
Abstract
Although adenosine is an important neuromodulator, its role in modulating motor functions at the level of the spinal cord is poorly understood. In the present study, we investigated the effects of adenosine on excitatory synaptic transmission and neuronal death induced by experimental ischaemia by using whole-cell patch-clamp recordings from lamina IX neurones in spinal cord slices. Adenosine significantly decreased the frequency of miniature excitatory postsynaptic currents (mEPSCs) in almost all neurones examined that could be mimicked by an A1 receptor agonist, N 6-cyclopentyladenosine (CPA), and inhibited by an A1 receptor antagonist, 8-cyclopentyl-1, 3-dipropylxanthine (DPCPX). Interestingly, adenosine increased mEPSC frequency in the presence of DPCPX in a subpopulation of neurones. In these neurones, an A2A receptor agonist, 2-[4-(2-carbonylethyl)-phenethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680), increased mEPSC frequency. Adenosine also induced an outward current that was blocked by the addition of Cs+ and tetraethylammonium into the patch-pipette solution and inhibited in the presence of Ba2+. The adenosine-induced outward current was mimicked by CPA, but not CGS21680, and inhibited by DPCPX. Moreover, superfusing with ischaemia simulating medium (ISM) generated an agonal inward current in all of the neurones tested. The latencies of the inward currents induced by ISM were significantly prolonged by adenosine or CPA, but not by CGS21680. These results suggest that adenosine receptors are functionally expressed in both the pre- and postsynaptic sites of lamina IX neurones and that their activation may exert multiple effects on motor function. Moreover, this study has provided a cellular basis for an involvement of A1 receptors in the neuroprotective actions of adenosine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adenosine is an endogenous neurotransmitter, which modulates synaptic transmission [5, 7]. The action of adenosine is mediated by specific receptors located on cell membranes, which belong to the family of G-coupled protein receptors. To date, four adenosine receptors have been cloned and characterised by classified G-coupled proteins: A1, A2A, A2B and A3 [12, 16, 38]. Functional roles of adenosine in the brain have been well investigated at the cellular level. Adenosine acting on pre- and postsynaptic A1 receptors elicits inhibitory neuromodulation and neuroprotection via activation of Gi proteins, which inhibit adenylate cyclase activity [48]. Presynaptic A1 receptor activation decreases excitatory transmitter release, and postsynaptic A1 receptor activation causes membrane hyperpolarisation through opening of potassium channels [29, 51]. On the other hand, adenosine A2A receptors produce excitatory actions via activation of Gs proteins that stimulate adenylate cyclase activity [7]. Activation of adenosine A2A receptors facilitates glutamatergic transmission [42]. A low concentration of adenosine is normally present in the extracellular fluid in the brain, but it increases dramatically during excitotoxicity, hypoxia, ischaemia or epileptic seizures [14, 45].
Adenosine receptors are widely expressed in the spinal cord as well as the brain. Ligand-binding studies have demonstrated the intense binding of radioactive A1 receptor agonists in the dorsal and ventral horn of the lumbar spinal cord [8, 17, 18]. Furthermore, in situ hybridisation studies have shown concentrated labelling of A1 receptor mRNA in ventral horn neurones and moderate labelling throughout the spinal grey matter [44]. The immunoreactivity of the A2A receptor is also present throughout the spinal cord. Strong staining is apparent in fibres and somata of presumptive motoneurones, and a similar pattern is found in the superficial dorsal horn [4]. It is well known that adenosine regulates pain transmission at the level of the spinal cord, particularly by activating A1 receptors. Intrathecal adenosine analogues or A1 receptor agonists produce antinociception in behavioural studies [20, 46]. Consistent with this, electrophysiological studies have demonstrated that adenosine directly hyperpolarises dorsal horn neurones [26, 41] and inhibits glutamatergic transmission in the dorsal horn through activation of presynaptic A1 receptors [22, 23, 41]. Furthermore, adenosine plays an important role in determining the level of activity of the sympathetic nervous system at the level of the spinal cord. A1 receptors are located in excitatory presynaptic terminals innervating neurones in the intermediolateral cell column (IML) of the lateral horn, and their activation reduces excitatory transmitter release [13]. In addition, the activation of A2A receptors on inhibitory presynaptic terminals innervating sympathetic preganglionic neurones and interneurones in the IML increases inhibitory transmitter release [4].
Several reports have described the direct actions of supraspinal adenosine on motor function [1, 6], while others have provided evidence for an indirect role of supraspinal adenosine in altering locomotor responses through its interaction with dopaminergic systems in the brain [52]. However, little is known about functional roles of adenosine in the spinal ventral horn, especially spinal motoneurones. In the present study, we therefore investigated the effects of adenosine on excitatory synaptic transmission and ischaemia-induced neuronal death by using whole-cell patch-clamp recordings from lamina IX neurones of rat spinal cord slices.
Materials and methods
All the experimental procedures involving the use of animals were approved by the Ethics Committee on Animal Experiments, Wakayama Medical University, and were in accordance with the UK Animals (Scientific Procedures) Act 1986 and associated guidelines.
Spinal cord slice preparation
The methods used for obtaining rat spinal cord slice preparations have been described previously [35]. In brief, Sprague–Dawley rats (8–12 days of age) were deeply anaesthetised with pentobarbital sodium (60 mg/kg, intraperitoneal), and then lumbosacral laminectomy was performed. The lumbosacral spinal cord (L1–S3) was removed and placed in preoxygenated Krebs solution at 1–3°C. Immediately after the removal of the spinal cord, the rats were given an overdose of pentobarbital sodium and were then killed by exsanguination. The pia-arachnoid membrane was removed after cutting all the ventral and dorsal roots near the root entry zone. The spinal cord was mounted on a microslicer, and then a 500-μm-thick transverse slice was cut. A spinal cord slice was transferred to a recording chamber (∼1 ml) and placed on the stage of an upright microscope equipped with an infrared-differential interference contrast (IR-DIC) system (BX51WI; Olympus, Tokyo, Japan). The spinal cord slice was superfused at a rate of 5–10 ml min−1 with Krebs solution saturated with 95% O2 and 5% CO2 and maintained at 36 ± 1°C. The Krebs solution contained (in millimolar) 117 NaCl, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3 and 11 glucose (pH = 7.4).
Patch-clamp recordings from lamina IX neurones
Lamina regions were identified under lower magnification (with a ×5 objective), and individual neurones were identified with a ×40 objective under the IR-DIC microscope and monitored by CCD camera (C2741-79; Hamamatsu Photonics, Hamamatsu, Japan) on a video monitor screen (Fig. 1a). Whole-cell patch-clamp recordings were made from lamina IX neurones with microelectrodes (4–8 MΩ), which were made from thin-walled filament-containing glass (1.5 mm o.d.). The patch-pipette solution used to examine the presynaptic actions of adenosine or adenosine receptor agonists was composed of (in millimolar) 110 Cs2SO4, 5 tetraethylammonium (TEA), 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES and 5 ATP-Mg (pH = 7.2). The patch-pipette solution used in the other experiments was composed of (in millimolar) 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES and 5 ATP-Mg (pH = 7.2). Signals were acquired with a patch-clamp amplifier (Axopatch 200B; Molecular Devices, Sunnyvale, CA, USA). Data were digitised with an A/D converter (Digidata 1322, Molecular Devices), stored and analysed with a personal computer using the pCLAMP data acquisition program (Version 8.2, Molecular Devices).

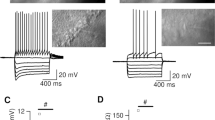
Actions of adenosine on excitatory synaptic transmission in lamina IX neurones. a Spinal cord slice preparation viewed under a ×5 objective lens (left) and a ×40 objective lens (right) of an IR-DIC microscope. Lamina IX regions were identified under a ×5 objective lens. The tip of a patch-pipette is inside the tissue ∼100 μm from the surface, and its location is indicated by a box (left). A neurone in the boxed region can be seen under a ×40 objective lens (right). b A continuous chart recording of glutamatergic miniature excitatory postsynaptic currents (mEPSCs) before and during the action of adenosine (1 mM; upper). Two consecutive traces of mEPSCs are shown in an expanded time scale, before (lower left) and during the action of adenosine (lower right). c Cumulative distribution of the inter-event interval of mEPSCs, before (continuous line) and during (dotted line) the action of adenosine. Note that adenosine shifted the inter-event interval to a longer one (P < 0.05; Kolmogorov–Smirnov test). d Glutamatergic mEPSC frequency following the application of adenosine, which is plotted against time; each bar indicates data calculated from the mEPSCs measured for 10 s. Data in b, c and d were obtained from the same neurone. The holding potential (V H) was −70 mV. Krebs solution contained TTX (1 μM)
Drug application and ischaemia simulation
Drugs were dissolved in Krebs solution and applied by perfusion via a three-way stopcock without any change in the perfusion rate or the temperature. The time necessary for the solution to flow from the stopcock to the surface of the spinal cord slice was approximately 20 s. The drugs used in this study were adenosine, N 6-cyclopentyladenosine (CPA), 8-cyclopentyl-1, 3-dipropylxanthine (DPCPX), 2-[4-(2-carbonylethyl)-phenethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680), bicuculline, strychnine, and dl-2-amino-5-phosphonopentanoic acid (AP5; Sigma, St. Louis, MO, USA), tetrodotoxin (TTX; Wako, Osaka, Japan), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Ballowin, MO, USA). CNQX was first dissolved in dimethyl sulfoxide (DMSO) at 1,000 times the concentration to be used. The other drugs were first dissolved in distilled water at 1,000 times the concentration to be used, and then these drugs were diluted to the final concentration in Krebs solution immediately before use. Ischaemia was mimicked by superfusing a Krebs solution [ischaemia simulating medium (ISM)] equilibrated with 95% N2–5% CO2 where glucose was replaced with an equimolar concentration of sucrose. ISM was also applied by perfusion via a three-way stopcock without any change in the perfusion rate or the temperature.
Statistical analysis
All numerical data were expressed as mean ± SEM. Statistical significance was determined as P < 0.05 using either the Kolmogorov–Smirnov test or the Student's paired t test. In electrophysiological data, n refers to the number of neurones studied. In analysing the change in frequency of postsynaptic currents following bath application of adenosine and adenosine receptor agonists, the time course of postsynaptic current frequency before and after agonist application was first constructed with a time bin of 10 s by the Mini Analysis Program 5.6.7 (Synaptosoft, Decatur, GA, USA). Cells were deemed to be responsive to the testing compounds when there was a >20% decrease or increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs). Then, the average response in 30 s around the peak was used to calculate the percentage change from control.
Results
Presynaptic actions of adenosine in lamina IX neurones
In order to inhibit a postsynaptic effect of adenosine through activation of K+ channels, Cs+ and TEA were added to the patch-pipette solution. Focussing on a presynaptic action of adenosine, we investigated its action on mEPSCs under this condition. After establishing the whole-cell patch-clamp configuration, lamina IX neurones exhibited mEPSCs in the presence of TTX (1 μM) at a holding potential of −70 mV where no inhibitory postsynaptic currents (IPSCs) occurred. These mEPSCs were completely blocked in the presence of a non-NMDA receptor antagonist, CNQX (10 μM; data not shown), indicating that mEPSCs were mainly mediated by glutamate released from presynaptic terminals innervating lamina IX neurones. Superfusing adenosine (1 mM) for 2 min resulted in a rapid and reversible reduction in mEPSC frequency in 41 (91.1%) of 45 neurones recorded, as shown in Fig. 1b and d. Figure 1c demonstrates the effects of adenosine (1 mM) on the cumulative distribution of the inter-event interval of the mEPSCs. While adenosine increased the proportion of mEPSCs having a significantly longer inter-event interval (P < 0.05; Kolmogorov–Smirnov test) compared to control, it had no effect on the cumulative distribution of mEPSC amplitude (P > 0.05). When measured for 30 s around the peak of its effect, mEPSC frequency and amplitude were 47.6 ± 4.7% (P < 0.05; n = 41; Fig. 4d) and 96.8 ± 2.9% (P > 0.05; n = 41) of control, respectively.
To clarify which subtype of adenosine receptor is involved in the actions of adenosine on mEPSC frequency, the effects of adenosine receptor agonists and antagonists were examined in the same neurones in which adenosine (1 mM) significantly decreased mEPSC frequency. A selective A1 receptor agonist, CPA (1 μM), exhibited an inhibitory effect similar to that of adenosine in 24 (96%) of 25 neurones examined (Fig. 2a,c). Figure 2b demonstrates the effects of CPA (1 μM) on the cumulative distribution of the inter-event intervals of the mEPSCs. CPA increased the proportion of mEPSCs having a significantly longer inter-event interval (P < 0.05) compared to control. Under the inhibitory actions of CPA, the average decrease in mEPSC frequency was 47.4 ± 5.2% (n = 24; Fig. 4d). Moreover, an adenosine-induced inhibition in mEPSC frequency was markedly suppressed in the presence of an A1 receptor antagonist, DPCPX (1 μM), in 17 of 27 neurones recorded (Fig. 3a). Interestingly, superfusion of adenosine (1 mM) significantly increased mEPSC frequency in the presence of DPCPX (1 μM) in the other ten neurones in which adenosine significantly decreased mEPSC frequency in the absence of DPCPX. The adenosine-induced decrease in mEPSC frequency in the presence of DPCPX averaged 12.3 ± 2.4% (n = 17); this value was significantly smaller than that in the absence of DPCPX (P < 0.05). On the other hand, the adenosine-induced increase in mEPSC frequency in the presence of DPCPX averaged 56.2 ± 8.0% (n = 10). With respect to an involvement of A2A receptors, the facilitatory effect of adenosine on mEPSC frequency in the presence of DPCPX was mimicked by a selective A2A receptor agonist, CGS21680. CGS21680 (1 μM) increased the mEPSC frequency in 11 (55%) of 20 neurones examined (Fig. 4a,c). Figure 2b demonstrates the effects of CGS21680 (1 μM) on the cumulative distributions of the inter-event intervals of the mEPSCs. CGS21680 increased the proportion of mEPSCs having a significantly shorter inter-event interval (P < 0.05) compared to control. The average increase in mEPSC frequency by CGS21680 was 46.7 ± 19.5% (n = 11; Fig. 4d).

Actions of CPA, an A1 receptor agonist, on excitatory synaptic transmission. a A continuous chart recording of glutamatergic mEPSCs before and during the action of CPA (1 μM; upper). Two consecutive traces of mEPSCs are shown in an expanded time scale, before (lower left) and during the action of CPA (lower right). b Cumulative distribution of the inter-event interval of mEPSCs, before (continuous line) and during (dotted line) the action of CPA. CPA shifted the inter-event interval to a longer one (P < 0.05; Kolmogorov–Smirnov test). c Glutamatergic mEPSC frequency following the application of CPA, which is plotted against time; each bar indicates data calculated from the mEPSCs measured for 10 s. Data in a, b and c were obtained from the same neurone. V H was −70 mV. Krebs solution contained TTX (1 μM)

Actions of adenosine on excitatory synaptic transmission in the presence of DPCPX, an A1 receptor antagonist. a, b A continuous chart recording of glutamatergic mEPSCs before and during the action of adenosine (1 mM) in the presence of DPCPX (1 μM; upper). Two consecutive traces of mEPSCs are shown in an expanded time scale, before (lower left) and during the action of adenosine (lower right). An adenosine-induced reduction in mEPSC frequency was blocked in the presence of DPCPX in 17 of 27 neurones recorded (a), while adenosine increased mEPSC frequency in the presence of DPCPX in the other neurones examined (b). V H was −70 mV. Krebs solution contained TTX (1 μM)

Actions of CGS21680, an A2A receptor agonist, on excitatory synaptic transmission. a A continuous chart recording of glutamatergic mEPSCs before and during the action of CGS21680 (1 μM; upper). Two consecutive traces of mEPSCs are shown in an expanded time scale, before (lower left) and during the action of CGS21680 (lower right). b Cumulative distribution of the inter-event interval of mEPSCs, before (continuous line) and during (dotted line) the action of CGS21680. CGS21680 shifted the inter-event interval to a shorter one (P < 0.05; Kolmogorov–Smirnov test). c Glutamatergic mEPSC frequency following the application of CGS21680, which is plotted against time; each bar indicates data calculated from the mEPSCs measured for 10 s. Data in a, b and c were obtained from the same neurone. d Summary of mEPSC frequency under the action of adenosine (n = 41), CPA (n = 24) and CGS21680 (n = 11), relative to control. Horizontal lines accompanied by bars show SEM; statistical significance between data shown by bars is indicated by an asterisk; *P < 0.05. V H was −70 mV. Krebs solution contained TTX (1 μM)
Postsynaptic actions of adenosine in lamina IX neurones
Postsynaptic effects of adenosine were investigated at a holding potential of −50 mV by using patch-pipettes containing potassium gluconate. Since this potential is farther than −70 mV to the equilibrium potential (−97.3 mV) for K+, as calculated from the Nernst equation using the K+ concentrations (3.6 and 140 mM, respectively) of normal Krebs and patch-pipette solutions, K+ currents could become larger. Although no synaptic activity was observed in the presence of CNQX (20 μM), AP5 (2 μM), bicuculline (20 μM) and strychnine (2 μM), superfusion of adenosine (1 mM) for 2 min induced an outward current in 33 (62%) of 54 neurones recorded under this condition (Fig. 5a). The average peak amplitude of the adenosine-induced outward current was 17.8 ± 2.1 pA (n = 33; Fig. 6c). On the other hand, adenosine did not affect the holding membrane currents when Cs+ and TEA were added to the patch-pipette solution to inhibit the activation of K+ channels (n = 6; Fig. 5b). Moreover, the adenosine-induced outward currents were significantly suppressed in amplitude by the K+ channel blocker Ba2+ (1 mM; Fig. 5c). The average amplitude of slow IPSCs was 4.8 ± 1.8 pA in the presence of Ba2+; this value was significantly smaller than that in the absence of Ba2+ (16.9 ± 5.8 pA, n = 8; Fig. 5d). These findings suggest that the adenosine-induced outward currents are mediated by the activation of K+ channels. We further investigated which subtype of adenosine receptor is involved in the postsynaptic actions of adenosine on holding membrane currents. DPCPX (1 μM) markedly reduced the adenosine-induced outward current in all nine neurones examined (Fig. 6a). The average peak amplitude of the adenosine-induced outward current in the presence of DPCPX was 0.6 ± 0.4 pA (n = 9); this value was significantly smaller than that in the absence of DPCPX (P < 0.05). Moreover, CPA (1 μM) induced an outward current in those neurones in which adenosine (1 mM) generated an outward current (n = 22; Fig. 6b). The average peak amplitude of the CPA-induced outward current was 16.4 ± 1.6 pA (n = 22; Fig. 6c). On the other hand, CGS21680 (1 μM) did not induce any outward current in those neurones in which adenosine (1 mM) generated an outward current (n = 14).

Adenosine induces an outward current through activation of K+ channels in lamina IX neurones. a Adenosine (1 mM) produces a slow outward current in the presence of CNQX (20 μM), AP5 (2 μM), bicuculline (20 μM) and strychnine (2 μM; upper). b Adenosine (1 mM) did not induce any outward current with the addition of Cs+ and TEA into the patch-pipette solution. c Adenosine (1 mM) was administrated in the absence (upper) and presence (lower) of Ba2+ (1 mM). The adenosine-induced outward current was significantly reduced in the presence of Ba2+. d The average amplitudes of the outward currents induced by adenosine in the absence and presence of Ba2+ (n = 8). Horizontal lines accompanied by bars show SEM; statistical significance between data shown by bars is indicated by an asterisk; *P < 0.05. V H = −50 mV

Effects of adenosine receptor agonist and antagonist on the adenosine-induced outward currents. a The adenosine-induced outward current was significantly inhibited by DPCPX (1 μM; lower). b Slow outward currents were produced by superfusing adenosine (1 mM; upper) and CPA (1 μM; lower) for 2 min in the same neurone. c The average amplitudes of the outward currents induced by adenosine in the absence (n = 33) and presence of DPCPX (n = 9) and by CPA (n = 22). Horizontal lines accompanied by bars show SEM; statistical significance between data shown by bars is indicated by an asterisk; *P < 0.05. V H = −50 mV
Effects of adenosine on membrane dysfunction induced by experimental ischaemia
In order to investigate whether adenosine has a neuroprotective effect in lamina IX neurones, we further observed the actions of adenosine on membrane dysfunction induced by experimental ischaemia. As we reported previously [36, 37], ISM exposure for several minutes generated an agonal inward current at a holding potential of −70 mV in all 59 lamina IX neurones recorded (Fig. 7a). When continuously superfused with ISM, the synaptic activity disappeared, and then the holding current became unstable and irreversible even if oxygen and glucose were reintroduced, thus indicating that ISM resulted in irreversible membrane dysfunction [36]. This agonal inward current consisted of a slow and subsequent rapid inward current (Fig. 7a). The latency of the rapid inward current was measured from the onset of superfusion with the ISM to the onset of the rapid inward current, as estimated by extrapolating the slope of the rapid inward current to the slope of the slow current (Fig. 7a). The average latency of the rapid inward current was 281.1 ± 11.1 s in control neurones (n = 17; Fig. 7c). Adenosine (1 mM) or CPA (1 μM) markedly prolonged the latencies of the inward currents induced by the ISM when compared to control (Fig. 7b), while CGS21680 did not significantly change the latency. The average latencies of the rapid inward currents in the presence of adenosine (1 mM), CPA (1 μM) and CGS21680 (1 μM) were 362.5 ± 15.6 (n = 10), 347.9 ± 18.2 (n = 23) and 283.9 ± 8.9 s (n = 9), respectively (Fig. 7c).

Adenosine prolonged the latency of inward currents induced by ISM. a A continuous chart recording of mEPSCs before and during the application of ISM. ISM produced an agonal inward current, which consisted of a slow and subsequent rapid inward current. The onset of the rapid inward current, as estimated by extrapolating the slope of the rapid inward current to the slope of the slow inward current. b A continuous chart recording of mEPSCs before and during the application of ISM in control (upper) and in the presence of adenosine (1 mM; middle) and CPA (1 μM; lower) at the same scale. Arrows indicate the onsets of the ISM-induced rapid inward currents. c Summary of the latencies of the rapid inward currents after ISM exposure in control (n = 17) and in the presence of adenosine (n = 10), CPA (n = 23) and CGS21680 (n = 9). Horizontal lines accompanied by bars show SEM; statistical significance between data shown by bars is indicated by asterisk; *P < 0.05; n.s., not significant. V H was −70 mV
Discussion
It is well known that adenosine modulates synaptic transmission throughout the central nervous system (CNS). However, intracellular recordings examining adenosine modulation of lumbar spinal motoneurones have not been documented. In the present study, we have demonstrated that adenosine receptors are functionally expressed in both the pre- and postsynaptic sites of lamina IX neurones of the spinal cord and that their activation exerts multiple effects on synaptic transmission.
Adenosine was shown, in the present study, to decrease glutamatergic mEPSC frequency in almost all lamina IX neurones recorded, and this effect could be mimicked by CPA, an A1 receptor agonist, and inhibited by DPCPX, an A1 receptor antagonist. These results indicate that this inhibitory effect of adenosine in lamina IX neurones is presynaptically mediated by A1 receptors; This presynaptic inhibition by the A1 receptor is widespread in the CNS [5]. Consistent with our electrophysiological findings, the presence of A1 receptors in the spinal ventral horn has been reported previously. Ligand-binding studies have demonstrated the intense binding of radioactive A1 receptor agonists in the ventral horn as well as the dorsal horn [17, 18, 8]. On the other hand, adenosine increased glutamatergic mEPSC frequency in the presence of DPCPX in a subpopulation of lamina IX neurones. CGS21680, an A2A receptor agonist, also increased glutamatergic mEPSC frequency in those neurones. These results indicate the involvement of A2A receptors in the facilitatory effect of adenosine on glutamate release from presynaptic terminals onto lamina IX neurones. This is consistent with the facilitatory effect of A2A receptor activation on excitatory synaptic transmission observed in other areas of the CNS [47]. Furthermore, A1 and A2A receptors are often expressed in the same regions [11] and indeed on the same nerve terminals at the neuromuscular junction [9]. Therefore, A1 and A2A receptors may be co-expressed in the same presynaptic terminals innervating lamina IX neurones of the spinal cord. However, further investigations will be required to clarify whether A1 and A2A receptors are co-expressed in the same presynaptic terminals in the spinal ventral horn. Since the activation of A1 and A2A receptors have opposite effects in the present study, the coexistence of these receptors leads to the question of which subtype of adenosine receptor is preferentially activated. Although adenosine presynaptically activates both A1 and A2A receptors in this region, adenosine itself significantly inhibited mEPSC frequency in almost all neurones examined. Thus, adenosine preferentially activates A1 receptors, with the result that the overall adenosine effect is a decrease in glutamate release from presynaptic terminals onto lamina IX neurones. This may be due to the difference in adenosine affinity between the subtypes of adenosine receptor since adenosine has a higher affinity for A1 receptors than A2A receptors [14].
Although it is well known that adenosine hyperpolarises membranes of CNS neurones [51], this effect has never been examined in lumbar spinal motoneurones. The present study demonstrated that adenosine produces an outward current at −50 mV in about 60% of lamina IX neurones recorded. The adenosine-induced outward current is mimicked by CPA, but not by CGS21680, and is inhibited by DPCPX. These results suggest that A1 receptors are expressed in the postsynaptic sites of lamina IX neurones and that their activation directly hyperpolarises the majority of lamina IX neurones. Consistent with this, in situ hybridisation studies have shown the concentrated labelling of A1 receptor mRNA in ventral horn neurones [44].
Previous behavioural studies have reported that intrathecal delivery of adenosine receptor agonists produces a reversible motor dysfunction [21, 25]. Although motor flaccidity was induced by both of A1 or A2A receptor agonists, this motor effect was antagonised by an A2A receptor antagonist [25]. Therefore, it has been considered that motor function in the spinal cord is mainly mediated by the activation of A2A receptors. On the other hand, we have demonstrated in the present study that excitatory synaptic transmission is presynaptically modulated by both inhibitory A1 and facilitatory A2A receptors and that the activation of postsynaptic A1 receptors directly hyperpolarises the majority of lamina IX neurones. Consistent with this, A1 receptors have been shown to be present in embryonic mouse motoneurones, and their activation decreased the excitability of motoneurones [34]. Also, acetylcholine release from rat motor nerve terminals is also modulated by both inhibitory A1 and facilitatory A2A receptors. Interestingly, intrathecal administration of an adenosine kinase inhibitor caused a significant reduction in locomotor activity as did intrathecal delivery of adenosine receptor agonists [32]. However, the site of the effect of an adenosine kinase inhibitor on locomotor activity did not appear to be the spinal cord, but was in supraspinal areas. Hypomobility after intrathecal delivery of an adenosine kinase inhibitor was likely to be due to its diffusion to supraspinal sites since its effect was not antagonised by intrathecal administration of an adenosine receptor antagonist but was reversed by direct administration of an adenosine receptor antagonist into the lateral ventricles. Therefore, the discrepancy between the previous behavioural studies and the present study may be due to the activation of A2A receptors in the supraspinal region.
Post-traumatic degeneration of the spinal cord is caused by a secondary injury process, which occurs during the first minutes, hours, and days after spinal cord injury [10, 28]. The mechanism of the secondary injury includes a variety of processes such as ischaemia, glutamate excitotoxicity, free radical-mediated cell death, ATP released from damaged tissues and cytoskeletal degradation [40]. As the primary injury is immediate and irreversible, it is not well suited for therapeutic intervention. Due to the delayed processes, the secondary injury is the most appropriate target for therapeutic intervention [2]. A high-dose regimen of steroid drugs such as methylprednisolone is often delivered to reduce the secondary injury [3, 19]. However, the effect of steroid drugs is not satisfactory, and their complications cannot be ignored [31]. Many other neuroprotective interventions have been tested in spinal cord injury patients and model animals [2]. Unfortunately, none has produced a major improvement in neurological recovery or a meaningful increase in function, although a lot of effort and resources have been expended. Adenosine levels markedly increase in response to ischaemia and hypoxia [14, 45]. Elevating extracellular adenosine levels by inhibiting adenosine degradation or uptake reduces hypoxic–ischaemic neuronal injury [24]. Glutamate is a key neurotransmitter in neuronal injury and damages postsynaptic neurones by allowing Ca2+ entry through activation of NMDA receptors, and adenosine decreases the spontaneous and electrically evoked release of glutamate in the brain [15, 43, 27]. These findings support an important role for adenosine in modulating neuronal injuries [33, 39]. However, it is still unknown whether adenosine acts predominantly as a neuroprotectant in spinal motoneurones. We further investigated the effects of adenosine or adenosine receptor agonists on ischaemia-induced neuronal death. In the present study, ischaemia was simulated by superfusing an oxygen- and glucose-deprived medium (ISM), which has been well established in spinal and brain slices [30, 49, 50]. ISM superfusion for several minutes induced an agonal inward current in lamina IX neurones, as we demonstrated previously [36, 37]. The latency of the ISM-induced rapid inward current was significantly prolonged by adenosine or an A1 receptor agonist CPA, but not affected by an A2A receptor agonist CGS21680. These results suggest that ischaemic neuronal death of spinal motoneurones could be reduced by activation of A1 receptors. Although adenosine activates both inhibitory A1 and facilitatory A2A receptors in the lamina IX neurones, adenosine preferentially activates A1 receptors, with the result that adenosine acts as a neuroprotectant in spinal motoneurones.
In summary, we have demonstrated for the first time that adenosine receptors are functionally expressed in both the pre- and postsynaptic sites of lamina IX neurones and that their activation may exert multiple effects on motor function. Moreover, this study has provided a cellular basis for the involvement of A1 receptors in the neuroprotective effects of adenosine in the spinal ventral horn.
References
Barraco RA, Martens KA, Parizon M, Normile HJ (1993) Adenosine A2a receptors in the nucleus accumbens mediate locomotor depression. Brain Res Bull 31:397–404
Blight AR, Zimber MP (2001) Acute spinal cord injury: pharmacotherapy and drug development perspectives. Curr Opin Investig Drugs 2:801–808
Bracken MB (2001) Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine 26:S47–54
Brooke RE, Deuchars J, Deuchars SA (2004) Input-specific modulation of neurotransmitter release in the lateral horn of the spinal cord via adenosine receptors. J Neurosci 24:127–137
Brundege JM, Dunwiddie TV (1997) Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv Pharmacol 39:353–391
Bruns RF, Katims JJ, Annau Z, Snyder SH, Daly JW (1983) Adenosine receptor interactions and anxiolytics. Neuropharmacology 22:1523–1529
Burnstock G (1997) The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology 36:1127–1139
Choca JI, Proudfit HK, Green RD (1987) Identification of A1 and A2 adenosine receptors in the rat spinal cord. J Pharmacol Exp Ther 242:905–910
Correia-de-Sá P, Ribeiro JA (1996) Adenosine uptake and deamination regulate tonic A2a receptor facilitation of evoked [3H]acetylcholine release from the rat motor nerve terminals. Neuroscience 73:85–92
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3:73–76
Cunha RA, Milusheva E, Vizi ES, Ribeiro JA, Sebastião AM (1994) Excitatory and inhibitory effects of A1 and A2A adenosine receptor activation on the electrically evoked [3H]acetylcholine release from different areas of the rat hippocampus. J Neurochem 63:207–214
Daly JW, Butts-Lamb P, Padgett W (1983) Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell Mol Neurobiol 3:69–80
Deuchars SA, Brooke RE, Deuchars J (2001) Adenosine A1 receptors reduce release from excitatory but not inhibitory synaptic inputs onto lateral horn neurons. J Neurosci 21:6308–6320
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55
Faden AI, Simon RP (1988) A potential role for excitotoxins in the pathophysiology of spinal cord injury. Ann Neurol 23:623–626
Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M (1994) Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143–156
Geiger JD, LaBella FS, Nagy JI (1984) Characterization and localization of adenosine receptors in rat spinal cord. J Neurosci 4:2303–2310
Goodman RR, Synder SH (1982) Autoradiographic localization of adenosine receptors in rat brain using [3H]cyclohexyladenosine. J Neurosci 2:1230–1241
Hall ED (1993) Neuroprotective actions of glucocorticoid and nonglucocorticoid steroids in acute neuronal injury. Cell Mol Neurobiol 13:415–432
Holmgren M, Hedner J, Mellstrand T, Nordberg G, Hedner T (1986) Characterization of the antinociceptive effects of some adenosine analogues in the rat. Naunyn Schmiedebergs Arch Pharmacol 334:290–293
Karlsten R, Gordh T Jr, Hartvig P, Post C (1990) Effects of intrathecal injection of the adenosine receptor agonists R-phenylisopropyl-adenosine and N-ethylcarboxamide-adenosine on nociception and motor function in the rat. Anesth Analg 71:60–64
Lao LJ, Kumamoto E, Luo C, Furue H, Yoshimura M (2001) Adenosine inhibits excitatory transmission to substantia gelatinosa neurons of the adult rat spinal cord through the activation of presynaptic A1 adenosine receptor. Pain 94:315–324
Lao LJ, Kawasaki Y, Yang K, Fujita T, Kumamoto E (2004) Modulation by adenosine of Ad and C primary-afferent glutamatergic transmission in adult rat substantia gelatinosa neurons. Neuroscience 125:221–231
Latini S, Bordoni F, Pedata F, Corradetti R (1999) Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. Br J Pharmacol 127:729–739
Lee YW, Yaksh TL (1996) Pharmacology of the spinal adenosine receptor which mediates the antiallodynic action of intrathecal adenosine agonists. J Pharmacol Exp Ther 277:1642–1648
Li J, Perl ER (1994) Adenosine inhibition of synaptic transmission in the substantia gelatinosa. J Neurophysiol 72:1611–1621
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622
Lu J, Ashwell KW, Waite P (2000) Advances in secondary spinal cord injury: role of apoptosis. Spine 25:1859–1866
Lupica CR, Proctor WR, Dunwiddie TV (1992) Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci 12:3753–3764
Matsumoto N, Kumamoto E, Furue H, Yoshimura M (2003) GABA-mediated inhibition of glutamate release during ischemia in substantia gelatinosa of the adult rat. J Neurophysiol 89:257–264
Matsumoto T, Tamaki T, Kawakami M, Yoshida M, Ando M, Yamada H (2001) Early complications of high-dose methylprednisolone sodium succinate treatment in the follow-up of acute cervical spinal cord injury. Spine 26:426–430
McGaraughty S, Chu KL, Wismer CT, Mikusa J, Zhu CZ, Cowart M, Kowaluk EA, Jarvis MF (2001) Effects of A-134974, a novel adenosine kinase inhibitor, on carrageenan-induced inflammatory hyperalgesia and locomotor activity in rats: evaluation of the sites of action. J Pharmacol Exp Ther 296:501–509
Mitchell HL, Frisella WA, Brooker RW, Yoon KW (1995) Attenuation of traumatic cell death by an adenosine A1 agonist in rat hippocampal cells. Neurosurgery 36:1003–1007
Mynlieff M, Beam KG (1994) Adenosine acting at an A1 receptor decreases N-type calcium current in mouse motoneurons. J Neurosci 14:3628–3634
Nakatsuka T, Ataka T, Kumamoto E, Tamaki T, Yoshimura M (2000) Alteration in synaptic inputs through C-afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience 99:549–556
Nishi H, Nakatsuka T, Takeda D, Miyazaki N, Sakanaka J, Yamada H, Yoshida M (2007) Hypothermia suppresses excitatory synaptic transmission and neuronal death induced by experimental ischemia in spinal ventral horn neurons. Spine 32:E741–747
Nohda K, Nakatsuka T, Takeda D, Miyazaki N, Nishi H, Sonobe H, Yoshida M (2007) Selective vulnerability to ischemia in the rat spinal cord—a comparison between ventral and dorsal horn neurons. Spine 32:1060–1066
Olah ME, Stiles GL (1995) Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol 35:581–606
Ongini E, Adami M, Ferri C, Bertorelli R (1997) Adenosine A2A receptors and neuroprotection. Ann N Y Acad Sci 825:30–48
Park E, Velumian AA, Fehlings MG (2004) The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma 21:754–774
Patel MK, Pinnock RD, Lee K (2001) Adenosine exerts multiple effects in dorsal horn neurones of the adult rat spinal cord. Brain Res 920:19–26
Rebola N, Sebastião AM, de Mendonca A, Oliveira CR, Ribeiro JA, Cunha RA (2003) Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J Neurophysiol 90:1295–1303
Regan RF, Choi DW (1991) Glutamate neurotoxicity in spinal cord cell culture. Neuroscience 43:585–591
Reppert SM, Weaver DR, Stehle JH, Rivkees SA (1991) Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol 5:1037–1048
Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB (1992) Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci 13:439–445
Sawynok J (1998) Adenosine receptor activation and nociception. Eur J Pharmacol 347:1–11
Sebastião AM, Ribeiro JA (1996) Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol 48:167–189
Stiles GL (1992) Adenosine receptors. J Biol Chem 267:6451–6454
Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H (1997) Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78:891–902
Thompson RJ, Zhou N, MacVicar BA (2006) Ischemia opens neuronal gap junction hemichannels. Science 312:924–927
Trussell LO, Jackson MB (1985) Adenosine-activated potassium conductance in cultured striatal neurons. Proc Natl Acad Sci U S A 82:4857–4861
Xie X, Ramkumar V, Toth LA (2007) Adenosine and dopamine receptor interactions in striatum and caffeine-induced behavioral activation. Comp Med 57:538–545
Acknowledgements
This work was supported by a grant from The General Insurance Association of Japan and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miyazaki, N., Nakatsuka, T., Takeda, D. et al. Adenosine modulates excitatory synaptic transmission and suppresses neuronal death induced by ischaemia in rat spinal motoneurones. Pflugers Arch - Eur J Physiol 457, 441–451 (2008). https://doi.org/10.1007/s00424-008-0542-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0542-1