
Abstract
Previous studies suggested that four transmembrane domains 5, 6, 11, 12 make the greatest contribution to forming the pore of the CFTR chloride channel. We used excised, inside-out patches from oocytes expressing CFTR with alanine-scanning mutagenesis in amino acids in TM6 and TM12 to probe CFTR pore structure with four blockers: glibenclamide (Glyb), glipizide (Glip), tolbutamide (Tolb), and Meglitinide. Glyb and Glip blocked wildtype (WT)-CFTR in a voltage-, time-, and concentration-dependent manner. At V M = −120 mV with symmetrical 150 mM Cl− solution, fractional block of WT-CFTR by 50 μM Glyb and 200 μM Glip was 0.64 ± 0.03 (n = 7) and 0.48 ± 0.02 (n = 7), respectively. The major effects on block by Glyb and Glip were found with mutations at F337, S341, I344, M348, and V350 of TM6. Under similar conditions, fractional block of WT-CFTR by 300 μM Tolb was 0.40 ± 0.04. Unlike Glyb, Glip, and Meglitinide, block by Tolb lacked time-dependence (n = 7). We then tested the effects of alanine mutations in TM12 on block by Glyb and Glip; the major effects were found at N1138, T1142, V1147, N1148, S1149, S1150, I1151, and D1152. From these experiments, we infer that amino acids F337, S341, I344, M348, and V350 of TM6 face the pore when the channel is in the open state, while the amino acids of TM12 make less important contributions to pore function. These data also suggest that the region between F337 and S341 forms the narrow part of the CFTR pore.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The gene defective in the chronic inherited disease cystic fibrosis (CF) encodes a large chloride channel protein, CFTR, which plays an important role in chloride secretion in epithelioid tissues such as the lung, pancreas, and intestine. The putative domain architecture of CFTR includes two membrane-spanning domains (MSD1 and MSD2), each containing six transmembrane (TM) helices, two nucleotide-binding domains (NBD1 and NBD2), and a regulatory (R) domain bearing multiple phosphorylation sites related to channel gating [36]. CFTR channel activity is regulated by protein kinase A (PKA)-mediated phosphorylation of the R-domain and the binding and hydrolysis of ATP at NBD1 and NBD2 [5, 7, 22].
The crystal structure of CFTR has not yet been solved. While multiple homology models are now available, all are based on the crystal structure of the Sav1866 transporter protein under conditions not consistent with the open CFTR channel state. Hence, it remains unclear what portions of the CFTR protein contribute to the open channel pore [2, 31, 32, 38]. Our CFTR working model suggests that the pore is comprised of amino acids contributed by TMs 5, 6, 11, and 12 [27]. Structure–function investigations of the CFTR Cl− channel pore have focused on the sixth transmembrane helix (TM6), which clearly plays a key role in forming the pore and determining its permeation properties. Mutation of residues within TM6, including R334, K335, F337, T338, S341, R347, T351, R352, and Q353, has been associated with changes in relative anion selectivity, single-channel kinetics, unitary conductance, interaction with pore blockers, and rate of reaction with sulfhydryl-modifying reagents (when replaced by cysteine) [6, 13, 15, 18, 19, 46–53].
Pore-blocking drugs have been used as probes of pore structure by assessing the effects of mutagenesis upon blocking behavior. Blockade of heterologously expressed CFTR by four classes of organic molecules has been described: disulfonic stilbenes, arylaminobenzoates, sulfonylureas, and glycine hydrazide analogs (e.g., GlyH-101). GlyH-101 is different from other classes of CFTR blockers from its water-solubility and membrane-impermeance (K i is ∼4 μM); it behaves as a fast open pore blocker of CFTR that occludes the extracellular vestibule of the CFTR pore [13, 33, 39]. The hypoglycemic sulfonylureas, including Tolb, Glip, and Glyb, and the non-sulfonylurea hypoglycemic agent Meglitinide, are used clinically to control the release of insulin from pancreatic β-cells by interaction with the sulfonylurea receptor, SUR, a member of the ATP-binding cassette (ABC) transporter superfamily [1, 30, 34]. Block of CFTR by Glyb [37, 40, 41, 47, 50], Tolb [45], and Meglitinide [8] has been described, although the binding sites for these drugs remain to be identified. We have shown previously that Glyb interacts with the CFTR pore in a complicated manner [47, 50]. Glyb introduces multiple classes of intraburst blocked states in single-channel recordings of CFTR activity, suggesting multiple sites of interaction that differ in voltage-dependence and pH-dependence. These complex interactions at the single-channel level result in block of macroscopic currents that is characterized by two components: one very rapid and one that exhibits significant time-dependence. These observations led us to speculate that Glyb may interact with the intracellular vestibule of the CFTR pore in more than one manner or at more than one site [40]. If the latter were true, one would not expect to find a single-point mutation that is capable of strongly altering the affinity of the pore for the blocker. Consistent with this notion, a mutagenesis study of part of CFTR’s TM6 and TM12 [21], albeit incomplete, reported mutations which increased the K d for block by Glyb only approximately twofold for seven CFTR mutations in TM6 or TM12 and suggested that there is a direct interaction between Glyb and F337 and T338 [21].
In the present study, we used Glyb, Glip, and Tolb as probes of pore structure, focusing on the 40 amino acids between R334 and Q353 in TM6 and between T1134 and V1153 in TM12. We used excised inside-out macropatches and single-channel patches from Xenopus oocytes expressing CFTR and performed alanine-scanning to identify residues that affect block significantly for the three blockers. We show here that Glyb and Glip block the pore by interacting with multiple residues. The pattern of interacting residues allows us to predict that F337, S341, M348, and V350 are pore-facing residues when the channel is in the open state. These data also strongly suggest that TM12 is less important to the function of the CFTR pore compared to TM6.
Materials and Methods
Reagents and chemicals
Unless otherwise noted, all reagents were obtained from Sigma Chemical (St. Louis, MO). L-15 media was purchased from Gibco/BRL (Gaithersburg, MD). PKA was from Promega Corp. (Madison, WI). Glyb, Glip, Tolb, and Meglitinide were initially prepared as 0.5 M stocks in dimethyl sulfoxide (DMSO), stored at −20°C, and thawed and diluted to the desired final concentration at the time of the experiment. At all dilutions used, DMSO was without effect on CFTR currents (data not shown).
Preparation of oocytes and cRNA injections
The methods used were similar to those described previously [16, 17, 29, 47–51]. All experimental protocols involving Xenopus frog care and oocytes harvesting were approved by the Institutional Animal Care and Use Committee of Emory University (Protocol number DAR-2000446-121513). Briefly, stage V–VI oocytes were prepared as described [29] and were incubated at 18°C in a modified Leibovitz’s L-15 medium with the addition of HEPES (pH 7.5), penicillin, and streptomycin. cRNA was prepared from a construct carrying the full coding region of CFTR in the pGEMHE high-expression vector, generously provided by D. Gadsby (Rockefeller University). Oocytes were injected with 30–100 ng CFTR cRNA for macropatch experiments, or 5–10 ng cRNA for single-channel experiments. Recordings were performed at room temperature 2–5 days after injection.
Mutagenesis
All the mutants used in this study were prepared with the QuikChange protocol (Stratagene, La Jolla, CA) using oligonucleotide-mediated mutagenesis with the pGEMHE-CFTR construct. All mutant constructs were verified by sequencing across the entire open-reading frame before use. cRNA was prepared by in vitro transcription and quantified as described [46, 49, 51]. All amino acids predicted to contribute to the transmembrane portions of TM6 and TM12 according to the available homology models were mutated individually to alanine and the endogenous alanine was mutated to serine. For simplification, names of mutant channels are often abbreviated (e.g., R334A is R334A-CFTR).
Excised patch clamp experiments
All recordings were obtained using excised inside-out patches under controlled conditions. Oocytes were prepared for study by manually removing the vitelline membrane after shrinking in hypertonic solution (in millimolar): 200 monopotassium aspartate, 20 KCl, 1 MgCl2, 10 EGTA, and 10 HEPES-KOH, pH 7.2. Pipettes were pulled from borosilicate glass (Sutter Instrument Company, Novato, CA) and fire polished. Normal pipette solution contained (in millimolar): 150 NMDG-Cl, 5 MgCl2, and 10 TES (N-tris (hydroxymethyl) methyl-2-aminoethanesulfonic acid; pH 7.45). Bath solution for excised, inside-out patches contained (in millimolar): 150 NMDG-Cl, 1.1 MgCl2, 2 Tris–EGTA, 1 MgATP, 10 TES (pH 7.45). Channels were activated by the catalytic subunit of PKA (50 U/mL) following excision. Patch pipette resistances were 1–2 MΩ for macropatch experiments and ∼10 MΩ for single-channel recording. Macropatch recordings were performed with an Axopatch 200B amplifier operated by pClamp 8.2 software (Axon Instruments, Union City, CA), filtered at 100 Hz with a four-pole Bessel filter (LPF-100B, Warner Instruments, Hamden, CT) and acquired at 2 kHz. A fast-perfusion system (Model SF-77B, Warner Instrument Corporation, Hamden, CT) was employed to switch between bath solutions containing 0 MgATP, 1 mM MgATP, and 1 mM MgATP plus different blockers. The time resolution of this system is <30 ms as judged by activation of endogenous calcium-activated chloride channels (data not shown). Due to the rapid switch of solutions, rundown of current did not contribute to the apparent effects of the drugs.
Single CFTR channel currents were measured with an Axopatch 200B amplifier and were recorded at 10 kHz to DAT tape, using symmetrical 150 mM Cl− solutions as described above. Data were subsequently played back and filtered with a four-pole Bessel filter at 100 Hz and acquired using a Digidata 1322A interface and computer at 500 Hz with pClamp 9.0.
Analysis of macropatch and single-channel recording experiments
pClamp 9.0 was used to analyze macropatch data after subtraction of background current (obtained in the absence of ATP). Three protocols for macropatch experiments were used. (1) To measure fractional block and kinetics of macroscopic block: holding potential 0 mV, switch to V M = +80 mV, to V M = −120 mV, to V M = +100 mV, then back to 0 mV. Each of the segments at a specific potential lasted 160 ms (see Fig. 2a for an example). Fractional block in the presence of blocker was measured from the steady-state currents at V M = −120 mV. (2) For I–V plots: holding potential was 0 mV, switch to V M = −100 mV for 260 ms, then ramp up to V M = +100 mV over a period of 300 ms. Voltage ramps were run in triplicate and averaged. (3) To measure voltage-dependence of block: holding potential was 0 mV, membrane potential was stepped to values ranging from −80 mV to +100 mV in 20 mV increments, holding at each for a period of 160 ms.
Fractional block (F b) at steady-state was calculated according to the equation:
Initial block and time-dependent block were calculated as:
where I i represents the peak current immediately following the jump to V M = −120 mV in the presence of blockers, and I c and I b represent the control currents and currents in the presence of blocker at steady-state, respectively.
For single-channel experiments, pClamp 9.0 was used to make all-points amplitude histograms which had bin widths of 0.01 pA and were fit with Gaussian distributions. Open and closed current levels were first identified manually, and then transition analysis was employed using a 50% cut-off between open and closed levels.
Details regarding methods for two-electrode voltage clamp experiments are provided in the Supplementary Materials.
Molecular modeling
Calculation of Glyb, Glip, Tolb, and Meglitinide structures and display of the models was performed with Hyperchem 6.0 (Hypercube, Inc., Gainesville, FL), running under Windows 2000. Solution of the above blocker structures was performed by molecular dynamics and energy minimization using a periodic box size 37.57 × 37.57 × 37.57 Å containing water molecules with simulating temperature 300°K and molecular mechanics force field Amber 94. The 3-D isopotential surface structures of the blockers were obtained with charge density equaling 0.05.
The CFTR model (shown in Fig. 11) was visualized using Pymol software (http://pymol.sourceforge.net). Specific residues are shown in space filling representation whereas backbone atoms are shown in diagram ribbon representation. All images were rendered in Pymol.
Statistics
Unless otherwise noted, values are expressed as mean ± S.E.M. Statistical analysis was performed using the Student’s t test for paired or unpaired measurements (SigmaStat 2.03, Jandel Scientific; San Rafael, CA). Differences were considered statistically significant when p < 0.05 (*) or p < 0.01 (**).
Results
Concentration- and time-dependent block of WT-CFTR by three members of the sulfonylurea family
We have reported that Glyb blocked WT-CFTR in a complicated way but provided important information regarding the pore of this channel [47, 50]. Other members of the sulfonylurea family also block the CFTR channel [8, 37, 41, 45]. Therefore, we designed experiments to probe both TM 6 and TM12 alanine mutants by using blockers of this family. Three chemicals used in this study are shown in Fig. 1 and exhibited similar chemical structure. The core of the molecular structure is a sulfonylurea moiety coupled to the sulfonyl phenyl. Tolb includes only a para-methyl substituent on the sulfonyl phenyl. Glyb and Glip contain a cyclohexyl group coupled to the other end of the sulfonylurea backbone; the cyclohexyl group is missing from Tolb. Glyb includes a 5-chloro-2-methoxybenzamido group linked to the sulfonyl phenyl by a short aliphatic chain. Glip is similar to Glyb except that it has a 5-methylpyrazine-2-carboxamido group linked to the sulfonyl phenyl by a short aliphatic chain. The charge distribution differs between Glyb and Glip (Fig. 1). The surface areas of Glyb, Glip, and Tolb were 887.08, 821.95 and 737.47 Å2, respectively. The dimensions of Glyb, Glip, and Tolb calculated using Hyperchem were 16.01 × 7.79 × 2.79 Å, 19.33 × 8.62 × 2.41 Å, and 13.51 × 4.33 × 2.50 Å, respectively.

Three blockers used in this study: Glyb, Glip, and Tolb. The three structures were solved in water using Hyperchem 6.0 (Gainesville, FL), and are shown as the 3D isopotential surfaces with charge density of 0.05. Blue indicates positive potential and red indicates negative potential
We investigated the ability of these three chemicals to block WT-CFTR. Figure 2 shows representative currents for WT-CFTR blocked by 50 μM Glyb (a), 200 μM Glip (b), and 300 μM Tolb (c). In this voltage clamp protocol, the pre-pulse to V M = +80 mV drives the charged drug out of the channel pore, and the subsequent hyperpolarization drives it in. At V M = −120 mV, block by Glyb and Glip developed over a time course of tens of milliseconds, producing substantial relaxations in the current trace. These relaxations were fit best using a first-order exponential (red line in Fig. 2a) [46, 47]. Following the subsequent step to V M = +100 mV, the current increased slowly during time-dependent dissociation of the Glyb or Glip from the pore; the time constants for dissociation of both drugs were several milliseconds (data not shown). Tolb did not show time-dependent block of WT-CFTR, reaching maximum inhibition immediately after the jump to −120 mV (Fig. 2c). Similarly, Tolb showed no time-dependent relief from block at +100 mV; currents in the presence of Tolb returned to unblocked levels immediately upon stepping to a depolarizing potential suggesting that it is a non-specific blocker. The three chemicals all blocked WT-CFTR macropatch currents in a concentration-dependent manner (Fig. 2d). Glyb had the strongest effect. K ds for block of WT-CFTR by Glyb, Glip, and Tolb were 26.5, 227.0, and 201.1 μM, respectively, with maximum fractional block of 0.87, 0.90, and 0.68, respectively.

Concentration-dependent block of WT-CFTR by Glyb, Glip and Tolb. Block of CFTR macropatch currents by 50 μM Glyb (a) and 200 μM Glip (b) was time-dependent while block by 300 μM Tolb (c) was not. Each panel shows current in the absence of drug (1 mM MgATP alone) and in the presence of drug (plus 1 mM MgATP) applied using the fast-perfusion system, during voltage steps from V M = 0 mV to +80 mV, then to −120 mV, then to +100 mV before returning to 0 mV. Solutions contained symmetrical 150 mM [Cl−], pH 7.50. CFTR channels were previously phosphorylated with PKA and 1 mM MgATP. Blue dashed lines zero current level; red line single-exponential fit of the current. d Dose–response curves for steady-state block by Glyb (triangles), Glip (squares), and Tolb (circles). K ds for block were obtained from a fit to the Hill equation, with adjusted R 2 of 0.97–0.99 for each curve (n = 5–8 each; p < 0.001); K ds for Tolb, Glip, and Glyb were 201.1, 227.0, and 26.5 μM, respectively
Mutations in TM6 differentially affect the behavior of the three blockers
In order to localize sites in TM6 that contribute to the interaction with Glyb, Glip, and Tolb, we performed alanine-scanning mutagenesis between R334, predicted to lie in the outer vestibule, and Q353, predicted to lie in the inner vestibule [2, 27, 38], one site at a time. The endogenous alanine at position 349 was mutated to serine (S). We then tested the effects of each drug at concentrations near the calculated K d for block of WT-CFTR: 50 μM for Glyb, 200 μM for Glip, and 300 μM for Tolb. The major effects of increasing or decreasing sensitivity to Glyb were seen with mutations R334A, K335A, F337A, S341A, I344A, R347A, M348A, V350A, and R352A (Fig. 3 left). The results were nearly identical for block by Glip (Fig. 3 middle) with one important distinction that significant effects were seen with mutations I344A (for Glyb) and V345A (for Glip), suggesting that these two highly related molecules share binding sites. Many substitutions affected block by Tolb (Fig. 3 right), suggesting that this is a fairly non-specific blocker, lacking intimate interactions with specific residues. This is consistent with the high measured K d and low efficacy for block of WT-CFTR, and with the reported brevity of Tolb-induced blocked states in single-channel records [45]. However, the mutations that showed the largest effects on block by Tolb were the same as those that had the largest effects on block by Glyb and Glip. These data with three different pore blockers suggest that the pore domain of CFTR, including the inner and outer vestibules, spans from R334 to at least V350.

Alanine-scanning in TM6 to identify the amino acids that interact with the three blockers. In 150 mM Cl− symmetrical solution, using the protocol as shown in Fig. 2, fractional block was measured at steady-state with V M = −120 mV. Fractional steady-state block by 50 μM Glyb (left), 200 μM Glip (middle), and 300 μM Tolb (right) is indicated for WT-CFTR and for alanine substitutions at each of the 20 residues predicted to comprise TM6 (bars show mean ± SEM, n = 5–10). In the single case where the wildtype residue was alanine, a serine was introduced. Asterisk indicates significantly different compared to WT-CFTR (p < 0.05). Double asterisks indicate significantly different compared to WT-CFTR (p < 0.01)
Mutations R334A and K335A lie in the outer vestibule of the pore of CFTR; surprisingly, the two mutations destroying positive charge exhibited opposite effects on block by Glyb and Glip. R334A weakened block while K335A strengthened block by both blockers. Results from groups including ours have suggested that both R334 and K335 provide very important positive charge to attract Cl− into CFTR’s pore [2, 13, 46]. Also, we reported that R334A and R334C exhibited multiple single-channel conductance levels, including subconductance 1 (s1), subconductance 2 (s2), and full conductance states (f). R334C can be modified by methanethiosulfonate reagents in a state-dependent manner suggesting that its position moves during channel gating [46]. On the contrary, K335A single-channel behavior is very similar to that of WT-CFTR except the single-channel conductance is slightly decreased (−0.56 ± 0.01 pA, n = 5; see Fig. 9), as would be expected for an amino acid that affects attraction of Cl− ions. These results led to us suggest that R334 exhibits multiple functions compared to K335: it might be involved in stabilizing the outer pore of CFTR architecture. Therefore, the effects of mutagenesis at position R334 on block of the pore by Glyb may reflect a highly indirect effect.
Mutations R347A and R352A also represent a separate category from the rest. The basic arginine residue at position 347 previously was shown to contribute to a salt bridge in combination with its acidic partner in TM8 (D924); mutations at R347 which destroyed or reversed side-chain charge led to destabilization of the channel open state [11]. We have found recently that the arginine at position 352 serves a similar role, forming a salt bridge in combination with D993 in TM9 [12]. Charge-reversing and charge-destroying mutations at R352 destabilized the open state, reduced single-channel amplitude, altered ion selectivity, and greatly reduced block by Glip. These functional characteristics were returned to approximately their wildtype behavior in R352E/D993R-CFTR, perhaps because the salt bridge was retained in this second-site reversion [12]. The present data show that mutations R347A and R352A significantly reduced block by all three blockers; for Glyb and Glip, block became strictly time-independent, perhaps reflecting the gross loss of pore architecture leading to loss of the binding site underlying slow pore block. These results strongly support the notion that R347 and R352 stabilize the open pore architecture of CFTR and are inconsistent with other proposed functions for these sites [9, 20, 25, 26, 43, 44].
TM12 alanine mutants are differentially blocked by Glyb and Glip
Unlike for TM6, we know very little about the function of TM12 so far, although it was suggested that TM6 and TM12 make equivalent contributions to the pore of CFTR [2, 27, 31, 32, 37]. We therefore asked whether alanine mutations at 20 amino acids in TM12 exhibited the same sequences on block by Glyb and Glip as these mutations at equivalent positions in TM6. Figure 4 summarizes WT-CFTR and TM12 mutants blocked by 50 μM Glyb (left) and 200 μM Glip (right). Surprisingly, nine mutations of TM12, including N1138A, M1140A, T1142A, V1147A, N1148A, S1149A, S1150A, I1151A, and D1152A, exhibited significantly altered block by Glyb; the pattern was not consistent with either α-helix or β-strand secondary structure along the full length of the region studied. The surprising finding that mutations at six adjacent positions all affected block by Glyb suggests that this portion of CFTR’s TM12 is not α-helical or it might move during channel opening and closing. Block by Glip exhibited a similar pattern as block by Glyb for mutations of TM12, but the major differences between effects of mutations on block by Glyb and Glip were at positions N1138 and M1140 that both mutants were blocked by Glyb significantly different from WT-CFTR while they were not distinctly different from WT-CFTR blocking by Glip. Out of 20 mutants in TM6 and 20 mutants in TM12, only two in TM6 (S341A and F337A) induced rectification in macropatch currents which were suggested to form the narrow part of the pore (see below, Fig. 7, Supplementary Fig. 3). Among the 20 single amino acid mutants of TM12 that we tested in this paper, none of them exhibited significant change in their single-channel conductance compared to WT-CFTR, while we know that mutations R334A, F337A, S341A, R347A, and R352A in TM6 all exhibited significant change in their single-channel conductance [11, 12, 29, and the present manuscript]; these data strongly suggest that TM6 and TM12 do not equally contribute to the pore of CFTR.

Alanine-scanning in TM12 to identify amino acids that interact with Glyb and Glip. In 150 mM Cl− symmetrical solution, using the protocol as shown in Fig. 2, fractional block was measured at steady-state with V M = −120 mV. Fractional steady-state block by 50 μM Glyb (left) and 200 μM Glip (right) is indicated for WT-CFTR and for alanine substitutions at each of the 20 residues predicted to comprise TM12 (bars show mean ± SEM, n = 5–10). Asterisk indicates significantly different compared to WT-CFTR (p < 0.05). Double asterisks indicate significantly different compared to WT-CFTR (p < 0.01)
Glyb and Glip block alanine mutants of TM6 and TM12 with different kinetics
For those mutations that significantly affected steady-state block, we determined the mechanism by which block by Glyb and Glip were altered, first by asking whether the mutation affected more the time-dependent or the time-independent (initial) component of block. We also determined how these mutations affected the voltage-dependence of steady-state block. These results are shown in Figs. 5–7.

Initial block of WT-CFTR and selected TM6 mutants by 50 μM Glyb (left) and 200 μM Glip (right) in symmetrical 150 mM Cl− solution. Data are shown only for those mutants which exhibited significant changes in steady-state fractional block according to Fig. 3 (bars show mean ± SEM, n = 5–10). Asterisk indicates significantly different compared to WT-CFTR (p < 0.05). Double asterisks indicate significantly different compared to WT-CFTR (p < 0.01)
Although mutations R334A and K335A exhibited opposite effects on steady-state block by Glyb and Glip, neither mutation altered initial block (Fig. 5). Effects on time-dependent block by mutations R334A and K335A were similar for Glyb and Glip, although the effect of R334A on Glyb was larger than for Glip and the effect of K335A was larger for Glip than Glyb (Fig. 6). The minor differences suggest that these two basic amino acids function differently as mentioned above and may not form direct interactions with these blockers (Figs. 5 and 6) [2, 13, 46]. Mutations M348A and V350A at sites predicted to lie in the inner vestibule strongly increased steady-state block of CFTR by Glyb and Glip (Fig. 3). In M348A, the initial component of block was nearly lost for both Glyb and Glip; this effect was somewhat smaller in V350A (Fig. 5). In both cases, the increase in overall block (Fig. 3) arose from a substantial increase in time-dependent block (Fig. 6).

Time-dependent block of WT-CFTR and selected TM6 mutants by 50 μM Glyb (left) and 200 μM Glip (right) in symmetrical 150 mM Cl− solution. Data are shown only for those mutants which exhibited significant changes in fractional block according to Fig. 3 (bars show mean ± SEM, n = 5–10). Asterisk indicates significantly different compared to WT-CFTR (p < 0.05). Double asterisks indicate significantly different compared to WT-CFTR (p < 0.01)
Mutation S341A caused the largest decrease in block by Glyb and Glip (aside from R347A and R352A, which have non-canonical effects as described above; Fig. 3). The initial component of block was not affected (Fig. 5), while time-dependent block decreased substantially for both blockers (Fig. 6). Mutation F337A caused a significant decrease in block by Glyb but a significant increase in block by Glip (Fig. 3). In contrast to the effects of mutation S341A, the reduction in block by Glyb in F337A reflected a substantial decrease in initial block without a change in the magnitude of time-dependent block (Figs. 5 and 6). Meanwhile, the increase in block of F337A by Glip reflected a clear decrease in initial block with a dramatic increase in the magnitude of time-dependent block (Figs. 5 and 6).
The apparent voltage-dependence of block by Glyb and Glip was not altered in any of the TM6 mutants other than in V350A suggesting that although several of these mutations affected the magnitude and kinetics of inhibition, the position of the blocker molecule in the voltage field at steady-state was not significantly different from that in the wildtype channel except in V350A (Fig. 8a, b). Both M348A and V350A single-channel full conductances are similar to that of WT-CFTR (Fig. 9, data not shown). Surprisingly, several mutations in TM12 Q1144A, V1147A, N1148A, S1149A, S1150A, and I1151A affected the voltage-dependence of block by Glyb (Fig. 8b). This suggests, again, that TM6 and TM12 do not make equivalent contributions to the pore of the CFTR channel.
S341 and F337 are predicted to lie on opposite ends of the narrowest region of the pore and likely comprise the region of strongest anion selectivity [18, 28]. F337C-and F337E-CFTR exhibited significantly altered reversal potential, relative permeability, and relative conductance compared to WT-CFTR (Supplementary Tables 1, 2, 3), as did F337A-, S-, Y-, and L-CFTR [24]. Both mutations S341A and F337A significantly decreased single-channel conductance (Fig. 9 and Ref. [29]). Consistent with this designation, macroscopic chloride currents in S341A exhibited inward rectification while F337A/C/E exhibited outward rectification (Fig. 7; Supplementary Fig. 1) [28, 29]. This may contribute to the differential effects of these mutations on initial vs. time-dependent block although the voltage-dependences of steady-state block were not altered (Fig. 8a, b).

I–V relationships for WT-CFTR and two important mutants, from inside-out macropatches in symmetrical 150 mM Cl− solution. Data were obtained by ramping the membrane potential from V M = −100 mV to +100 mV over 300 ms. Data are shown for macroscopic currents measured before (black) and after (red) exposure to 50 μM Glyb, 200 μM Glip, and 300 μM Tolb. Data for each CFTR variant is from a single patch expressing WT-, S341A-, or F337A-CFTR

Voltage-dependent block of WT-CFTR and some important mutants in TM6 and TM12. a Voltage-dependence of block of WT-CFTR, F337A-CFTR, and T1142A-CFTR by 50 μM Glyb, and WT-CFTR by 100 μM Glyb, at V M = −100 mV to −20 mV. Fractional block was calculated from the steady-state currents at each potential. Lines shown are from linear regression. b Summary of fractional block by 50 μM Glyb on some important mutants of TM6 (upper) and TM12 (lower). The Δ fractional block from −20 mV to −100 mV was calculated by subtracting the fractional blocking ratio at −20 mV from the fractional blocking ratio at −100 mV for each mutant. *p < 0.05 compared to WT-CFTR blocked by 50 μM Glyb; # p < 0.05 compared to WT-CFTR blocked by 100 μM Glyb
For both Glyb and Glip, initial block was more sensitive to mutations at the cytoplasmic end of TM6 while time-dependent block was more sensitive to mutations deeper in the pore (toward the extracellular end, especially at R334 or K335). This is consistent with our previous finding that time-dependent block of WT-CFTR by Glyb exhibits greater voltage-dependence than does initial block [47–50].
Due to the dramatic differences of effects of TM6 and TM12 mutations on block by Glyb and Glip, we measured single-channel conductance of multiple mutants that exhibited changes in strength of block, emphasizing those for which the single-channel conductance has not yet been published (Fig. 9, and data not shown). All of these mutants except D1152A showed stable open–closed behavior similar to that of human WT-CFTR with subconductance states as rare events. D1152A exhibited increased single-channel conductance for the full open state while it also showed frequent transitions to subconductance states (−0.98 ± 0.02 pA, n = 4; Fig. 9). S1141A (−0.83 ± 0.02 pA, n = 5) increased and T1134A (−0.59 ± 0.02 pA, n = 4) decreased single-channel full open state amplitude compared to WT-CFTR (−0.70 ± 0.03 pA, n = 10; Fig. 9).

Representative single-channel traces for WT-, K335A-, F337A-, M348A-, T1134A-, S1141A-, and D1152A-CFTR (left) from excised inside-out membrane patches with symmetrical 150 mM Cl− solution, and their all-points amplitude histograms (right). All traces were recorded at V M = −100 mV. c closed state; f full open state; s2 subconductance 2 state. The solid lines in the histograms are fit results to a Gaussian function. The scale of the abscissa (current amplitude) in the current trace for F337A is different from the others
Probable orientation of drugs in the pore
Glyb and Glip are identical molecules along most of their lengths, differing only in the substituents on the ring at the non-sulfonylurea end (Fig. 1). From the differences in the effects of mutations S341A and F337A on block by Glyb and Glip, and the similarity of effects of mutations M348A and V350A on block by the two drugs, we can infer that both drugs bind in the pore with the sulfonylurea-linked cyclohexamide end facing toward the cytoplasm. To test this hypothesis, we studied block of CFTR by the non-sulfonylurea hypoglycaemic agent Meglitinide [8], the structure of which is nearly identical to the non-sulfonylurea half of Glyb (Fig. 10). We reasoned that if Meglitinide interacted with the same pore-lining residues as the non-sulfonylurea end of Glyb, block by Meglitinide should be sensitive to the same mutations as block by Glyb.

Structure of Meglitinide and concentration-dependent block of WT-CFTR. a Meglitinide structure was solved under the same conditions as in Fig. 1, and is shown as the 3D isopotential surface structure with charge density of 0.05. Blue indicates positive potential and red indicates negative potential. b Dose–response curve for steady-state block by Meglitinide. K d obtained from a fit to the Hill equation was 271.3 μM (n = 6; p < 0.001)
Meglitinide blocked WT-CFTR with a K d of 271.3 μM (Fig. 10); at V M = −120 mV, fractional block by 300 μM Meglitinide was 0.64 ± 0.02 (n = 6). In the presence of 300 μM Meglitinide, block of WT-CFTR at negative voltages was only slightly time-dependent and relief from block at positive voltages was very rapid, consistent with weak interactions between drug and channel. Similar to their effects on block by Glyb, both the S341A and F337A mutations decreased the efficacy of block by Meglitinide (fractional block was 0.35 ± 0.04 and 0.45 ± 0.04, p < 0.01, respectively). These data are consistent with a model that has Glyb and Glip interacting with multiple residues along the length of the CFTR pore and penetrating deep into the intracellular vestibule of the CFTR pore, with the non-sulfonylurea end pointing toward the external mouth of the channel.
Discussion
The goal of this study was to further our understanding of CFTR channel pore structure by using members of the sulfonylurea family of blockers, Glyb, Glip, and Tolb, together with alanine-scanning mutagenesis, to probe the contributions to pore structure made by individual amino acids in TM6 and TM12. We found that these three blockers, despite their structural differences, all showed similar voltage- and concentration-dependent block. Both Glyb and Glip also showed time-dependent block.
Each of the functional parameters comprising the biophysical signature of CFTR (single-channel conductance, rectification, selectivity, blocker pharmacology, etc.) can be compared between wildtype and site-directed mutants to infer channel structure. Studies of this sort have identified amino acids that contribute to anion-binding sites and to sites that play direct or indirect roles in determining anion selectivity [13]. Inhibition of CFTR-mediated whole-cell currents by Glyb was first described by Sheppard and Welsh [41]. Later studies using single-channel measurements demonstrated that application of Glyb to the cytoplasmic face of excised patches containing CFTR channels resulted in a dose-dependent, reversible decrease in open probability (P O) [37, 40]. Tolb blocks single CFTR channels with extremely rapid kinetics, typical of a fast-type blocker [45]. The concentration of Tolb required to reduce P O by 50% was 540 μM, indicative of very weak interactions between the drug and the channel pore [45]. Glip is an ATP-dependent K+ channel blocker and has the same structure as Glyb aside from different substituents at the non-sulfonylurea end [4]. It has been reported that these three sulfonylureas had different affinities for interaction with the rat pancreatic SUR, falling in the order of Glyb > Glip > Tolb [14]. The affinities of these three drugs for block of WT-CFTR in the present study exhibited the same rank order. Sheppard and coworkers also showed that CFTR can be inhibited with low affinity by non-sulfonylurea hypoglycemic drugs such as Mitiglinide and Meglitinide, which are related in structure to the benzamido (non-sulfonylurea) portion of Glyb [8].
The data presented here show that block of CFTR channels by Glyb, Glip, and Tolb is sensitive to mutations at R334, K335, F337, S341, R347, M348, V350, and R352 of TM6. Gupta and Linsdell reported that mutation T338A reduced block by Glyb [21], but this was not the case in our experiments with either Glyb or Glip. Moreover, block by neither drug was affected by mutations T351A or Q353A, suggesting that these sites do not contribute to the region comprising the anion selectivity filter as previously suggested [9, 20]. The effects of mutations R334A and K335A are indirect, likely related to the movement of chloride within the pore, or the stabilization of the outer vestibule. Likewise, the effects of mutations R347A and R352A are also indirect, because charge-destroying substitutions at these sites alter the gross architecture of the pore, with pleiotropic effects [11, 12]. Mutations at the four remaining sites (F337, S341, M348, and V350) appear to affect block by all three drugs directly, either by increasing or by decreasing the energy of interaction between drug and channel walls. At V350, M348, and S341, alanine substitutions affected block by Glyb and Glip in an identical manner; the effects of the F337A mutation were opposite for Glyb and Glip. Since these two drugs differ only at the non-sulfonylurea end of the molecular structure, it seems reasonable to conclude that it is this end of Glyb and Glip that approaches the F337 site at the extracellular limit of the pore’s narrow region. This conclusion is bolstered by the finding that the effects of mutations S341A and F337A on block by Glyb were the same as their effects on block by Meglitinide, which shares structure with the non-sulfonylurea end of Glyb. In conclusion with these results, for the following reasons, we believe that the narrow region in TM6 of the CFTR pore is located between F337 and S341: (1) mutations F337A/S/C/E/Y/L and S341A/E/T dramatically altered the relative permeability of different anions in the channel (Supplementary Tables 2, 3; Refs. [26, 28]). (2) Both F337 and S341 mutations dramatically decreased the single-channel conductance (Fig. 9, Ref. [29]). (3) Both F337 and S341 mutations exhibited outward or inward rectification, respectively; and (4) both S341A and F337A affected block by all four sulfonylurea family blockers [8, 21, 40, 42, 50, 53].
Tolb lacks the benzamido (non-sulfonylurea) moiety of Glyb and Glip, and block of WT-CFTR by Tolb is time-independent, suggesting that strong time-dependence of block (and relief from block) by Glyb and Glip may arise from interactions of this portion of the drug molecule with specific binding sites in the pore. However, Meglitinide essentially consists of the non-sulfonylurea moiety alone, and yet block by this drug exhibits very limited time-dependence. Therefore, we cannot conclude that one part of the Glyb molecule binds exclusively to one section of the pore because: (a) mutations along the full length of the pore affected block by Tolb, and (b) mutations S341A and F337A affected block by both Tolb and Meglitinide, which represent the two disparate halves of the Glyb structure. Hence, strong time-dependent block of macropatch currents, and the appearance of multiple drug-induced closed states in single-channel recordings, may not arise from interactions of a specific moiety with distinct binding sites in the pore [46–51].
In terms of the 20 mutations of TM12 studied here, we were unable to recognize a clear trend for block similar to that in TM6; for example, block was significantly altered by mutations at every other amino acid between N1148 and T1142 for Glyb, and between T1142 and Q1144 for Glip, which might suggest a β-strand structure at this point; however, the published homology models showed clearly that TM12 was α-helical over this predicted range. For both Glyb and Glip, block was significantly altered by each residue in the series from V1147 to D1152, suggesting that this segment of TM12 may be disordered. Alternatively, these phenomena may arise from sequential interactions with multiple sites in the pore. One part of the flexible Glyb molecule may bind initially, which then facilitates tighter binding of the rest of the molecule by interactions with other parts of the pore. From single-channel and macropatch electrophysiology recording, we previously determined that initial binding may be responsible for the brief Glyb-induced blocked states in single-channel records and time-independent block in macropatches, while subsequent stabilization of binding through additional interactions, underlies the longer duration drug-induced blocked states and time-dependent block [46–51]. In this study, we have focused on 20 amino acids in TM6 and 20 amino acids in TM12; it is quite reasonable to believe that other amino acids contributed by the remaining ten TM helices of CFTR would contribute to the function of these blockers, such as K95 of TM1 [23]. The crystal structure of P-glycoprotein, an ABC transporter superfamily member related to CFTR, showed that the small molecule verapamil interacts with 16 amino acids and QZ-59-SSS interacts with 17 amino acids in the substrate binding pocket of this protein. Similarly, the small molecule antagonist eticlopride interacts with 18 amino acids in the D2R/D3R dopamine receptor [3, 10, 34, 35]. Hence, it is not surprising to find a distributed binding site for sulfonylurea blockers in the CFTR pore, where the energy and kinetics of binding arise from contributions from many different residues.
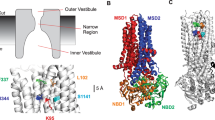
The recently published CFTR homology models from different groups showed that 12 TM helices form the pore of CFTR and they are entirely α-helical, although the various models reflect slightly different alignments [2, 31, 32, 38]. We selected the amino acids of TM6 and TM12 that we have studied in this paper based upon the model from the Dawson and Sansom groups (Fig. 11). It is important to note that these specific amino acids in TM6 and TM12 do not take up equivalent positions across the pore in all homology models and, of course, their positions may change during channel gating. Specifically, R334 to Q353 in TM6 appears to be located more to the extracellular side while T1134 to V1153 in TM12 appears to be located relatively more toward the intracellular side; this difference might partially underlie the differences in Glyb- and Glip-mediated block of TM6 and TM12 mutants in this study. In fact, even though the TM12 amino acids from T1134 to about T1142 appear to be homologous to S341 to R352 in the Riordan model [38], F337 to R352 in the Callebaut model [31, 32], and S341 to Q353 in the Dawson model (see Fig. 11), effects of mutations at these sites were not similar at all. The alanine-scanning mutagenesis results indicate that every third or fourth residue appears to interact with the drug molecule, at least from F337 to M348 of TM6, which supports the α-helical structure of TM6 and indicates that the amino acids F337, S341, I334, and M348 are pore-facing residues when CFTR is in the open state [6]. We cannot recognize a similar pattern in TM12. Consequently, we believe that TM6 is the major TM that affects blocker interactions (along with ion conduction and selectivity) while TM12 makes less important contributions to the channel function of CFTR. Meanwhile, it is not surprising to find that our open pore blocker data are not fully interpretable based upon any one of the homology models, because all the homology models were developed from the crystal structure of Sav1866 which is in the equivalent of the closed state [2, 31, 32, 38].

Homology models of CFTR shown in side-views. The models were made by using Pymol software from the homology model by the Dawson group published in 2009 [2]. Dawson model: CFTR model at 0 ns; Dawson 5 ns model: CFTR model after a 5 ns molecular dynamics simulation. The two models show all TM domains (left) and TM6 and TM12 alone (right). The models were labeled in the same color for the same TM and amino acids: TM1–TM5 pale green; TM7–TM11 pale yellow; TM6 red except 20 amino acids tested in the paper which are shown in blue color; TM12 blue except the 20 amino acids tested in the paper which are shown in red color. In TM6, cyan indicates three basic amino acids R334, R347, and R352; magenta indicates three amino acids K335, M348, and V350 where mutations strengthened block by 50 μM Glyb; yellow indicates three amino acids F337, S341, and I344 where mutations weakened block by 200 μM Glip. In TM12, magenta indicates amino acids N1138, T1142, N1148, and D1152 where mutations strengthened block by 50 μM Glyb; yellow indicates amino acids M1140, V1147, S1149, S1150, and I1151 where mutations weakened block by 200 μM Glip
References
Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP IV, Boyd AE III, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA (1995) Cloning of the β-cell high-affinity sulfonlyurea receptor: a regulator of insulin secretion. Science 268:423–426
Alexander C, Ivetac A, Liu X, Norimatsu Y, Serrano JR, Landstrom A, Sansom M, Dawson DC (2009) Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochem 48:10078–10088
Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G (2009) Structure of P-glycoprotein reveals a molecular basis for polyspecific drug binding. Science 323:1718–1722
Amalric M, Heurteaux C, Nieoullon A, Lazdunski M (1992) Behavioral effects of modulators of ATP-sensitive K+ channels in the rat dorsal pallidum. Eur J Pharmacol 217:71–77
Anderson MP, Berger HA, Rich DP, Gregory RJ, Smith AE, Welsh MJ (1991) Nucleoside triphosphates are required to open the CFTR chloride channel. Cell 67:775–784
Bai Y, Li M, Hwang TC (2010) Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J Gen Physiol 136:393–309
Baukrowitz T, Hwang TC, Nairn AC, Gadsby DC (1994) Coupling of CFTR Cl− channel gating to ATP hydrolysis cycle. Neuron 12:473–482
Cai Z, Lansdell KA, Sheppard DN (1999) Inhibition of heterologously expressed cystic fibrosis transmembrane conductance regulator Cl− channels by non-sulphonylurea hypoglycaemic agents. Br J Pharmacol 128:108–118
Cheung M, Akabas MH (1997) Locating the anion-selectivity filter of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. J Gen Physiol 109:289–299
Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newan AH, Javitch JA, Cherezov V, Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095
Cotton JF, Welsh MJ (1999) Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture for CFTR. Evidence for disruption of a salt bridge. J Biol Chem 274:5429–5435
Cui G, Zhang ZR, O’Brien AR, Song B, McCarty NA (2008) Mutations at arginine 352 alter the pore architecture of CFTR. J Membr Biol 222:91–106
Dawson DC, Liu X, Zhang Z-R, McCarty NA (2003) Anion conduction in CFTR: mechanisms and models. In: Kirk K, Dawson DC (eds) The CFTR chloride channel. Landes, Georgetown, pp 1–34
Dorschner H, Brekardin E, Uhde I, Schwanstecher C, Schwanstecher M (1999) Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol Pharmacol 55:1060–1066
Fatehi M, St Aubin CN, Linsdell P (2007) On the origin of asymmetric interactions between permeant anions and the cystic fibrosis transmembrane conductance regulator chloride channel pore. Biophys J 92:1241–1253
Fuller MD, Zhang Z-R, Cui G, Kubanek J, McCarty NA (2004) Inhibition of CFTR channels by a peptide toxin of scorpion venom. Am J Physiol Cell Physiol 287:C1328–C1341
Fuller MD, Zhang ZR, Cui G, McCarty NA (2005) The block of CFTR by scorpion venom is state dependent. Biophys J 89:3960–3975
Gong X, Linsdell P (2003) Mutation-induced blocker permeability and multiion block of the CFTR chloride channel pore. J Gen Physiol 122:673–687
Gong X, Linsdell P (2003) Molecular determinants and role of an anion binding site in the external mouth of the CFTR chloride channel pore. J Physiol 549:387–397
Guinamard R, Akabas MH (1999) Arg 352 is a major determinant of charge selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel. Biochem 38:5528–5537
Gupta J, Linsdell P (2002) Point mutations in the pore region directly or indirectly affect glibenclamide block of the CFTR chloride channel. Pflugers Arch 443:739–747
Hwang TC, Nagel G, Nairn AC, Gadsby DC (1994) Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proc Natl Acad Sci USA 91:4698–4702
Linsdell P (2005) Location of a common inhibitor binding site in the cytoplasmic vestibule of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem 280:8945–8950
Linsdell P, Evagelidis A, Hanrahan JW (2000) Molecular determinants of anion selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel pore. Biophys J 78:2973–2982
Linsdell P, Hanrahan JW (1996) Flickery block of single CFTR chloride channels by intracellular anions and osmolytes. Am J Physiol 271:C628–C634
Linsdell P, Hanrahan JW (1996) Disulphonic stilbene block of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a mammalian cell line and its regulation by a critical pore residue. J Physiol 496:687–693
McCarty NA (2000) Permeation through the CFTR chloride channel. J Exp Biol 203:1947–1962
McCarty NA, Zhang Z-R (2001) Identification of a region of strong discrimination in the pore of CFTR. Am J Physiol Lung Cell Mol Physiol 281:L852–L867
McDonough S, Davidson N, Lester HA, McCarty NA (1994) Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron 13:623–634
McNicholas CM, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME (1996) Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc Natl Acad Sci USA 93:8083–8088
Monron JP, Lehn P, Callebaut I (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci 66:3469–3486
Mornon JP, Lehn P, Callebaut I (2008) Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci 65:2594–2612
Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS (2004) Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124:125–137
Raeburn D, Brown TJ (1991) RP49356 and cromakalim relax airway smooth muscle in vitro by opening a sulfonylurea-sensitive K+ channel: a comparison with nifedipine. J Pharmacol Exp Ther 256:480–485
Reyes CL, Chang G (2005) Structure of the ABC transporter MsbA in complex with ADP.vanadate and lipopolysaccharide. Science 308:1028–1031
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FC, Tsui L-C (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073
Schultz BD, DeRoos AD, Venglarik CJ, Singh AK, Frizzell RA, Bridges RJ (1996) Glibenclamide blockade of CFTR chloride channels. Am J Physiol 271:L192–L200
Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA 105:3256–3261
Sheppard DN (2004) CFTR channel pharmacology: novel pore blockers identified by high-throughput screening. J Gen Physiol 124:109–113
Sheppard DN, Robinson KA (1997) Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane regulator Cl− channels expressed in a murine cell line. J Physiol 503:333–346
Sheppard DN, Welsh MJ (1992) Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J Gen Physiol 100:573–591
Smith SS, Liu X, Zhang Z-R, Sun F, Kriewall TE, McCarty NA, Dawson DC (2001) CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J Gen Physiol 118:407–431
Tabcharani JA, Linsdell P, Hanrahan JW (1997) Halide permeation in wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels. J Gen Physiol 110:341–354
Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW (1993) Multi-ion pore behavior in the CFTR chloride channel. Nature 366:79–82
Venglarik CJ, Schultz BD, DeRoos AD, Singh AK, Bridges RJ (1996) Tolbutamide causes open channel blockade of cystic fibrosis transmembrane conductance regulator Cl− channels. Biophys J 70:2696–2703
Zhang Z-R, Cui G, Liu X, Song S, Dawson DC, McCarty NA (2005) Determination of the functional unit of the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem 280:458–468
Zhang Z-R, Cui G, Zeltwanger S, McCarty NA (2004) Time-dependent interactions of glibenclamide with CFTR: kinetically complex block of macroscopic currents. J Membr Biol 201:139–155
Zhang Z-R, McDonough SI, McCarty NA (2000) Interaction between permeation and gating in a putative pore-domain mutant in CFTR. Biophys J 79:298–313
Zhang Z-R, Zeltwanger S, McCarty NA (2000) Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol 175:35–52
Zhang Z-R, Zeltwanger S, McCarty NA (2004) Steady-state interactions of glibenclamide with CFTR: evidence for multiple sites in the pore. J Membr Biol 199:15–28
Zhang Z-R, Zeltwanger S, Smith SS, Dawson DC, McCarty NA (2002) Voltage-sensitive gating induced by a mutation in the fifth transmembrane domain of CFTR. Am J Physiol Lung Cell Mol Physiol 282:L135–L145
Zhou JJ, Fatehi M, Linsdell P (2007) Direct and indirect effects of mutations at the outer mouth of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Membr Biol 216:129–142
Zhou Z, Hu S, Hwang TC (2002) Probing an open CFTR pore with organic anion blockers. J Gen Physiol 120:647–662
Acknowledgments
This work was supported by the National Institute for Diabetes, Digestive, and Kidney Diseases (DK056481 to N.A.M.). The authors thank Z.-R. Zhang for the comments.
Ethical standards
No specific ethical issues are related to reported experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM doc
(DOC 2.04 MB)
Rights and permissions
About this article
Cite this article
Cui, G., Song, B., Turki, H.W. et al. Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflugers Arch - Eur J Physiol 463, 405–418 (2012). https://doi.org/10.1007/s00424-011-1035-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-011-1035-1