specific NCX knockout mice show that NCX plays a new role in priming the Ca2+ entry in cardiomyocytes [50, 120, 127]. Namely, the rise in cytosolic [Na+] within the restricted space of diadic cleft (upon rapid depolarization of the membrane) transiently reverses NCX directionality to mediate Ca2+ entry, which “primes” diadic cleft so that subsequent Ca2+ entry through LTCC can more efficiently trigger Ca2+ release from the SR. An apparent drawback of this putative mechanism is that the time slot between the opening of voltage-sensitive Na+ and Ca2+ channels is so short (<2 ms) that it would be hardly enough to complete even a single turnover of the NCX transport cycle. Thus, computational modeling is required for evaluating the physiological relevance of the proposed mechanism, taking into account the cleft architecture and site-density of relevant ion-transport systems.
The creation of new knockout models for distinct NCX variants is required for elucidating the important mechanisms associated with the specific tissue/organ contribution of NCX variants in systemic function and regulation of the fundamental physiological processes. For example, tissue-specific ablation of mitochondrial NCLX may provide useful information on the contribution of this protein in the systemic regulation of blood pressure, insulin, or neurotransmitter secretion, long-term potentiation of brain activity, and learning, immune response, etc.
NCX, excitation-contraction (EC) coupling, and localized Ca2+-signaling
A major role of NCX in EC-coupling is to maintain a dynamic balance between Ca2+ entry and Ca2+ exit during the action potential by extruding nearly all Ca2+ that has entered through the LTCC into the cell during the depolarization upstroke [8–10, 15]. Thus, NCX represents a unique system that is responsible for fine-tuning and integration of rate/force relationships in cardiomyocytes [8, 9]. Interestingly, in intact cardiomyocytes, the activity of NCX is very low at resting [Ca2+]i, whereas ∼100 % of the maximal capacity of NCX is recruited at the [Ca2+]i peak [21]. Moreover, the [Ca2+]i-dependent allosteric activation of NCX has an unusually high degree of cooperativity in intact cardiomyocytes and shows a steep dependence on [Ca2+]i with a Hill coefficient of n H = 4–8 [21, 49, 95]. Most importantly, a time slot for this highly cooperative activation of NCX seems to be several-fold slower than the cardiac cycle, meaning that the activation of NCX by Ca2+ is a slow event, which does not occur on a beat-to-beat basis [49]. Therefore, the regulation of NCX and its dynamic contribution to EC-coupling should be considered in light of multi-beat modeling. An important physiological implication of these observations could be that cardiomyocyte functioning requires a highly sensitive but delayed response of NCX to cytosolic Ca2+ to precisely control the dynamic changes in the steady-state rates of Ca2+ extrusion that occur on a multi-beat time-scale [49]. However, paradoxically, it is impossible to explain the observed high values of cooperativity for Ca2+-dependent NCX activation even if one were to assume that all six Ca2+ binding sites of CBD12 are involved in allosteric regulation (which apparently is not the case, since only three Ca2+ sites of CBD12 are involved in allosteric regulation, see above). Interestingly enough, the high values of cooperativity (n H = 4–8) for [Ca2+]-dependent allosteric activation are observed only in intact cardiomyocytes [21, 49, 95], whereas much lower degrees of cooperativity (n H ∼ 2) are detected in excised patches of cardiomyocytes and in an oocyte expression system [13, 27, 118, 119]. A low degree of cooperativity is also observed for Ca2+ binding to isolated CBD1, CBD2, and CBD12 preparations [21, 23, 43, 44]. Since all these measurements seem to be reliable, the mechanisms supporting a steep and slow activation of NCX might operate only in intact cardiomyocytes. For example, the Ca2+-dependent slow oligomerization of NCX may occur only in the sarcolemma membranes of intact cardiomyoctes but not in “nonphysiological” preparations.
Recent advances in understanding, the regulatory mechanisms underlying CBD domains (see above) provide new clues regarding the dynamic regulation of EC-coupling. Namely, the CBD12 tandem represents a wide-range Ca2+ sensor, the dynamic properties of which are well-suited for sensing the rapid changes in [Ca2+]i within sub-cellular compartments (dyadic cleft, cytosolic bulk phase, etc.). Since the LTCCs are basically located in front of the junctional SR (jSR) across a nanoscopic subspace (∼150 Å), it is expected that when an LTCC opens, the “subspace” [Ca2+]SS increases briefly from a diastolic level of 100 nM to ∼10 μM [71, 72], whereas NCX near the jSR is exposed to high levels of [Ca2+]SS. Moreover, assuming that a Ca2+ spark is triggered by the LTCC opening, [Ca2+]SS can reach 30–100 μM levels [71, 72, 142]. Thus, for effective response to large [Ca2+] swings, NCX needs an integrative feedback sensor over a range of 10−7–10−4 M. By having CBD1 and CBD2 domains, NCX 1.1 fully covers this sensitivity range and thus, allows differential sensing of [Ca2+] within the dyadic cleft, the sub-membrane cavities, and bulk cytosol [43, 44, 47]. For example, transient occupation of the CaI site on CBD2 by Ca2+ (K d ∼ 5 μM) may relieve Na+-dependent inactivation of NCX within the dyadic cleft, although this putative mechanism has not yet been demonstrated. Nevertheless, the low affinity Ca1–C2 and CaII sites are not directly involved in Ca2+-sensing, and constitutive occupation of these sites by Mg2+ can adjust the affinity of the regulatory Ca3–Ca4 and CaI sites within the physiological range, thereby playing an important role in integrating the function of CBD1 and CBD2 [46, 47].
Interestingly, recent studies suggest that the high-affinity (Ca3–Ca4 sites) allosteric activation of NCX in cardiomyocytes involves slow inactivation of NCX with a time constant of ∼20 s [49]. This inactivation kinetics displays a striking similarity to the slow dissociation of “occluded” Ca2+ from the cardiac CBD12, showing a rate-constant of k s ∼ 0.05 s−1 [43, 47]. The rationale behind this is that slow inactivation of NCX is important for dynamic regulation on a multibeat time scale, whereas NCX might better sustain a long-standing Ca2+ balance while contributing to the ability of cardiomyocytes to generate Ca2+ transients over a wide range of amplitudes [49].
NCX and Ca2+-dependent pacemaker activity
Cardiac contractions are initiated and synchronized by pacemaker cells (SAN, AVN, and the bundle of His), representing a small fraction of total cardiac tissue that controls heartbeat (automaticity) in a sophisticated way. Although the heart pacemaker was discovered more than a hundred years ago, the underlying mechanisms are still under debate [87, 150]. In general, automaticity entails the existence of inward currents at diastolic potentials, and many ion-transport systems are dynamically matched and integrated to reach the threshold of the next action potential. It is thought that SR Ca2+ release activates the forward mode NCX (Ca2+-efflux), which generates an inward current that brings the late diastolic potential to the threshold of the next action potential; however, the contribution of NCX to pacemaker activity remains a controversial issue [87, 97]. A principal question is whether NCX contributes to basal heartbeat rates and/or to regulation of basal heartbeat rates in response to global regulatory modes (e.g., adrenergic activation). Recent studies revealed that genetic ablation of NCX-mediated ion currents in vivo, ex vivo, and in isolated SAN cells disables “fight” or “flight” SAN activity without affecting the resting heart rate, meaning that NCX1.1 is required for increasing sinus rates but not for maintaining the resting heart rate [42]. However, this conclusion has been challenged by a recent report suggesting that cardiac NCX1.1 is a key player in the initiation and maintenance of a stable heart rhythm [56]. More extensive research is required to resolve these issues.
The previously unrecognized Ca2+-activated K+ channel (SK4) has been identified recently in human embryonic stem cell-derived cardiomyocytes as a key player in pacemaker activity [152]. Interestingly, in some developmental cells, the NCX-mediated inward current (in the absence of If current) has the capacity to gradually reduce SK4-mediated outward current, which allows inward currents to take over in reaching the threshold of the next action potential [152]. Although the physiological relevance of this mechanism in adult heart pacemaker cells is currently unclear, it is obvious that a subtle balance between the outward SK4 currents, on the one hand, and the If/NCX inward currents, on the other hand, play a critical role in shaping the pacemaker activity at yearly stages of embryonic heart differentiation and development. Further research is required to resolve the underlying mechanisms involved in balancing the expression/regulation of SK4 and NCX in adult and developmental pacemaker cells.
NCX, neurotransmitter secretion, and synaptic transmission
Although NCX’s contribution to synaptic transmission was suspected for a long time, only a recent report indicated the involvement of reverse-mode NCX in synaptic activity at the parallel fiber-to-Purkinje neuron synapse in the mouse cerebellum [137]. Namely, the NCX-mediated Ca2+-efflux boosts the amplitude and duration of parallel fiber Ca2+ transients during short bursts of high-frequency action potentials, typical of their behavior in vivo, whereas the computer-aided simulations suggest transient accumulation of intracellular [Na+], which is sufficient to drive NCX-mediated Ca2+-efflux. It was suggested that this mechanism can feed additional Ca2+ influx into the parallel fibers to support synaptic transmission to Purkinje neurons for up to 400 ms after the burst [137]. The relevant mechanisms may shape the dynamics of presynaptic [Ca2+] swings to boost synaptic transmission with extra capacity to properly integrate and optimize the accuracy of cerebellar information transmission.
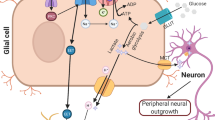
The general mechanism underlying Ca2+-dependent neurotransmitter release in neurons and neuroendocrine cells involves agonist-induced stimulation, which subsequently provokes a transient increase in cytosolic [Ca2+]i, the amplitude and duration of which dynamically promotes and integrates exocytosis of hormone or neurotransmitter (GABA, glycine, glutamate, etc.)-containing granules [112, 123, 135, 138]. The shape of the transient Ca2+ signal is regulated by multiple mechanisms, although the rising phase of the agonist-induced Ca2+ signal is mainly governed by the amount of Ca2+ entry via LTCCs and/or Ca2+ release from the ER stores through the IP3 receptor channels [112, 138]. Interestingly, exocytotic-like release, depending on the extracellular Ca2+, can be observed when cytosolic [Na+]i rises enough to allow the reversal of NCX [112, 135, 138]. Although NCX is involved in neurotransmitter release in both neuronal and glial tissues, NCX seems to play a major role in controlling ionic homeostasis when responding to mechanical and biochemical stimulations requiring integration of regulatory signals in the framework of a neuron-glia network [123, 135, 138]. This is true for both astrocytes and microglia, in which homeostatic responses are controlled by dynamic changes in the cytosolic [Ca2+]i and [Na+]i [112, 123, 135, 138]. The contribution of mitochondrial NCLX to Ca2+-dependent proliferation of the neuro/glial network is of special interest in terms of its contribution to the development of neurodegenerative diseases. Recent findings suggest that NCLX regulates Ca2+ signaling, gliotransmission, and proliferation of astrocytes and thus, links between the mitochondria and plasma membrane Ca2+ homeostasis to control a diverse array of astrocyte functions [123].
NCX and insulin secretion
Ca2+ homeostasis in β cells plays a critical role in regulating insulin secretion through multiple mechanisms involving Ca2+ release from ER via RyR, Ca2+ entry through LTCC, Ca2+ extrusion through NCX, and Ca2+ reuptake into ER via SERCA [29]. Stimulation of pancreatic β cells by glucose is associated with slow waves of membrane potential and with LTCC- and NCX-mediated oscillations of [Ca2+]i [29, 53]. The β cells express NCX1.3 and NCX1.7 splice variants, the regulatory specificity of which as well as their partial contributions to Ca2+ homeostasis and insulin secretion remain unclear. Interestingly, overall, the overexpression of NCX1 accelerates apoptosis of β cells, whereas apoptotic cell death in β cells significantly decelerates in the heterozygous Slc8a1 KO mouse model [129, 130]. Moreover, heterozygous Slc8a1 KO mice exhibit enhanced glucose-induced insulin release as well as increased proliferation and resistance to abnormal changes caused by diabetes [53–55]. The selective inhibition of NCX1.7 could be useful for improving insulin secretion. On the other hand, the pharmacological inhibition of NCX1.3 (the second NCX variant expressed in β cells) seems to be undesirable since NCX1.3 is chiefly expressed in the kidney, and its inhibition may reduce the renal Ca2+-reabsorption and thus, may induce systemic hypocalcemia.
The contributions of mitochondrial NCX in Ca2+ shuttling of pancreatic β cells was suspected for many years, but its contribution to Ca2+ signaling and to subsequent insulin secretion has remained controversial because the inhibitors that were used non-specifically modulate many other Ca2+ transporters. This certainly underscores the importance of a molecular-based approach to elucidate the role of mitochondrial NCLX in Ca2+-dependent insulin release in β cells. The molecular identification of mitochondrial NCLX [121, 122] provided new molecular tools for selective ablation of this transporter. The role of mitochondrial NCLX in Ca2+ signaling and insulin secretion is of major interest because of the dual role of mitochondrial Ca2+ shuttling in metabolism and global Ca2+ signaling. These new molecular tools were very instrumental in showing that NCLX plays a predominant role in shaping glucose-dependent cytosolic Ca2+ transients regulating the temporal pattern of insulin secretion [111]. Additional analyses of the expression/regulation of NCX1.3, NCX1.7, and NCLX may elucidate the partial contributions of these variants to altered homeostasis, which may help in pinpointing the best strategy for effective pharmacological targeting of relevant NCX variants.
NCX’s relationship to diseases
NCX, arrhythmias, heart failure, and myocardial ischemia–reperfusion injury
The process of human remodeling involves the altered expression/regulation of NCX in most cases of disease development [11, 14, 40, 51, 103, 150]. In heart failure, the overexpressed levels of NCX1.1 protein can significantly contribute to reduced levels of SR Ca2+ load (with an associated reduction in contractile force) as well as can mediate depolarizing arrhythmogenic currents (EAD or DAD) during the action potential that arises upon the unmatched “spontaneous” release of Ca2+ from the SR [14, 103, 150]. In animal models, NCX contributes to arrhythmogenesis and Ca2+ depletion during heart failure, owing to combined volume and pressure overload whereas, in compensated hypertrophy, NCX contributes to Ca2+ loading [92, 127–129]. Upregulation of NCX is extremely dangerous in combination with altered expression of the other important proteins. For example, in heart failure or hypertrophy, the concomitant overexpression of NCX and the downexpression of SERCA may cause SR Ca2+ depletion (with contractile malfunctions), whereas an affiliated overexpression of NCX and the downexpression of the K1-potassium channel may disturb the balance between the inward and outward currents during the repolarization, causing life-threatening arrhythmias [8–11, 103, 113, 127]. Although disorders in the expression/regulation of NCX accompany the clinical settings of arrhythmias, heart failure, and myocardial ischemia–reperfusion injury, it is not trivial to match the contributions of NCX to clinical states [11, 51, 92, 103, 150]. A major problem is that heart remodeling involves numerous proteins, the expression levels of which are controlled by many personal variables including the history and complexity of disease development and drug treatment.
NCX, cerebral ischemia, and stroke
A general feature of cerebral ischemia is the overload of Na+ and Ca2+ [4, 34]. This is a very harmful situation because the elevated levels of Ca2+ induce necrosis and/or apoptosis of vulnerable neurons, whereas the elevated levels of [Na+]i result in cell swelling and microtubular disorganization. In animal models of cerebral ischemia, consequently resulting in permanent vascular occlusion, the pharmacological activation of NCX reduces brain damage, whereas the NCX blockers worsen the infarct lesion [4, 98, 149]. This conclusion was supported in experiments with transgenic mouse models, whereas the ablation of NCX1, NCX2, or NCX3 protein leads to a worsening of brain damage after focal ischemia [17–19, 73]. An important message is that the pharmacological activation of neuronal NCX variants could be a beneficial approach for decreasing brain ischemia-associated damage.
NCX, blood pressure regulation, and hypertension
The transgenic mouse model, expressing altered levels of smooth muscle NCX1, demonstrated that NCX1 chiefly contributes to diverse signaling pathways involved in activating vascular smooth muscle contraction in response to stretch (i.e., myogenic response) and in activating certain G protein-coupled receptors [16, 155]. Moreover, overexpression of vascular NCX1 has been linked with human primary pulmonary hypertension and with several salt-dependent hypertensive animal models [155–158]. It was suggested that specific mechanisms governing arterial NCX1 expression and local levels of sub-sarcolemma [Na+]i, induced by Na+/K+ ATPase and TRPC6, assist in regulating arterial vasoconstriction and blood pressure [16, 155, 157]. According to this proposal, the Ca2+-influx through NCX1 is coupled with Na+ transport via store-operated channels, TRPC6, and Orai1, whereas TRPC6, Orai1, and NCX1 are clustered with α-2 Na+/K+ ATPases in cell-membrane “microdomains” in the vicinity of junctional SR [16, 156–158]. This putative cluster may integrate the local [Na+], Ca2+ signaling, and arterial tone to control vascular resistance. Interestingly, NCX1 and TRPC6 are vastly overexpressed in de-endothelialized mesenteric arteries in ouabain-induced hypertensive rats, Milan hypertensive strain rats, and Dahl salt-sensitive hypertensive rats [159].
NCX, apoptosis, and proliferation
It is well established that the Ca2+-dependent regulation of apoptotic and proliferative processes is a general mechanism in many cell types, although the particular contributions of specific Ca2+-transport systems (and their variants) in regulating cell apoptosis/proliferation under normal and disease conditions remain unresolved. Previous studies have shown that distinct NCX variants contribute to altered [Ca2+]i and [Na+]i homeostasis in neurodegenerative disorders [4, 17–19, 123, 138] and diabetes [53–55, 123]. Altered expression/regulation of specific NCX variants is a hallmark feature of disease-related remodeling of microglia migration and proliferation, meaning that the underlying mechanisms could be relevant in pathophysiological settings [112, 135]. Thus, NCX variants control the balance between apoptosis and proliferation under normal and disease conditions. Mitochondrial NCLX also regulates Ca2+-dependent proliferation of astrocytes, which could be related to neurodegeneration [123].
Recent studies have shown that NCX plays an essential role in regulating Ca2+ and Na+ homeostasis, cell migration, and proliferation of human gastric myofibroblasts, whereas all three isoforms (NCX1-3) contribute in handling ionic homeostasis and cellular functions [77]. The physiological significance of these findings is that gastrointestinal myofibroblasts are contractile, electrically non-excitable, transitional cells that participate in extracellular matrix formation and thus, contribute to ulcer healing, chronic inflammation, and tumor development. Inhibition of predefined NCX variant(s) could be a therapeutic target in combating hyperproliferative gastric diseases.
Pharmacological implications
Undoubtedly, there is a cumulative request by both researchers and clinicians for effective pharmacological targeting of disease-related NCX variants owing to the effect of high potency, selectivity, and the bioavailability of potential drugs at the cellular and systemic levels. Notably, both the selective blockers and activators of NCX variants are desired for intervening in diverse pathological scenarios. For example, activation of NCX1, NCX2, or NCX3 isoforms could be beneficial for brain ischemia or stroke [4, 17–19, 74, 75], whereas a potent inhibition of NCX (e.g., by SEA0400) can prevent dopaminergic neurotoxicity in the mouse model of Parkinson’s disease [1]. Potentially, the cardiac NCX blocker may have an anti-arrhythmic effect as well as improve cardiac contractility (e.g., in heart failure), but it may also worsen conditions such as recovery from ischemia or relaxation abnormalities [5, 14, 67, 103, 147]. The search for marginal anti-arrhythmic strategies is motivated by the shortcomings of currently available drugs as well as by new prospects arising from the screening and identification of new lead-structures for selective and effective targeting. In general, the overall outcome might be rather positive in comparison with its potential drawbacks, whereas the NCX blocker could be a part of a multi-target strategy for devising new drugs [5, 14, 20].
Drawbacks of currently available NCX blockers
During the last four decades, several organic compounds have been identified that affect NCX activity, but it appears that they also affect other ion-transport systems as well. For example, amiloride or bepridil analogs and isothiourea derivatives (KB-R7943) are non-selective inhibitors of NCX, but they exhibit relatively low potency (IC50 = 5–10 μM) for inhibition [5, 20, 68–70, 147]. Positively charged peptide inhibitors, XIP [90, 101], and FRCRCFa [64, 84, 102, 152] exhibit relatively high potency (IC50 < 1 μM) for inhibition in patch-clamp experiments (when added from the cytosolic side), but they are not selective to NCX isoforms and are unusable in most physiological experiments due to poor permeability into the cell membrane when added to an extracellular medium. SEA0400 exhibits remarkably high potency for inhibition [1, 104], although experiments with Slc8a1 knockout mice have shown that SEA0400 is a non-selective NCX blocker as well [5, 133]. Recently developed ethoxyaniline, quinazolinone, thiazolidine, phenoxypyridine, acylacetamide, benzofuran, and imidazoline derivatives (including SN-6 and YM-244769) exhibit improved pharmacological properties for NCX inhibition, although the development of selective blockers for tissue-specific NCX variants on the basis of these derivatives remains doubtful [5, 69, 147].
More advanced approaches are required to generate a new generation of NCX blockers/activators. There are perhaps two foremost motives why effective pharmacological targeting of NCX variants is unavailable at present: (1) The molecular and cellular mechanisms governing the regulatory diversity of isoform/splice variants are still poorly understood; (2) No analytical systems are available for in vitro high-throughput screening (HTS) of small “drug-like” compounds. Isolated protein preparations of CBD12 constructs obtained from the cardiac, brain, kidney, and pancreas NCX [43], in combination with fluorescent assays for Ca2+ binding [44], could be a useful analytical approach for serial HTS analyses of NCX variants, aiming for primary identification and development of potential drug candidates.
General perspectives toward future NCX pharmacology
The resolution of the structure–activity relationships underlying the interaction between the two CBD domains is especially interesting in terms of developing coherent strategies for selective targeting of NCX variants. The rationale behind this is that the wide-range of the Ca2+ sensing machinery is modulated by alternatively spliced segments on CBD2, thereby making CBD12 a highly versatile Ca2+ sensor [47]. This enables NCX to efficiently respond to and regulate a plethora of Ca2+-dependent signals and processes in distinct tissues and cellular compartments, consistent with the ubiquitous tissue distribution of NCX1. Drugs targeting the regulatory CBDs, rather than the ion translocation sites, have potential to target tissue-specific NCX variants since the alternative splicing region of NCX lies within CBD2. More specifically, drugs targeting the domain’s interface are of interest because the splicing region is adjacent to that area. More specifically, drugs directed at the interface can enhance NCX activity via domain stabilization or inhibit NCX by disrupting specific interactions between the two CBD domains, such as the salt-bridge network. Using computer-added virtual screening, numerous compounds may be found and thus, in vitro HTS tests are required to detect potential candidates. The identification of Ca2+ occlusion as a biochemical hallmark of interdomain interactions in CBD12 can serve as a selection criterion for drug discovery. Stopped-flow kinetics analyses are convenient because they are rapid and require minute amounts of protein sample [43–47], making this technique suitable for screening. In addition, knowledge regarding the structural consequences of Ca2+ binding allows the use of SAXS for defining the structural outcomes exerted by compounds binding to CBD12 [45–48]. Importantly, SAXS is much less time and resource consuming than is X-ray crystallography and allows analyses of dynamic conformational states, making it appropriate for screening drug-like compounds.
Future perspectives
Since 2006, significant progress has been made in better understanding the molecular and cellular mechanisms underlying NCX regulation in health and disease, and much more progress is expected in forthcoming years. Substantial progress has been achieved in deciphering the molecular basis for allosteric regulation of the NCX family, whereas the equilibrium, kinetic, and structural characterization of CBD12 isoforms, variants, orthologs, and their mutants have provided important mechanistic insights and a conceptual framework for better understanding the mechanisms underlying the allosteric regulation of mammalian NCX. Recent breakthroughs provided fundamental information on the crystal structure of archaebacterial NCX_Mj, which may serve as a template structure not only for eukaryotic NCX proteins, but also for numerous proteins belonging to the CaCA superfamily of Ca2+-transport proteins. The significance of these findings is that the general ion-transport mechanism may involve the sliding of the TM1/TM6 cluster, although it remains unclear how ion interaction with the binding pocket drives the alternating excess (sliding) of the TM1/TM6 cluster and what are the dynamic features of the involved helix movements underlying alternative access. Despite these similarities between archaebacterial and eukaryotic NCX, up to 103-fold differences in the turnover rate occur among phylogenetically distant NCX orthologs, the underlying mechanisms of which are challenging to resolve.
The present achievements provide new hopes for better understanding the molecular basis of ion transport and regulation in NCX proteins as well as for the rational design of a new generation of drugs. Application of advanced molecular approaches, including the silencing/overexpression of specific NCX variants in cellular systems and organ-specific KO mouse models provided useful information about the contribution of NCX proteins to physiological processes and diseases. It is expected that selective pharmacological targeting of predefined NCX isoforms/variants has the ability to rescue specific cell types from apoptosis, malfunction, or irreparable harm. The application of adequate in vitro procedures is required for HTS and testing of large libraries of “drug-like” synthetic compounds. Although this long-wanted intervention has not yet been realized and definitely requires long-term collaboration between academic staffs and industrial partners, the needed scientific knowledge and technological tools are now available to challenge the rational development of potential drug candidates for predefined NCX variants.
Abbreviations
- CALX:
-
Drosophila melanogaster NCX ortholog
- CAX:
-
Ca2+/anion exchanger
- CBD:
-
Ca2+ binding domain
- E Ca :
-
Equilibrium potential of Ca2+
- E NCX :
-
Equilibrium potential of NCX
- FRCRCFa:
-
NCX inhibitory cyclic hexapeptide
- FRET:
-
Fluorescence resonance energy transfer.
- I1-inactivation:
-
Na+-dependent inactivation of NCX
- I2-inactivation:
-
Ca2+-dependent inactivation of NCX
- NCLX:
-
Mitochondrial Na+/Ca2+ exchanger
- NCKX:
-
Na+/Ca2+-K+ exchanger
- NCX:
-
Na+/Ca2+ exchanger
- n H :
-
Hill coefficient
- NMR:
-
Nuclear magnetic resonance
- SAXS:
-
Small-angle X-ray scattering
- SLC8:
-
Solute carrier 8 gene family
- SLC24:
-
Solute carrier 24 gene family
- SR:
-
Sarcoplasmic reticulum
- TM:
-
Trans-membrane segment
- VCX:
-
Vacuolar Ca2+/H+ exchanger
- LTCC:
-
L-type voltage-dependent Ca2+ channel
- XIP:
-
NCX inhibitory peptide
References
Ago Y, Kawasaki T, Nashida T, Ota Y, Cong Y, Kitamoto M et al (2011) SEA0400, a specific Na+/Ca2+ exchange inhibitor, prevents dopaminergic neurotoxicity in an MPTP mouse model of Parkinson’s disease. Neuropharmacology 61:1441–1451
Altimimi HF, Fung EH, Winkfein RJ, Schnetkamp PP (2010) Residues contributing to the Na+-binding pocket of the SLC24 Na+/Ca2+-K+ exchanger NCKX2. J Biol Chem 285:15245–15255
Andrikopoulos P, Baba A, Matsuda T, Djamgoz MBA, Yaqoob MM, Eccles SA (2011) Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J Biol Chem 286:37919–37931
Annunziato L, Pignataro G, Di Renzo GF (2004) Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol Rev 56:633–654
Antoons G, Willems R, Sipido KR (2012) Alternative strategies in arrhythmia therapy: Evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther 134:26–42
Baazov D, Wang X, Khananshvili D (1999) Time-resolved monitoring of electrogenic Na+-Ca2+ exchange in the isolated cardiac sarcolemma vesicles by using a rapid-response fluorescent probe. Biochemistry 38:1435–1445
Berberián G, Forcato D, Beaugé L (2009) Key role of a PtdIns-4,5P2 micro domain in ionic regulation of the mammalian heart Na+/Ca2+ exchanger. Cell Calcium 45:546–553
Bers DM (2000) In: Excitation-contraction coupling and cardiac contractile force, 2nd edit., Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 133–333
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Bers DM, Grandi E (2011) Human atrial fibrillation: insights from computational electrophysiological models. Trends Cardiovasc Med 21:145–150
Besserer GM, Ottolia M, Nicoll DA, Chaptal V, Cascio D, Philipson KD, Abramson J (2007) The second Ca2+-binding domain of the Na+/Ca2+ exchanger is essential for regulation: Crystal structures and mutational analysis. Proc Natl Acad Sci U S A 104:18467–18472
Besserer GM, Nicoll DA, Abramson J, Philipson (2012) Characterization and purification of a Na+/Ca2+ exchanger from an archaebacterium. J Biol Chem 287:8652–8659
Biesmans L, Macquaide N, Heinzel FR, Bito V, Smith GL, Sipido KR (2011) Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One 6(10):e25100
Blaustein MP, Lederer WJ (1999) Sodium/calcium exchange: Its physiological implications. Physiol Rev 79:763–854
Blaustein MP, Leenen FH, Chen L, Golovina VA et al (2012) How NaCl raises blood pressure: A new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302:H1031–H1049
Boscia F, D’Avanzo C, Pannaccione A, Secondo A, Casamassa A, Formisano L, Guida N, Annunziato L (2011) Silencing or knocking out the Na+/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. doi:10.1038/cdd.2011.125
Boscia F, Gala R, Pignataro G, de Bartolomeis A, Cicale M et al (2006) Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J Cereb Blood Flow Metab 26:502–517
Boscia F, Gala R, Pannaccione A, Secondo A, Scorziello A, Di Renzo G, Annunziato L (2009) NCX1 expression and functional activity increase in microglia invading the infarct core. Stroke 40:3608–3617
Bourgonje VJ, Vos MA, Ozdemir S, Doisne N, Acsai K, Varro A et al (2013) Combined Na+/Ca2+ exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ Arrhythm Electrophysiol 6:371–379
Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ, Khananshvili D (2011) Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem 286:28811–28820
Boyman L, Hiller R, Lederer WJ, Khananshvili D (2008) Direct loading of the purified endogenous inhibitor into the cytoplasm of patched cardiomyocytes blocks the ion currents and calcium transport through the NCX1 protein. Biochemistry 47:6602–6611
Boyman L, Mikhasenko H, Hiller R, Khananshvili D (2009) Kinetic and equilibrium properties of regulatory calcium sensors of NCX1 protein. J Biol Chem 284:6185–6193
Boyman L, Williams GSB, Khananshvili D, Sekler I, Lederer WJ (2013) NCLX: The mitochondrial sodium calcium exchanger. J Mol Cell Cardiol 59:205–213
Breukels V, Konijnenberg A, Nabuurs SM, Touw WG, Vuister GW (2011) The second Ca2+-binding domain of NCX1 binds Mg2+ with high affinity. Biochemistry 50:8804–8812
Cai X, Lytton J (2004) The cation/Ca2+ exchanger superfamily: phylogenetic analysis and structural implications. Mol Biol Evol 21:1692–1703
Chaptal V, Besserer GM, Ottolia M, Nicoll DA, Cascio D, Philipson KD, Abramson J (2007) How does regulatory Ca2+ regulate the Na+-Ca2+ exchanger? Channels 1:397–403
Chaptal V, Ottolia M, Mercado-Besserer G, Nicoll DA, Philipson KD, Abramson J (2009) Structure and functional analysis of a Ca2+ sensor mutant of the Na+/Ca2+ exchanger. J Biol Chem 284:14688–14692
Chen L, Koh DS, Hille B (2003) Dynamics of calcium clearance in mouse pancreatic beta-cells. Diabetes 52:1723–1731
Cheung JY, Rothblum LI, Moorman JR, Tucker AL, Song J, Ahlers BA, Carl LL, Wang J, Zhang XQ (2007) N Y Acad Sci 1099:119–134
Cheung JY, Zhang XQ, Song J, Gao E, Chan TO, Rabinowitz JE, Koch WJ, Feldman AM, Wang J (2013) Coordinated regulation of cardiac Na+/Ca2+ exchanger and Na+-K+-ATPase by phospholemman (FXYD1). Adv Exp Med Biol 961:175–190
Cheung JY, Zhang XQ, Song J, Gao E, Rabinowitz JE, Chan TO, Wang J (2010) Phospholemman: A novel cardiac stress protein. Clin Transl Sci 4:189–196
Cook NJ, Kaupp UB (1988) Solubilization, purification and functional reconstitution of the sodium-calcium exchanger from bovine rod outer segments. J Biol Chem 263:11382–11388
Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey NW (2010) Modes of neuronal calcium entry and homeostasis following cerebral ischemia. Stroke Res Treat 2010:316862
DiPolo R, Beauge L (2006) Sodium/calcium exchanger: Influence of metabolic regulation on ion carrier interactions. Physiol Rev 86:155–203
Doering AE, Eisner DA, Lederer WJ (1996) Cardiac Na-Ca exchange and pH. Ann N Y Acad Sci 779:182–198
Doering AE, Lederer WJ (1994) The action of Na+ as a cofactor in the inhibition by cytoplasmic protons of the cardiac Na+-Ca2+ exchanger in the guinea-pig. J Physiol 480:9–20
Dunn J, Elias CL, Le HD, Omelchenko A, Hryshko LV, Lytton J (2002) The molecular determinants of ionic regulatory differences between brain and kidney Na+/Ca2+ exchanger (NCX1) isoforms. J Biol Chem 277:33957–33962
Dyck C, Omelchenko A, Elias CL, Quednau BD, Philipson KD, Hnatowich M, Hryshko LV (1999) Ionic regulatory properties of brain and kidney splice variants of the NCX1 Na+-Ca2+ exchanger. J Gen Physiol 114:701–711
Eisner D, Sipido K (2004) Sodium calcium exchange in the heart—Necessity or luxury? Circ Res 95:549–551
Forrest LR, Krämer R, Ziegler C (2011) The structural basis of secondary active transport mechanisms. Biochem Biophys Acta 1807:167–188
Gao Z, Rasmussen TP, Li Y, Kutschke W, Koval OM, Wu Y et al (2013) Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ Res 112:309–317
Giladi M, Bohbot H, Buki T, Schulze DH, Hiller R, Khananshvili D (2012) Dynamic features of allosteric Ca2+ sensor in tissue-specific NCX variants. Cell Calcium 51:478–485
Giladi M, Boyman L, Mikhasenko H, Hiller R, Khananshvili D (2010) Essential role of the CBD1-CBD2 linker in slow dissociation of Ca2+ from the regulatory two-domain tandem of NCX1. J Biol Chem 285:28117–28125
Giladi M, Friedberg I, Fang X, Hiller R, Wang YX, Khananshvili D (2012) G503 is obligatory for coupling of regulatory domains in NCX proteins. Biochemistry 51:7313–7320
Giladi M, Hiller R, Hirsch JA, Khananshvili D (2013) Population shift underlies Ca2+-induced regulatory transitions in the sodium-calcium exchanger (NCX). J Biol Chem 288:23141–23149
Giladi M, Khananshvili D (2013) Molecular determinants of allosteric regulation in NCX proteins. Adv Exp Med Biol 961:35–48
Giladi M, Sasson Y, Fang X, Hiller R, Buki T, Wang Y-X, Hirsch JA, Khananshvili D (2012) A common Ca2+-driven interdomain module governs eukaryotic NCX regulation. PloS One 7(6):e39985
Ginsburg KS, Weber CR, Bers DM (2013) Cardiac Na+–Ca2+ exchanger: Dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J Physiol 591:2067–2086
Goldhaber JI, Philipson KD (2013) Cardiac sodium-calcium exchange and efficient excitation-contraction coupling: Implications for heart disease. Adv Exp Med Biol 961:355–364
Greiser M, Lederer WJ, Schotten U (2011) Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc Res 89:722–733
Guo K, Wang X, Gao G, Huang C, Elmslie KS, Peterson BZ (2010) Amino acid substitutions in the FXYD motif enhance phospholemman-induced modulation of cardiac L-type calcium channels. Am J Physiol Cell Physiol 99:C1203–C1211
Herchuelz A, Kamagate A, Ximenes H, Van Eylen F (2007) Role of Na/Ca exchange and the plasma membrane Ca2+-ATPase in beta cell function and death. Ann N Y Acad Sci 1099:456–467
Herchuelz A, Nguidjoe E, Jiang L, Pachera N (2012) β-Cell preservation and regeneration in diabetes by modulation of β-cell Ca2+ homeostasis. Diabetes Obes Metab 14(Suppl 3):136–142
Herchuelz A, Nguidjoe E, Jiang L, Pachera N (2013) Na+/Ca2+ exchange and the plasma membrane Ca2+-ATPase in β-cell function and diabetes. Adv Exp Med Biol 961:385–394
Herrmann S, Lipp P, Wiesen K, Stieber J, Nguyen H, Kaiser E, Ludwig A (2013) The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc Res 99(4):780–788. doi:10.1093/cvr/cvt154
Hilge M, Aelen J, Vuister GW (2006) Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol Cell 22:15–25
Hilge M, Aelen J, Foarce A, Perrakis A, Vuister GW (2009) Ca2+ regulation in the Na+/Ca2+ exchanger features a dual electrostatic switch mechanism. Proc Natl Acad Sci U S A 106:14333–14331
Hilgemann DW (1990) Regulation and deregulation of cardiac Na+-Ca2+ exchange in giant excised sarcolemmal membrane patches. Nature 344:242–245
Hilgemann DW, Ball R (1996) Regulation of cardiac Na+/Ca2+ exchange and MgATP potassium channels by PIP2. Science 273:956–959
Hilgemann DW, Collins A, Matsuoka S (1992) Steady-state and dynamic properties of cardiac Na/Ca. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol 100:933–961
Hilgemann DW, Matsuoka S, Nagel GA, Collins A (1992) Steady-state and dynamic properties of cardiac sodium-calcium exchange. Sodium-dependent inactivation. J Gen Physiol 100:905–932
Hilgemann DW, Nicoll DA, Philipson KD (1991) Charge movement during Na+-translocation by native and cloned Na+/Ca2+ exchanger. Nature 352:715–718
Hobai IA, Khananshvili D, Levi AJ (1997) The peptide ‘FRCRCFa’, dialyzed intracellularly, inhibits the Na/Ca exchange with high affinity in rabbit ventricular myocytes. Plugers Arch 433:455–463
Hryshko L (2008) What regulates Na+/Ca2+ exchange? Focus on “sodium-dependent inactivation of sodium/calcium exchange in transfected Chinese hamster ovary cells. Am J Physiol Cell Physiol 295:C869-871
Hryshko LV, Matsuoka S, Nicoll DA, Weiss JN, Schwarz EM et al (1996) Anomalous regulation of the Drosophila Na+-Ca2+ exchanger by Ca2+. J Gen Physiol 108:67–74
Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E (2005) Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res 97:916–921
Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T (2004) Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med 10:1193–1199
Iwamoto T, Watanabe Y, Kita S, Blaustein MP (2007) Na+/Ca2+ exchange inhibitors: a new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets 3:188–198
Iwamoto T, Watano T, Shigekawa M (1996) A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem 271:22391–22397
Jayasinghe ID, Baddeley D, Kong CH, Wehrens XH, Cannell MB, Soeller C (2012) Nanoscale organization of junctophilin-2 and ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophys J 102:L19–L21
Jayasinghe I, Dj C, Soeller C, Cannell M (2012) Comparison of the organization of T-tubules, sarcoplasmic reticulum and ryanodine receptors in rat and human ventricular myocardium. Clin Exp Pharmacol Physiol 39:469–476
Jeffs GJ, Meloni BP, Sokolow S, Herchuelz A, Schurmans S, Knuckey NW (2008) NCX3 knockout mice exhibit increased hippocampal CA1 and CA2 neuronal damage compared to wild-type mice following global cerebral ischemia. Exp Neurol 210:268–273
Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS (2003) Enhanced learning and memory in mice lacking Na+-Ca2+ exchanger 2. Neuron 38:965–976
John SA, Liao J, Jiang Y, Ottolia M (2013) The cardiac Na+-Ca2+ exchanger has two cytoplasmic ion permeation pathways. Proc Natl Acad Sci U S A 110:7500–7505
John SA, Ribalet B, Weiss JN, Philipson KD, Ottolia M (2011) Ca2+-dependent structural rearrangements within Na+-Ca2+ exchanger dimers. Proc Natl Acad Sci U S A 108:1699–1704
Kemeny LV, Schnur A, Czepan M, et al (2013) Na+/Ca2+ exchangers regulate the migration and proliferation of human gastric myofibroblasts. Am J Physiol Gastrointest Liver Physiol 305:G552-G563
Khananshvili D (2013) The SLC8 gene family of sodium-calcium exchangers (NCX)—Structure, function, and regulation in health and disease. Mol Aspects Med 34:220–235
Khananshvili D (1998) Structure, mechanism and regulation of the cardiac sarcolemma Na+-Ca2+ exchanger. Mol Cell Biol 23B:309–356
Khananshvili D (1990) Distinction between the two basic mechanisms of cation transport in the cardiac Na+-Ca2+ exchange system. Biochemistry 29:2437–2442
Khananshvili D (1991) Voltage-dependent modulation of ion binding and translocation in the cardiac Na+-Ca2+ exchange system. J Biol Chem 266:13764–13769
Khananshvili D (1991) Mechanism of partial reactions in the cardiac Na+-Ca2+ exchanger. Ann N Y Acad Sci 639:85–98
Khananshvili D, Shaulov G, Weil-Maslansky E (1995) Rate-limiting mechanisms of exchange reactions in the cardiac sarcolemma Na+-Ca2+ exchanger. Biochemistry 34:10290–10297
Khananshvili D, Shaulov G, Weil-Maslansky E, Baazov D (1995) Positively charged cyclic hexapeptides, novel blockers for the cardiac sarcolemma Na+-Ca2+ exchange. J Biol Chem 270:16182–16188
Khananshvili D, Weil-Maslansky E, Baazov D (1996) Kinetics and mechanism: modulation of ion transport in the cardiac sarcolemma sodium-calcium exchanger by protons, monovalent, ions, and temperature. Ann N Y Acad Sci 779:217–235
Kofuji P, Lederer WJ, Schulze DH (1994) Mutually exclusive and cassette exons underlie alternatively spliced isoforms of the Na/Ca exchanger. J Biol Chem 269:5145–5149
Lakatta EG, DiFrancesco D (2009) What keeps us ticking: A funny current, a calcium clock, or both? J Mol Cell Cardiol 47:157–170
Laüger P (1987) Voltage dependence of sodium-calcium exchange: Predictions from kinetic models. J Membr Biol 99:1–11
Lee SL, Yu AS, Lytton J (1994) Tissue-specific expression of Na+-Ca2+ exchanger isoforms. J Biol Chem 269:14849–14852
Li Z, Nicoll DA, Collins A, Hilgemann DW et al (1991) Identification of a peptide inhibitor of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem 266:1014–1020
Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y (2012) Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335:686–690
Linck B, Bokník P, Huke S, Kirchhefer U et al (2000) Functional properties of transgenic mouse hearts overexpressing both calsequestrin and the Na+/Ca2+exchanger. J Pharmacol Exp Ther 294:648–657
Linck B, Qiu Z, He Z, Tong Q, Hilgemann DW, Philipson KD (1998) Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3). Am J Physiol 274:C415–C423
Lytton J (2007) Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+-transport. Biochem J 406:365–382
Maack C, Ganesan A, Sidor A, O’Rourke B (2005) Cardiac sodium-calcium exchanger is regulated by allosteric calcium and exchanger inhibitory peptide at distinct sites. Circ Res 96:91–99
Ma B, Tsai C-J, Haliloğlu T, Nussinov R (2011) Dynamic allostery: Linkers are not merely flexible. Structure 19:907–917
Mangoni ME, Nargeot J (2008) Genesis and regulation of the heart automaticity. Physiol Rev 88:919–982
Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y et al (2001) SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther 298:249–256
Matsuoka S, Nicoll DA, Hryshko LV, Levitsky DO, Weiss JN, Philipson KD (1995) Regulation of the cardiac Na+-Ca2+ exchanger by Ca2+. Mutational analysis of the Ca2+-binding domain. J Gen Physiol 105:403–420
Matsuoka S, Nicoll DA, Reilly RF, Hilgemann DW, Philipson KD (1993) Initial localization of regulatory regions of the cardiac sarcolemmal Na+-Ca2+ exchanger. Proc Natl Acad Sci U S A 90:3870–3874
Matsuoka S, Nicoll DA, He Z, Philipson KD (1997) Regulation of cardiac Na+-Ca2+ exchanger by the endogenous XIP region. J Gen Physiol 109:273–286
Meszaros J, Khananshvili D, Hart G (2001) Mechanisms underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. Am J Physiol Heart Circ Physiol 281:H903–H914
Milberg P, Pott C, Frommeyer G, Fink M, Ruhe M, Matsuda T, Baba A et al (2012) Acute inhibition of the Na+/Ca2+ exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm 9:570–578
Nagy ZA, Virág L, Tóth A, Biliczki P, Acsai K, Bányász T, Nánási P, Papp JG, Varró A (2004) Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol 143:827–831
Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, Goldhaber JI (2010) Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J 99:755–764
Nicoll DA, Applebury ML (1989) Purification of the bovine rod outer segment Na+/Ca2+ exchanger. J Biol Chem 264:16207–16213
Nicoll DA, Longoni S, Philipson KD (1990) Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science 250:562–565
Nicoll DA, Sawaya MR, Kwon S, Cascio D, Philipson KD, Abramson J (2006) The crystal structure of the primary Ca2+ sensor of the Na+/Ca2+ exchanger reveals a novel Ca2+ binding motif. J Biol Chem 281:21577–21581
Niggli E, Lederer WJ (1991) Molecular operations of the sodium-calcium exchanger revealed by conformational currents. Nature 349:621–624
Nishizawa T, Kita S, Maturana AD, Furuya N, Hirata K, Kasuya G, Ogasawara S, Dohmae N, Iwamoto T, Ishitani R and Nureki O. (2013). Structural basis for the counter-transport mechanism of a H+/Ca2+ exchanger. Science www.sciencemag.org/content/early/recent
Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, Khananshvili D, Lewis EC, Sekler I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signaling linked to insulin secretion. PloS One 7(10):e46649
Noda M, Ifuku M, Mori Y, Verkhratsky A (2013) Calcium influx through reversed NCX controls migration of microglia. Adv Exp Med Biol 961:289–294
Ohtsuka M, Takano H, Suzuki M, Zou Y, Akazawa H et al (2004) Role of Na+-Ca2+ exchanger in myocardial ischemia/reperfusion injury: Evaluation using a heterozygous Na+-Ca2+ exchanger knockout mouse model. Biochem Biophys Res Commun 314:849–853
Omelchenko A, Dyck C, Hnatowich M, Buchko J, Nicoll DA et al (1998) Functional differences in ionic regulation between alternatively spliced isoforms of the Na+-Ca2+ exchanger from Drosophila melanogaster. J Gen Physiol 111:691–702
On C, Marshall CR, Chen N, Moyes CD, Tibbits GF (2008) Gene structure evolution of the Na+-Ca2+ exchanger (NCX) family. BMC Evol Biol 8:127–142
On C, Marshall CR, Perry SF, Le HD, Yurkov V, Omelchenko A, Hnatowich M, Hryshko LV, Tibbits GF (2009) Characterization of zebrafish (Danio rerio) NCX4: A novel NCX with distinct electrophysiological properties. Am J Physiol Cell Physiol 296:C173–C181
Ota Y, Kawanai T, Watanabe R, Nishimura A, Ago Y, Takuma K, Matsuda T (2013) Effect of overexpression of the brain-specific Na+/Ca2+ exchanger splice variant NCX1.5 on NO cytotoxicity in HEK293 cells. J Pharmacol Sci 121:351–354
Ottolia M, Nicoll DA, John S, Philipson KD (2010) Interactions between Ca2+ binding domains of the Na+-Ca2+ exchanger and secondary regulation. Channels 4:1–4
Ottolia M, Nicoll DA, Philipson KD (2009) Roles of two Ca2+-binding domains in regulation of the cardiac Na+-Ca2+ exchanger. J Biol Chem 284:32735–32741
Ottolia M, Torres N, Bridge JH, Philipson KD, Goldhaber JI (2013) Na/Ca exchange and contraction of the heart. J Mol Cell Cardiol 61:28–33
Palty R, Hershfinkel M, Sekler I (2012) Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J Biol Chem 287:31650–31657
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A 107:436–441
Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, Sekler I, Nolte CJ (2013) Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes. J Neurosci 33:7206–7219
Pavlovic D, Fuller W, Shattock MJ (2013) Novel regulation of cardiac Na pump via phospholemman. J Mol Cell Cardiol 61:83–93
Philipson KD, Nicoll DA (2000) Sodium-calcium exchange: A molecular perspective. Annu Rev Physiol 62:111–133
Pobarko D, Fameli N, Kuo KH, van Breemen C (2008) Ca2+ signaling in smooth muscle: TRPC6, NCX and Na+ in nanodomains. Channels 2:10–12
Pott C, Eckardt L, Goldhaber JI (2011) Triple threat: the Na+/Ca2+ exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr Drug Targets 12:737–747
Pott C, Philipson KD, Goldhaber JI (2005) Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: Reduced transsarcolemmal Ca2+ flux. Circ Res 97:1288–1295
Puglisi JL, Negroni JA, Chen-Izu Y, Bers DM (2013) The force-frequency relationship: Insights from mathematical modeling. Adv Physiol Educ 37:28–34
Reeves JP, Hale CC (1984) The stoichiometry of the cardiac sodium-calcium exchange system. J Biol Chem 259:7733–7739
Reeves JP, Condrescu M (2008) Ionic regulation of the cardiac sodium-calcium exchanger. Channels 2:322–328
Ren X, Philipson K (2013) The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J Mol Cell Cardiol 57:68–71
Reuter H, Henderson SA, Han T, Matsuda T, Baba A, Ross RS, Goldhaber JI, Philipson KD (2002) Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ Res 91:90–92
Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, Goldhaber JI, Philipson KD (2003) Cardiac excitation-contraction coupling in the absence of Na+-Ca2+ exchange. Cell Calcium 34:19–26
Reyes RC, Verkhratsky A, Parpura V (2012) Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neurol 4(1):e00075
Rispoli G, Navangione A, Vellani V (1995) Transport of K + by Na + -Ca2+, K + exchanger in isolated rods of lizard retina. Biophys J 69:74–81
Roome CJ, Power EM, Empson RM (2013) Transient reversal of the sodium/calcium exchanger boosts presynaptic calcium and synaptic transmission at a cerebellar synapse. J Neurophysiol 109:1669–1680
Rose CR, Karus C (2013) Two sides of the same coin: Sodium homeostasis and signaling in astrocytes under physiological and pathophysiological conditions. Glia 61:1191–1205
Salinas PK, Brüschweiler-Li L, Johnson E, Brüschweiler R (2011) Ca2+ binding alters the inter-domain flexibility between the cytoplasmic Ca-binding domains in the Na/Ca exchanger. J Biol Chem 286:32123–32131
Schnetkamp PP (2013) The SLC24 gene family of Na+/Ca2+-K+ exchangers: From sight and smell to memory consolidation and skin pigmentation. Mol Asp Med 34:455–464
Sirabella R, Secondo A, Pannaccione A, Molinaro P, Formisano L, Guida N, Di Renzo G, Annunziato L, Cataldi M (2012) ERK1/2, p38, and JNK regulate the expression and the activity of the three isoforms of the Na+/Ca2+ exchanger, NCX1, NCX2, and NCX3, in neuronal PC12 cells. J Neurochem 122:911–922
Sobie EA, Song LS, Lederer WJ (2002) Termination of cardiac Ca2+ sparks: An investigative mathematical model of calcium-induced calcium release. Biophys J 83:59–78
Stein WD (1986) Transport and diffusion across cell membranes. Acad. Press, NY, pp 55–120
Svensson KJ, Kucharzewska P, Christianson HC et al (2011) Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc Natl Acad Sci U S A 108:13147–13152
Takuma K, Ago Y, Matsuda T (2013) The glial sodium-calcium exchanger: A new target for nitric oxide-mediated cellular toxicity. Curr Protein Pept Sci 14:43–50
Takuma K, Tanaka T, Takahashi T, Hiramatsu N, Ota Y, Ago Y, Matsuda T (2012) Neuronal nitric oxide synthase inhibition attenuates the development of L-DOPA-induced dyskinesia in hemi-Parkinsonian rats. Eur J Pharmacol 683:166–173
Tóth A, Kiss L, Varró A, Nánási PP (2009) Potential therapeutic effects of Na+/Ca2+ exchanger inhibition in cardiac diseases. Curr Med Chem 16:3294–3321
Tsai CJ, del Sol A, Nussinov R (2008) Allostery: Absence of a change in shape does not imply that allostery is not at play. J Mol Biol 378:1–11
Valsecchi V, Pignataro G, Del Prete A, Sirabella R et al (2011) NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke 42:754–763
Voigt N, Li N, Wang Q, Wang W, Trafford AW et al (2012) Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125:2059–2070
Waight AB, Pedersen BP, Schlessinger A, Bonomi M, Chau BH, Roe-Zurz Z, Risenmay AJ, Sali A, Stroud RM (2013) Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499:107–110
Weisbrod D, Peretz A, Ziskind A, Menaker N, Oz S, Barad L, Itskovitz-Eldor J, Dascal N, Khananshvili D, Binah O, Attali B (2013) The Ca2+-activated K+ channel IKCa/SK4: A critical new player in human embryonic cardiac pacemaker. Proc Natl Acad Sci U S A 110:1685–1694
Wu M, Tong S, Gonzalez J, Jayaraman V, Spudich JL, Zheng L (2011) Structural basis of the Ca2+ inhibitory mechanism of Drosophila Na/Ca exchanger CALX and its modification by alternative splicing. Structure 19:1509–1517
Wu M, Tonga S, Walterspergerb S, Diederichsc K, Wang M and Zheng L (2013) Crystal structure of Ca2+/H+ antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc Natl Acad Sci doi: 10.1073/pnas.1302515110
Zhang J (2013) New insights into the contribution of arterial NCX to the regulation of myogenic tone and blood pressure. Adv Exp Med Biol 961:329–343
Zhang S, Dong H, Rubin LJ, Yuan JX (2007) Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292:C2297–C2305
Zhang J, Ren C, Chen L, Navedo MF, Antos LK et al (2010) Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol 298:H1472–H1483
Zhang J, Ren C, Chen L, Navedo MF et al (2010) Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol 298:H1472–H1483
Zulian A, Baryshnikov SG, Linde CI, Hamlyn JM, Ferrari P, Golovina VA (2010) Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am J Physiol Heart Circ Physiol 299:H624–H633
Acknowledgments
This work was supported by the Israeli Ministry of Health grant #2010-3-6266, the USA-Israeli Binational Research grant # 2009-334, and the Israel Science Foundation grant #23/10. Financial support from the Fields Foundation is highly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
In memoriam John Reeves
Rights and permissions
About this article
Cite this article
Khananshvili, D. Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch - Eur J Physiol 466, 43–60 (2014). https://doi.org/10.1007/s00424-013-1405-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1405-y