
Abstract
In cardiovascular diseases and during aging, endothelial dysfunction is due in part to the release of endothelium-derived contracting factors that counteract the vasodilator effect of the nitric oxide. Endothelium-dependent contractions involve the activation of endothelial cyclooxygenases and the release of various prostanoids, which activate thromboxane prostanoid (TP) receptors of the underlying vascular smooth muscle. The stimulation of TP receptors elicits not only the contraction and the proliferation of vascular smooth muscle cells but also diverse physiological/pathophysiological reactions, including platelet aggregation and activation of endothelial inflammatory responses. TP receptor antagonists curtail endothelial dysfunction in diseases such as hypertension and diabetes, are potent antithrombotic agents, and prevent vascular inflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: the history
At the very beginning of the endothelial saga, the comparison of the endothelium-dependent responses of canine arteries and veins yielded the surprising finding that in the latter endothelial cells not only release relaxing factors, but also can initiate endothelium-dependent contractions of the underlying vascular smooth muscle cells [13]. A pivotal finding was that those endothelium-dependent contractions can be prevented by various inhibitors of cyclooxygenases [54]. The initial observations made in canine veins were soon extended to the basilar artery of the same species and the aorta of the rat [37, 38, 46]. Later bioassay studies demonstrated that the endothelium-dependent contractions were indeed caused by vasoconstrictor prostanoids [endothelium-derived contracting factor (EDCF)] produced by the endothelial cells and diffusing to the underlying vascular smooth muscle [101]. The EDCF-mediated responses evoked by stretching and agonists that elevate the endothelial intracellular calcium concentration shared the characteristic to be abrogated by inhibitors of cyclooxygenase. Thus, the increase in endothelial intracellular calcium must stimulate phospholipase A2, which frees arachidonic acid for further metabolism by cyclooxygenase. The breakdown of the fatty acid by this enzyme generates endothelium-derived constrictor prostanoids. They ultimately activate thromboxane prostanoid (TP) receptors of the smooth muscle to evoke contractions [5, 88]. Over the years, oxygen-derived free radicals [39, 100], thromboxane A2 [26, 75], endoperoxides [23], prostacyclin [25], and prostaglandin F2α [97] have been identified as cyclooxygenase-derived mediators of endothelium-dependent contractions.
To exemplify this phenomenon, this brief review will highlight the pathological role of endothelium-dependent contractions, especially in aging, hypertension, and diabetes, and in view of the central role of cyclooxygenases in these EDCF-mediated responses, will focus in particular on the two endothelial isoforms of the enzyme, cyclooxygenase-1 (COX-1), and cyclooxygenase-2 (COX-2).
Arachidonic acid metabolism
Arachidonic acid, the most common precursor of prostaglandins, is generally released from the cell membrane phospholipids by phospholipases and can be metabolized by several enzymatic systems including cyclooxygenases, lipoxygenases, and cytochrome P450 monooxygenases [73].
The first cyclooxygenase (COX-1) was purified in 1976 and subsequently cloned in 1988 [16, 53, 55, 104]. In 1991, the product of a second gene (COX-2), possessing also both cyclooxygenase and peroxidase activities, was identified [33, 63]. It is generally assumed that COX-1, in most tissues, is expressed constitutively while COX-2 is induced mainly at sites of inflammation [12, 87]. However, COX-2 is also expressed constitutively in several organs and cell types, including the endothelial cells where its expression is regulated by shear stress [83]. In the vascular wall, both endothelial and vascular smooth muscle cells contain COXs; however, in healthy blood vessels, endothelial cells contain much more of the enzyme than the surrounding smooth muscle cells [15]. In most blood vessels, prostacyclin, first described as a potent anti-aggregating agent and as a vasodilator, is the principal metabolite of arachidonic acid, the endothelium being the major site of its synthesis [55, 56]. In the rat aorta, where both COXs isoforms are detected, the amount of COX-2 transcripts in endothelial or smooth muscle cells is markedly less than that of COX-1 [81]. However, the COX isoform expressed by the human vasculature has been a matter of controversy. Nevertheless, several lines of evidence indicate that in humans, although COX-2 is the predominant contributor of the systemic generation of prostacyclin, endothelial COX-1, in both healthy and diseased blood vessels, appears to be also a major source of vascular prostaglandins [19, 21, 51, 68].
Various biologically active eicosanoids are formed from the short-lived, but biologically active endoperoxides [prostaglandin H2 (PGH2)], through the action of a set of synthases namely PGD, PGE, PGF, PGI, and thromboxane synthases. Prostaglandins interact with specific seven transmembrane, G-protein-coupled receptors, which are classified in five subtypes DP, EP, FP, IP, and TP receptors in function of their sensitivity to the five primary prostanoids, prostanglandins D2, E2, F2α, I2 (prostacyclin), and thromboxane A2, respectively [84].
The stimulation of TP receptors elicits diverse physiological/pathophysiological responses, including platelet aggregation and smooth muscle contraction. Furthermore, the activation of endothelial TP receptors promotes the expression of adhesion molecules and favors adhesion and infiltration of monocytes/macrophages [61]. Although thromboxane A2 is the preferential physiological ligand of the TP receptor [42], PGH2 and the other prostaglandins, with a various range of potency, also can activate this receptor [25] (Fig. 1). Additionally, isoprostanes (prostaglandin isomers that are generally produced non-enzymatically from the oxidative modification of polyunsaturated fatty acids [59] but also in endothelial cells, in a COX-dependent manner [91]) as well as hydroxyeicosatetraenoic acids (HETEs, generated by lipoxygenases and cytochrome P450 monoxygenases or formed by nonenzymatic lipid peroxidation in endothelial cells and leukocytes) are also potent endogenous agonists at TP receptors [9, 20, 82, 86, 103].

Endothelium-dependent responses in WKY and SHR isolated aortic rings. The measurement of the changes in isometric tension in isolated aortic rings contracted with phenylephrine (PE) shows that the acetylcholine-induced endothelium-dependent relaxation is blunted in SHR rings when compared to that of normotensive WKY. In SHR quiescent rings treated with an inhibitor of NO synthase (L-nitroarginine, L-NA), acetylcholine produces endothelium- and concentration-dependent contractions, which are blocked by valeryl salicylate, a preferential COX-1 inhibitor or S 18886, a specific TP receptor antagonist, but are only partially inhibited by NS-398, a preferential COX-2 inhibitor (modified from Yang et al. [100])
Prostacyclin is a potent inhibitor of platelet adhesion to the endothelial cell surface and of platelet aggregation, and is generally described as an endothelium-derived vasodilator [55–57, 65, 66]. Prostacyclin is the preferential ligand of IP receptors and most of its effects involve the activation of adenylyl cyclase and the subsequent elevation of intracellular cyclic-AMP [90, 94].
Spontaneously hypertensive rats: the archetypal model
The first endothelium-dependent contractions associated with an endothelial dysfunction were observed in the isolated aorta of the spontaneously hypertensive rats (SHR) [17, 46]. In this artery, the endothelium-dependent relaxations are impaired because the generation of a diffusible EDCF opposes the relaxing effect of nitric oxide [46, 101]. These endothelium-dependent contractions are correlated with the severity of hypertension. They increase during the aging process and also occur in aging normotensive WKY [34, 43]. Endothelium-dependent contractions and the associated endothelial dysfunction are less pronounced in female SHR [29, 40].
Endothelium-dependent responses are associated with an increase in endothelial intracellular calcium concentration ([Ca2+]i). Indeed, acetylcholine causes a rapid increase in [Ca2+]i in endothelial cells of SHR and to a much lesser extent in that of WKY. By contrast the calcium ionophore A 23187, which allows the free entry of extracellular calcium into endothelial cells, produces a similar increase in [Ca2+]i in the endothelial cells of both WKY and SHR. Acetylcholine- and more generally receptor-mediated endothelium-dependent contractions are larger in SHR than in WKY while the maximal amplitude of these responses, when elicited by A 23187, are similar in the aortae of the two strains [27, 80, 103]. These results illustrate the first endothelial dysfunction associated with endothelium-dependent contractions, i.e., an abnormal calcium signaling in the endothelial cells of SHR in response to neuro-humoral agents.
Phospholipase A2 catalyzes the breakdown of membrane phospholipids to arachidonic acid. There are two major cytosolic types of the enzyme, calcium-dependent (cPLA2) and calcium-independent (iPLA2) phospholipase A2. The increase in endothelial [Ca2+]i, irrespective of the stimulus, activates cPLA and provokes the mobilization of arachidonic acid. However, in response to acetylcholine, iPLA2 is involved by producing lysophospholipids, which open store-operated calcium channels, permitting the influx of extracellular calcium and the subsequent activation of cPLA2. By contrast, the calcium ionophore bypasses the cell membrane receptors and causes increase in endothelial calcium and a direct activation of cPLA2 [95].
The subsequent steps involve the activation of cyclooxygenase and the production of reactive oxygen species along with that of EDCFs and finally the activation of TP receptors [23, 25, 26, 46, 80, 100, 102]. In the SHR aorta, COX-1 is the preponderant enzyme involved in the generation of EDCF since endothelium-dependent contractions are blocked by specific inhibitors of COX-1 and minimally affected by specific inhibitors of COX-2 [23, 25, 26, 80, 100–102]. Indeed, aortic endothelial cells of various species express preferentially COX-1 versus COX-2 [41, 64] and, in SHR endothelial cells, the mRNA and protein expression of COX-1 is enhanced when compared to that of WKY [23, 81]. In agreement with a preponderant role for COX-1 in endothelium-dependent contractions, these responses are abolished in aortae taken from COX-1 knockout mice while they are maintained in aortic rings of COX-2 knockout animals [79].
However, in both WKY and SHR endothelial cells, the induction of COX-2, especially in resistance arteries and during aging, is also associated with the generation of endothelium-derived contractile prostanoids. In these arteries, COX-2 contributes to the endothelial dysfunction [2, 7, 8, 22, 32, 71, 90, 99, 105]. Therefore, the enhanced endothelial expression of COX-1 and/or COX-2 is the second endothelial dysfunction associated with endothelium-dependent contractions.
Additionally, COX is also involved in the endothelial generation of reactive oxygen species. The enhanced COX-dependent generation of reactive oxygen species is the third endothelial abnormality associated with endothelium-dependent contractions observed in the SHR aorta [80]. Reactive oxygen species decrease NO bioavailability [30, 69] and activate COX [31]. This may involve a positive feedback loop on the endothelial cells by further activating COX and, since reactive oxygen species diffuse toward the vascular smooth muscle cells, they can stimulate COX in smooth muscle cells and produce more contractile prostanoids [4, 39, 100]. Reactive oxygen species can also favor contraction of the vascular smooth muscle cells. Superoxide anions stimulate Ca2+ release from the sarcoplasmic reticulum of these cells [74]. In addition, exogenous hydrogen peroxide and/or the reactive oxygen species generated by the activation of TP receptors itself, enhances the stability and increases the density of functional TP receptors at the cell membrane [85, 93]. Thus, while the activated TP receptors are being internalized and degraded (a key component in limiting the action of agonists at these receptors), a reactive oxygen species-dependent pathway induces the enhanced biogenesis of new TP receptors. Activation of this positive feedback mechanism may underlie the augmented TP expression observed in cardiovascular diseases [36]. Finally, TP receptors are also expressed in endothelial cells and their stimulation can induce the inhibition of NO production [45]. This feed-forward loop involving a reactive oxygen species-dependent post-transcriptional stabilization of TP receptors associated with a decrease production of NO, further altering the unbalance between relaxing and contracting factors and exacerbating the endothelial dysfunction, suggest that TP receptors are very likely to play a pivotal role in cardiovascular diseases [93].
EDCFs diffuse toward the vascular smooth muscle cells and directly activate the TP receptors [101]. Inhibition of thromboxane A2 synthesis does not affect the endothelium-dependent contractions to acetylcholine but partially inhibits those in response to A23187, ADP, or endothelin, indicating that thromboxane A2 is only one of the EDCFs that can be released from SHR aortic endothelial cells [5, 23, 25–27, 35, 44, 46, 75, 100, 103]. PGH2 is the second most potent agonist at TP receptors and is more effective in activating TP receptor in vascular smooth muscle from SHR than in that of WKY. Therefore, PGH2 is also a suitable candidate as EDCF [5, 23, 25–27, 35]. However, in SHR aortic endothelial cells, the massive expression of prostacyclin synthase [81] and its close association with COX-1 [41] are not in favor of a large PGH2 spill over. Paradoxically, prostacyclin is likely to be a major EDCF in SHR aorta. Because of an early and specific dysfunction of the IP receptors of the vascular smooth muscle [28], prostacyclin does not produce relaxations but evokes TP-receptor-dependent contractions [25, 67]. Furthermore, prostacyclin, as PGH2, is also more potent in producing contraction in SHR than in WKY [25]. Therefore, the fourth dysfunction associated with endothelium-dependent contractions involves changes in the responses of the IP and TP receptors of the vascular smooth muscle without major changes in their respective expression [62, 74]. Prostacyclin is also a major contributing factor accounting for the endothelial dysfunction in the aorta and mesenteric artery of WKY and SHR treated with aldosterone [7, 99]. Finally, PGE2 and PGF2α can also act as EDCF when prostacyclin synthase is inhibited and the metabolism of PGH2 diverted [25], a phenomenon that may occur when severe oxidative stress leads to the tyrosine nitration of prostacyclin synthase [107]. Thus, in the SHR aorta, thromboxane A2, PGH2, PGI2, and, depending on the circumstances, PGE2 and PGF2α can all act as EDCF (Fig. 2).

Mechanisms of endothelium-dependent contractions in WKY and SHR aortic rings. M muscarinic receptor, AA arachidonic acid, eNOS endothelial nitric oxide synthase, NO nitric oxide, O
2
− superoxide anion, PGS prostaglandin synthases, COX-1 cyclooxygenase-1, PGI
2
prostacyclin, TXA
2
thromboxane A2, TP TP receptor, IP IP receptor, SR sarcoplasmic reticulum, Sol GC soluble guanylyl cyclase, GTP guanosine triphosphate, cGMP cyclic guanosine monophosphate. The number  ,
,  ,
,  , and
, and  indicates identified abnormalities which contribute to the exacerbated endothelium-dependent contractions in SHR aorta, i.e., endothelial calcium handling, enhanced endothelial COX-1 expression and activity, increased generation of endothelial reactive oxygen species, and dysfunctional smooth muscle TP and IP receptors, respectively (modified from Vanhoutte et al. [88])
indicates identified abnormalities which contribute to the exacerbated endothelium-dependent contractions in SHR aorta, i.e., endothelial calcium handling, enhanced endothelial COX-1 expression and activity, increased generation of endothelial reactive oxygen species, and dysfunctional smooth muscle TP and IP receptors, respectively (modified from Vanhoutte et al. [88])
The contribution of EDCF- and TP-receptor-mediated responses in endothelial dysfunction, first observed in the SHR, has been reported in numerous other models of hypertension and is likely to occur in patients with essential hypertension [18]. Additionally, non-endothelium-derived contractile prostanoids can contribute to vascular dysfunction. For instance, in the aorta of the hypertensive eNOS knockout mice, smooth muscle- and COX-2-derived thromboxane A2 contributes to the enhanced contractions in response to endothelin-1 [106].
Enhanced action of COX-derived EDCF in aging
Aging favors a shift of the fine balance between NO-mediated endothelium-dependent relaxations and COX-dependent contractions towards the latter. Impaired endothelium-dependent relaxations to acetylcholine have been demonstrated in the aorta and superior mesenteric arteries of aged rats. This is accompanied by an increased expression of COX isoforms. The relaxations are potentiated by a non-selective COX inhibitor indomethacin, suggesting a critical role of COX-derived vasoconstrictor prostanoids in impairing endothelium-dependent relaxations [49]. Similar findings are reported in small mesenteric arteries [1], in which indomethacin and a specific COX-2 inhibitor NS-398 restored the attenuated endothelium-dependent relaxations and eliminated contractions caused by high concentrations of acetylcholine.
Endothelium-dependent contractions are usually unveiled in pathological models, owing to a reduction of NO bioavailability that allows the emergence of endothelium-dependent contractions. However, there are exceptions, one being the occurrence of endothelium-dependent contractions in the aorta of young and healthy hamsters [96, 97]. In this preparation, COX-2 is expressed constitutively and incubation with an inhibitor of eNOS unmasks the ability of acetylcholine to elicit endothelium-dependent contractions which are sensitive to COX-2 inhibition and TP receptor antagonism, while these responses are unaffected by COX-1 inhibitors and reactive oxygen species scavengers. By contrast to the SHR aorta in which the proposed EDCFs are prostacyclin, PGH2, and thromboxane A2, the endothelium-derived vasoconstrictor prostanoid responsible in the hamster aorta appears to be PGF2α [96, 97]. In the hamster, aging not only exaggerates endothelium-dependent contractions, which are again attenuated by COX-2 inhibitors, but also increases COX-2 expression and augments the release of and the vascular sensitivity to PGF2α [97]. One distinctive feature of the endothelium-dependent contraction in the aorta of aged hamsters compared with their younger counterparts is that the response can be observed in the absence of inhibitors of eNOS. On the other hand, the endothelium-dependent relaxations are diminished in preparations from aging hamsters, reinforcing the interpretation that endothelium-dependent contractions are unmasked by a reduction in NO bioavailability in aged animals.
The endothelial alterations observed in various animal models of aging suggest that the endothelial dysfunction observed in hypertension can be considered as the consequence of the premature aging of the blood vessel wall [17].
Role of prostanoids in endothelial dysfunction in diabetes
Micro- and macrovascular diseases are currently the major causes of morbidity and mortality in patients with diabetes mellitus and endothelial dysfunction plays also a key role in the pathogenesis of these diabetic vascular diseases. Impaired endothelium-dependent vasodilatation has been demonstrated in various vascular beds of different animal models of diabetes and in humans with types 1 and 2 diabetes. However, the mechanisms of endothelial dysfunction appear to differ according to the diabetic model and the vascular bed under study and include impaired signal transduction or substrate availability, impaired release of NO, increased destruction of NO, decreased sensitivity of the vascular smooth muscle to NO, and enhanced release of endothelium-derived constricting factors. These dysfunctions are again generally associated with the over-generation of reactive oxygen species, lipid peroxidation, and elevated production of adhesion molecules [14, 50].
Streptozotocin-induced diabetes leads to a diminished endothelium-dependent NO-mediated relaxation in rat conduit arteries. TP receptor antagonism, again, restores the impaired relaxation and prevents the endothelium-dependent contraction. Neither thromboxane A2 nor prostacyclin plays a significant role as the relaxation is unaffected by thromboxane A2 synthesis inhibition and since prostacyclin does not cause contractions in those arteries [72]. Likewise, indomethacin inhibits the occurrence of endothelium-dependent contraction in the femoral artery of streptozotocin-treated rats whereby COX-1-derived products appear to play a dominant role [70]. Thromboxane A2 does not play a direct role in reducing endothelial function in type 1 diabetes, but endothelium-derived thromboxane A2 may be involved in the enhanced contractile response to endothelin-1 as thromboxane A2 synthesis inhibition attenuates the exaggerated contraction to endothelin-1 in the mesenteric arteries of streptozotocin-treated rats [3]. In the mesenteric artery of type 2 diabetic OLETF rats, the impaired endothelium-dependent relaxation to acetylcholine is normalized and the contraction to the muscarinic agonist inhibited by indomethacin [48]. The acetylcholine-stimulated release of thromboxane A2 and PGE2 are greater in OLETF than non-diabetic rats; but it is not clear which COX-derived prostanoid is most likely involved [48]. The impaired endothelium-dependent relaxations in the mesenteric vascular bed of streptozotocin-induced diabetic mice is opposed by a compensatory up-regulation of both expression and activity of COX-2 and selective inhibition of COX-2 unmasks endothelial dysfunction [60], suggesting a vascular benefit of COX-2-derived products.
Despite a clearly demonstrated role of COX-derived prostaglandins in the regulation of vascular reactivity in conduit arteries, their link to endothelium-dependent hyperpolarization of vascular smooth muscle (EDHF-mediated responses) in resistance blood vessels is unclear. Indomethacin augments the endothelium-dependent EDHF-mediated relaxation in mesenteric resistance arteries from streptozotocin-induced diabetic but not in those from control mice [58], suggesting that COX-derived prostanoids inhibit either the release of EDHF from the endothelium or its action on vascular smooth muscle [58].
Clinical relevance
The information available in animal models demonstrates that in aging and in a number of diseases such as hypertension, diabetes, and atherosclerosis, as the endothelium becomes dysfunctional, the release of EDCF is favored and endothelium-dependent contractions become more prominent [11, 24, 44, 46, 48]. The indirect evidence available in people suggests that the same is true. Indeed, indomethacin potentiates the relaxations to acetylcholine in isolated renal arteries of aged patients [47] and the vasodilator response to the muscarinic agonist in the forearm of people with essential hypertension [76–78]. The comparison of the effect of the non-selective inhibitor of cyclooxygenases in different age groups further suggests that the contribution EDCF augments with advancing age [77, 78], as it does in the animal. In patients with endothelial dysfunction an improvement was observed with selective COX-2 inhibitors [10, 92], which may imply an important role for that isoform of the enzyme. The TP-receptor blocker S18886 improves endothelial function in patients with coronary disease [6], which further illustrates the role of vasoconstrictor prostanoids in human endothelial dysfunction.
The role of the specific COX isoforms and arachidonic acid metabolites in the regulation of vascular function in human diabetes is less well defined. In young patients with type 1 diabetes, the impaired NO-dependent relaxation may be compensated by an increase in prostacyclin-mediated responses [52]. Indeed, indomethacin-sensitive blood flow is greater in patients with type 1 diabetes than in non-diabetic subjects [98], further suggesting a compensatory response for prostacyclin when the bioavailability of NO is declining. On the other hand, COX-derived prostanoids contribute to the appearance of endothelium-dependent contractions in arteries from older patients with diabetes and hypertension [97].
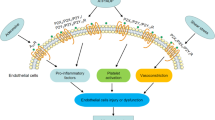
Since the stimulation of TP receptors elicits not only the contraction and the proliferation of vascular smooth muscle cells but also diverse physiological/pathophysiological reactions in platelets (adhesion and aggregation), and endothelial cells (expression of adhesion molecules associated with the subsequent adhesion and infiltration of monocytes/macrophages), TP receptor antagonists may have a unique potential for the treatment of cardiovascular disorders [61, 89] (Fig. 3). However, in contrast to many examples of animal models of cardiovascular diseases, in patients, no study is yet available demonstrating that reversal of endothelial dysfunction is independently associated with a better clinical outcome. The results of large-scale clinical trials are awaited in order to determine the proper therapeutic indication of TP antagonists and to confirm their potential beneficial effect.

Involvement of TP and IP receptors in vascular dysfunction. COXs cyclooxygenases, LOX lipoxygenase, P450 cytochrome P450 monooxygenase, ROS reactive oxygen species, PGS prostaglandin synthases, PGIS prostacyclin synthase, TXS thromboxane synthase, PGs prostaglandins, PGG 2 prostaglandin G2, PGH 2 prostaglandin H2, PGI 2 prostaglandin I2 (prostacyclin), TXA 2 thromboxane A2, HETE hydroxyeicosatetraenoic acid
References
Alvarez de Sotomayor M, Mingorance C, Andriantsitohaina R (2007) Fenofibrate improves age-related endothelial dysfunction in rat resistance arteries. Atherosclerosis 193:112–120
Alvarez Y, Briones AM, Balfagón G, Alonso MJ, Salaices M (2005) Hypertension increases the participation of vasoconstrictor prostanoids from cyclooxygenase-2 in phenylephrine responses. J Hypertens 23:767–777
Arikawa E, Cheung C, Sekirov I, Battell ML, Yuen VG, McNeill JH (2006) Effects of endothelin receptor blockade on hypervasoreactivity in streptozotocin-diabetic rats: vessel-specific involvement of thromboxane A2. Can J Physiol Pharmacol 84:823–833
Auch-Schwelk W, Katusic ZS, Vanhoutte PM (1989) Contractions to oxygen-derived free radicals are augmented in aorta of the spontaneously hypertensive rat. Hypertension 13:859–864
Auch-Schwelk W, Katusic ZS, Vanhoutte PM (1990) Thromboxane A2 receptor antagonists inhibit endothelium-dependent contractions. Hypertension 15:699–703
Belhassen L, Pelle G, Dubois-Rande J, Adnot S (2003) Improved endothelial function by the thromboxane a2 receptor antagonist s 18886 in patients with coronary artery disease treated with aspirin. J Am Coll Cardiol 41:1198–1204
Blanco-Rivero J, Cachofeiro V, Lahera V, Aras-Lopez R, Márquez-Rodas I, Salaices M, Xavier FE, Ferrer M, Balfagón G (2005) Participation of prostacyclin in endothelial dysfunction induced by aldosterone in normotensive and hypertensive rats. Hypertension 46:107–112
Camacho M, Lopez-Belmonte J, Vila L (1998) Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ Res 83:353–365
Cayatte AJ, Du Y, Oliver-Krasinski J, Lavielle G, Verbeuren TJ, Cohen RA (2000) The thromboxane receptor antagonist S18886 but not aspirin inhibits atherogenesis in apo E-deficient mice: evidence that eicosanoids other than thromboxane contribute to atherosclerosis. Arterioscler Thromb Vasc Biol 20:1724–1728
Chenevard R, Hürlimann D, Béchir M, Enseleit F, Spieker L, Hermann M, Riesen W, Gay S, Gay RE, Neidhart M, Michel B, Lüscher TF, Noll G, Ruschitzka F (2003) Selective COX-2 inhibition improves endothelial function in coronary artery disease. Circulation 107:405–409
Cohen RA (2002) Does EDCF contribute to diabetic endothelial cell dysfunction? Dialog Cardiovasc Med 7:225–231
Davidge ST (2001) Prostaglandin H synthase and vascular function. Circ Res 89:650–660
De Mey JG, Vanhoutte PM (1982) Heterogeneous behavior of the canine arterial and venous wall. Importance of the endothelium. Circ Res 51:439–447
De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM (2000) Endothelial dysfunction in diabetes. Br J Pharmacol 130:963–974
De Witt DL, Day JS, Sonnenburg WK, Smith WL (1983) Concentrations of prostaglandin endoperoxide synthase and prostaglandin I2 synthase in the endothelium and smooth muscle of bovine aorta. J Clin Invest 72:1882–1888
De Witt DL, Smith WL (1988) Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci U S A 85:1412–1416
Félétou M, Vanhoutte PM (2006) Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 291:H985–H1002
Félétou M, Vanhoutte PM, Verbeuren TJ (2010) The TP-receptor: the common villain. J Cardiovasc Pharmacol (in press)
Flavahan NA (2007) Balancing prostanoid activity in the human vascular system. Trends Pharmacol Sci 28:106–110
Fonlupt P, Croset M, Lagarde M (1991) 12-HETE inhibits the binding of PGH2/TXA2 receptor ligands in human platelets. Thromb Res 63:239–248
Funk CD, FitzGerald GA (2007) COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol 50:470–479
Garcia-Cohen EC, Marin J, Diez-Picazo LD, Baena AB, Salaices M, Rodriguez-Martinez MA (2000) Oxidative stress induced by tert-butyl hydroperoxide causes vasoconstriction in the aorta from hypertensive and aged rats: role of cyclooxygenase-2 isoform. J Pharmacol Exp Ther 293:75–81
Ge T, Hughes H, Junquero DC, Wu KK, Vanhoutte PM, Boulanger CM (1995) Endothelium dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ Res 76:1003–1010
Gendron ME, Thorin E (2007) A change in the redox environment and thromboxane A2 production precede endothelial dysfunction in mice. Am J Physiol Heart Circ Physiol 293:H2508–H2515
Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Félétou M (2005) Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol 146:834–845
Gluais P, Paysant J, Badier-Commander C, Verbeuren T, Vanhoutte PM, Félétou M (2006) In SHR aorta, calcium ionophore A-23187 releases prostacyclin and thromboxane A2 as endothelium-derived contracting factors. Am J Physiol Heart Circ Physiol 291:H2255–H2564
Gluais P, Vanhoutte PM, Félétou M (2007) Mechanisms underlying ATP-induced endothelium-dependent contractions in the SHR aorta. Eur J Pharmacol 556:107–114
Gomez E, Schwendemann C, Roger S, Simonet S, Paysant J, Courchay C, Verbeuren TJ, Félétou M (2008) Aging and prostacyclin responses in aorta and platelets from WKY and SHR rats. Am J Physiol Heart Circ Physiol 295:H2198–H2211
Graham DA, Rush JW (2009) Cyclooxygenase and thromboxane/prostaglandin receptor contribute to aortic endothelium-dependent dysfunction in aging female spontaneously hypertensive rats. J Appl Physiol 107:1059–1067
Gryglewski RJ, Palmer RM, Moncada S (1986) Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320:454–456
Harlan JM, Callahan KS (1984) Role of hydrogen peroxide in the neutrophil-mediated release of prostacyclin from cultured endothelial cells. J Clin Invest 74:442–448
Heymes C, Habib A, Yang D, Mathieu E, Marotte F, Samuel JL, Boulanger CM (2000) Cyclo-oxygenase-1 and –2 contribution to endothelial dysfunction in ageing. Br J Pharmacol 131:804–810
Hla T, Neilson K (1992) Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A 89:7384–7388
Iwama Y, Kato T, Muramatsu M, Asano H, Shimizu K, Toki Y, Miyazaki Y, Okumura K, Hashimoto H, Ito T, Satake T (1992) Correlation with blood pressure of the acetylcholine-induced endothelium-derived contracting factor in the rat aorta. Hypertension 19:326–332
Kato T, Iwama Y, Okumura K, Hashimoto H, Ito T, Satake T (1990) Prostaglandin H2 may be the endothelium-derived contracting factor released by acetylcholine in the aorta of the rat. Hypertension 15:475–481
Katugampola SD, Davenport AP (2001) Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br J Pharmacol 134:1385–1392
Katusic Z, Shepherd JT, Vanhoutte PM (1987) Endothelium-dependent contraction to stretch in canine basilar arteries. Am J Physiol 21:H671–H673
Katusic ZS, Shepherd JT, Vanhoutte PM (1988) Endothelium-dependent contractions to calcium ionophore A23187, arachidonic acid and acetylcholine in canine basilar arteries. Stroke 19:476–479
Katusic ZS, Vanhoutte PM (1989) Superoxide anion is an endothelium-derived contracting factor. Am J Physiol 257:H33–H37
Kauser K, Rubanyi GM (1995) Gender difference in endothelial dysfunction in the aorta of spontaneously hypertensive rats. Hypertension 25:517–523
Kawka DW, Ouellet M, Hétu PO, Singer II, Riendeau D (2007) Double-label expression studies of prostacyclin synthase, thromboxane synthase and COX isoforms in normal aortic endothelium. Biochim Biophys Acta 1771:45–54
Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S (1997) Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol 122:217–224
Koga T, Takata Y, Kobayashi K, Takeshita S, Yamashita Y, Fujishima M (1988) Ageing suppresses endothelium-dependent relaxation and generates contraction mediated by the muscarinic receptors in vascular smooth muscle of normotensive Wistar Kyoto and spontaneously hypertensive rats. J Hypertens 6:S243–S245
Koga T, Takata Y, Kobayashi K, Takishita S, Yamashita Y, Fujishima M (1989) Age and hypertension promote endothelium-dependent contractions to acetylcholine in the aorta of the rat. Hypertension 14:542–548
Liu CQ, Leung FP, Wong SL, Wong WT, Lau CW, Lu L, Yao X, Yao T, Huang Y (2009) Thromboxane prostanoid receptor activation impairs endothelial nitric oxide-dependent vasorelaxations: the role of Rho kinase. Biochem Pharmacol 78:374–381
Lüscher TF, Vanhoutte PM (1986) Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 8:344–348
Lüscher TF, Cooke JP, Houston DS, Neves RJ, Vanhoutte PM (1987) Endothelium-dependent relaxations in human arteries. Mayo Clin Proc 62:601–606
Matsumoto T, Kakami M, Noguchi E, Kobayashi T, Kamata K (2007) Imbalance between endothelium-derived relaxing and contracting factors in mesenteric arteries from aged OLETF rats, a model of Type 2 diabetes. Am J Physiol Heart Circ Physiol 293:H1480–H1490
Matz RL, de Sotomayor MA, Schott C, Stoclet JC, Andriantsitohaina R (2000) Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. Br J Pharmacol 131:303–311
Mazzone T, Chait A, Plutzky J (2008) Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet 371:1800–1809
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA (1999) Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A 96:272–277
Meeking D, Browne D, Allard S, Munday J, Chowienczyk P, Shaw KM, Cummings MH (2000) Effects of cyclo-oxygenase inhibition on vasodilatory response to acetylcholine in patients with type 1 diabetes and nondiabetic. Diabetes Care 23:1–4
Merlie JP, Fagan D, Mudd J, Needleman P (1988) Isolation and characterization of the complementary DNA for sheep seminal prostaglandins endoperoxide synthase (cyclooxygenase). J Biol Chem 263:3550–3553
Miller VM, Vanhoutte PM (1985) Endothelium-dependent contractions to arachidonic acid are mediated by products of cyclo-oxygenase in canine veins. Am J Physiol 248:H432–H437
Moncada S, Gryglewski RJ, Bunting S, Vane JR (1976) An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 263:663–665
Moncada S, Herman AG, Higgs EA, Vane JR (1977) Differential formation of prostacyclin (PGX or PGI2) by layers of the arterial wall. An explanation for the anti-thrombotic properties of vascular endothelium. Thromb Res 11:323–344
Moncada S, Vane JR (1979) Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2 and prostacyclin. Pharmacol Rev 30:293–331
Morikawa K, Matoba T, Kubota H, Hatanaka M, Fujiki T, Takahashi S, Takeshita A, Shimokawa H (2005) Influence of diabetes mellitus, hypercholesterolemia, and their combination on EDHF-mediated responses in mice. J Cardiovasc Pharmacol 45:485–490
Morrow JD, Hill KE, Burk RF, Nannour TM, Badr KF, Roberts LJ II (1990) A series of prostaglandins F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical catalysed mechanism. Proc Natl Acad Sci U S A 87:9383–9387
Nacci C, Tarquinio M, De Benedictis L, Mauro A, Zigrino A, Carratù MR, Quon MJ, Montagnani M (2009) Endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes is opposed by compensatory overexpression of cyclooxygenase-2 in the vasculature. Endocrinology 150:849–861
Nakahata N (2008) Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 118:18–35
Numaguchi Y, Harada M, Osanai H, Hayashi K, Toki Y, Okamura K, Ito T, Hayakawa T (1999) Altered gene expression of prostacyclin synthase and prostacyclin receptor in the thoracic aorta of spontaneously hypertensive rats. Cardiovasc Res 41:682–688
O’Banion MK, Winn VD, Young DA (1992) cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci U S A 89:4888–4892
Onodera M, Morita I, Mano Y, Murota S (2000) Differential effects of nitric oxide on the activity of prostaglandin endoperoxide H synthase-1 and -2 in vascular endothelial cells. Prostaglandins Leukot Essent Fatty Acids 62:161–167
Radomski MW, Palmer RMJ, Moncada S (1987) Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br J Pharmacol 92:181–187
Radomski MW, Palmer RMJ, Moncada S (1987) The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol 92:639–646
Rapoport RM, Williams SP (1996) Role of prostaglandins in acetylcholine-induced contraction of aorta from spontaneously hypertensive and Wistar-Kyoto rats. Hypertension 28:64–75
Rovati GE, Sala, A, Capra V, Dahlen SE, Folco G. (2010) Dual COXIB/TP antagonists: a possible new twist in NASID pharmacology and cardiovascular risk. Trends Pharmacol Sci 31:102–107
Rubanyi GM, Vanhoutte PM (1986) Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol 250:H222–H227
Shi Y, Feletou M, Ku DD, Man RYK, Vanhoutte PM (2007) The calcium ionophore A23187 induces endothelium-dependent contractions in femoral arteries from rats with streptozotocin-induced diabetes. Br J Pharmacol 150:624–632
Shi Y, Man RY, Vanhoutte PM (2008) Two isoforms of cyclooxygenase contribute to augmented endothelium-dependent contractions in femoral arteries of 1-year-old rats. Acta Pharmacol Sin 29:185–192
Shimizu K, Muramatsu M, Kakegawa Y, Asano H, Toki Y, Miyazaki Y, Okumura K, Hashimoto H, Ito T (1993) Role of prostaglandin H2 as an endothelium-derived contracting factor in diabetic state. Diabetes 42:1246–1252
Smith WL, Marnett LJ (1991) Prostaglandin endoperoxide synthase: structure and catalysis. Biochem Biophys Acta 1083:1–17
Suzuki YJ, Ford GD (1992) Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am J Physiol 262:H114–H116
Taddei S, Vanhoutte PM (1993) Role of endothelium in endothelin-evoked contractions in the rat aorta. Hypertension 21:9–15
Taddei S, Virdis A, Ghiadoni L, Magagna A, Salvetti A (1997) Cyclooxygenase inhibition restores nitric oxide activity in essential hypertension. Hypertension 29:274–279
Taddei S, Virdis A, Mattei P, Ghiadoni L, Fasolo CB, Sudano I, Salvetti A (1997) Hypertension causes premature aging of endothelial function in humans. Hypertension 29:736–743
Taddei S, Virdis A, Mattei P, Ghiadoni L, Gennari A, Fasolo CB, Sudano I, Salvetti A (1995) Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation 91:1981–1987
Tang EH, Ku DD, Tipoe GL, Félétou M, Man RY, Vanhoutte PM (2005) Endothelium-dependent contractions occur in the aorta of wild-type and COX2-/- knockout but not COX1-/- knockout mice. J Cardiovasc Pharmacol 46:761–765
Tang EH, Leung FP, Huang Y, Félétou M, So KF, Man RY, Vanhoutte PM (2007) Calcium and reactive oxygen species increase in endothelial cells in response to releasers of endothelium-derived contracting factor. Br J Pharmacol 151:15–23
Tang EH, Vanhoutte PM (2008) Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol Genomics 32:409–418
Tesfamariam B, Brown ML, Cohen RA (1995) 15-Hydroxyeicosatetraenoic acid and diabetic endothelial dysfunction in rabbit aorta. J Cardiovasc Pharmacol 25:748–755
Topper JN, Cai J, Falb D, Gimbrone MA Jr (1996) Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci U S A 93:10417–10422
Tsuboi K, Sugimoto Y, Ichikawa A (2002) Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat 68–69:535–556
Valentin F, Field MC, Tippins JR (2004) The mechanism of oxidative stress stabilization of the thromboxane receptor in COS-7 cells. J Biol Chem 279:8316–8324
Van Diest MJ, Verbeuren TJ, Herman AG (1991) 15-lipoxygenase metabolites of arachidonic acid evoke contractions and relaxations in isolated canine arteries: role of thromboxane receptors, endothelial cells and cyclooxygenase. J Pharmacol Exp Ther 256:194–203
Vane J, Bakhle YS, Botting RM (1998) Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 38:97–120
Vanhoutte PM, Félétou M, Taddei S (2005) Endothelium-dependent contractions in hypertension. Br J Pharmacol 144:449–458
Verbeuren TJ (2006) Terutroban and endothelial TP receptors in atherogenesis. Med Sci (Paris) 22:437–443
Virdis A, Colucci R, Versari D, Ghisu N, Fornai M, Antonioli L, Duranti E, Daghini E, Giannarelli C, Blandizzi C, Taddei S, Del Tacca M (2009) Atorvastatin prevents endothelial dysfunction in mesenteric arteries from spontaneously hypertensive rats: role of cyclooxygenase 2-derived contracting prostanoids. Hypertension 53:1008–1016
Watkins MT, Patton GM, Soler HM, Albadawi H, Humphries DE, Evans JE, Kadowaki K (1999) Synthesis of 8-epi-prostaglandinF2α by human endothelial cells: role of prostaglandin H2 synthase. Biochem J 344:747–775
Widlansky ME, Price DT, Gokce N, Eberhardt RT, Duffy SJ, Holbrook M, Maxwell C, Palmisano J, Keaney JF Jr, Morrow JD, Vita JA (2003) Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension 42:310–315
Wilson SJ, Cavanagh CC, Lesher AM, Frey AJ, Russell SE, Smyth EM (2009) Activation-dependent stabilization of the human thromboxane receptor: role of reactive oxygen species. J Lipid Res 50:1047–1056
Wise H, Jones RL (1996) Focus on prostacyclin and its novel mimetics. Trends Pharmacol Sci 17:17–21
Wong MS, Man RY, Vanhoutte PM. (2010) Calcium-independent phospholipase A2 plays a key role in the endothelium-dependent contractions to acetylcholine in the aorta of SHR. Am J Physiol Heart Circ Physiol (in press)
Wong SL, Leung FP, Lau CW, Vanhoutte P, Huang Y (2008) Endothelium-dependent contractions in hamster aorta: the essential role of COX-2 and prostaglandin-2α. Basic Clin Pharmacol Toxicol 102(suppl 1):15–15
Wong SL, Leung FP, Lau CW, Au CL, Yung LM, Yao X, Chen ZY, Vanhoutte PM, Gollasch M, Huang Y (2009) Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ Res 104:228–235
Wotherspoon F, Browne DL, Meeking DR, Allard SE, Munday LJ, Shaw KM, Cummings MH (2005) The contribution of nitric oxide and vasodilatory prostanoids to bradykinin-mediated vasodilation in Type 1 diabetes. Diabet Med 22:697–702
Xavier FE, Aras-López R, Arroyo-Villa I, Campo LD, Salaices M, Rossoni LV, Ferrer M, Balfagón G (2008) Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane A2 and prostacyclin. Br J Pharmacol 154:1225–1235
Yang D, Félétou M, Boulanger CM, Wu HF, Levens N, Zhang JN, Vanhoutte PM (2002) Oxygen-derived free radicals mediate endothelium-dependent contractions to acetylcholine in aortas from spontaneously hypertensive rats. Br J Pharmacol 136:104–110
Yang D, Félétou M, Levens N, Zhang JN, Vanhoutte PM (2003) A diffusible substance(s) mediates endothelium-dependent contractions in the aorta of SHR. Hypertension 41:143–148
Yang D, Levens N, Zhang JN, Vanhoutte PM, Félétou M (2003) Specific potentiation of endothelium-dependent contractions in SHR by tetrahydrobiopterin. Hypertension 41:136–142
Yang D, Gluais P, Zhang JN, Vanhoutte PM, Félétou M (2004) Endothelium-dependent contractions to acetylcholine, ATP and the calcium ionophore A 23187 in aortas from spontaneously hypertensive and normotensive rats. Fundam Clin Pharmacol 18:321–326
Yokohama C, Takai T, Tanabe T (1988) Primary structure of sheep prostaglandin endoperoxide synthase deduced from cDNA sequence. FEBS Lett 231:347–351
Zerrouk A, Auguet M, Chabrier PE (1998) Augmented endothelium-dependent contraction to angiotensin II in the SHR aorta: role of an inducible cyclooxygenase metabolite. J Cardiovasc Pharmacol 31:525–533
Zhou Y, Mitra S, Varadharaj S, Parinandi N, Zweier JL, Flavahan NA (2006) Increased expression of cyclooxygenase-2 mediates enhanced contraction to endothelin ETA receptor stimulation in endothelial nitric oxide synthase knockout mice. Circ Res 98:1439–1445
Zou MH, Shi C, Cohen RA (2002) High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 51:198–203
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Félétou, M., Huang, Y. & Vanhoutte, P.M. Vasoconstrictor prostanoids. Pflugers Arch - Eur J Physiol 459, 941–950 (2010). https://doi.org/10.1007/s00424-010-0812-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-010-0812-6