
Abstract
During metabolic acidosis (MA), urinary phosphate excretion increases and contributes to acid removal. Two Na+-dependent phosphate transporters, NaPi-IIa (Slc34a1) and NaPi-IIc (Slc34a3), are located in the brush border membrane (BBM) of the proximal tubule and mediate renal phosphate reabsorption. Transcriptome analysis of kidneys from acid-loaded mice revealed a large decrease in NaPi-IIc messenger RNA (mRNA) and a smaller reduction in NaPi-IIa mRNA abundance. To investigate the contribution of transporter regulation to phosphaturia during MA, we examined renal phosphate transporters in normal and Slc34a1-gene ablated (NaPi-IIa KO) mice acid-loaded for 2 and 7 days. In normal mice, urinary phosphate excretion was transiently increased after 2 days of acid loading, whereas no change was found in Slc34a1−/− mice. BBM Na/Pi cotransport activity was progressively and significantly decreased in acid-loaded KO mice, whereas in WT animals, a small increase after 2 days of treatment was seen. Acidosis increased BBM NaPi-IIa abundance in WT mice and NaPi-IIc abundance in WT and KO animals. mRNA abundance of NaPi-IIa and NaPi-IIc decreased during MA. Immunohistochemistry did not indicate any change in the localization of NaPi-IIa and NaPi-IIc along the nephron. Interestingly, mRNA abundance of both Slc20 phosphate transporters, Pit1 and Pit2, was elevated after 7 days of MA in normal and KO mice. These data demonstrate that phosphaturia during acidosis is not caused by reduced protein expression of the major Na/Pi cotransporters NaPi-IIa and NaPi-IIc and suggest a direct inhibitory effect of low pH mainly on NaPi-IIa. Our data also suggest that Pit1 and Pit2 transporters may play a compensatory role.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Body inorganic phosphate (Pi) homeostasis is the product of intestinal absorption and renal excretion or reabsorption, respectively. The bulk of filtered phosphate is reabsorbed at the brush border membrane (BBM) of proximal tubular cells via action of sodium-dependent phosphate (Na/Pi) transporters, NaPi-IIa (Slc34a1) [23], and to a smaller extent, NaPi-IIc (Slc34a3) [30]. Abundance and activity of NaPi-IIa are regulated by a variety factors, including dietary Pi intake, parathyroid hormone, as well as other hormones [18, 22, 24], and FGF23 [21]. In contrast, less is known about the regulation of NaPi-IIc, which appears to be modulated by dietary phosphate intake, growth, and FGF-23 [26, 30–32]. Intestinal Pi absorption appears to be mediated by one sodium-dependent cotransporter NaPi-IIb (Slc34a2) that may be also altered by dietary Pi intake, 1,25 (OH)2-dihydroxyvitamin D3, and FGF-23 [8, 14, 28].
Recently, a different class (type III) of Na+-dependent Pi transporters were identified in rat kidney: Pit1 (Glvr-1, Slc20a1) and Pit2 (Ram-1, Slc20a2) [15, 16]. Both Pit1 and Pit2 are widely distributed in many tissues, suggesting that they serve as housekeeping Na/Pi cotransporters in mammalian cells [15].The exact cellular and subcellular localization of Pit1 and Pit2 in the kidney remains to be determined, although in situ hybridization suggests expression along the proximal nephron [35].
The bulk of apical Na/Pi cotransport activity in the proximal tubule is accounted for by the type IIa Na/Pi cotransporter, as evident from murine NaPi-IIa gene ablation studies [3]. NaPi-IIc has been suggested to be growth-related and important only during weaning in rodents [30]. However, recent studies identified SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria, indicating that NaPi-IIc may play a more prominent role in maintaining body phosphate homeostasis than previously thought [4, 19].
During metabolic acidosis, phosphate is released from bone together with Ca2+ and carbonate [17] and is excreted into urine in large amounts [12]. Phosphate, together with ammonia and citrate, acts as so-called titratable acid that binds protons and increases the kidney’s ability to excrete protons. Ambuhl et al. reported previously that induction of metabolic acidosis in a rat model caused massive downregulation of sodium-dependent phosphate transport activity in brush border membrane vesicles associated with reduced NaPi-IIa messenger RNA (mRNA) and protein abundance [1]. In contrast, we failed to find a clear reduction in brush border membrane Na+/phosphate cotransport activity and NaPi-IIa expression both in rat and mouse kidney despite massive phosphaturia [33].
During complementary DNA (cDNA) microarray screening of mouse kidneys 2 and 7 days after the induction of metabolic acidosis, we found strong downregulation of NaPi-IIc mRNA abundance, which may contribute to the phosphaturia observed [25]. To further investigate the contribution of phosphate transporter regulation to phosphaturia, we induced metabolic acidosis for 2 and 7 days in C57BL/6 and NaPi-IIa KO mice and examined NaPi-IIa, NaPi-IIc, Pit1, and Pit2 mRNA expression, changes in NaPi-IIa and NaPi-IIc protein abundance, transport activity, and cotransporter localization.
Materials and methods
Animals
All experiments were performed on 12-week-old C57BL/6J and homozygous NaPi-IIa-deficient (Slc34a1−/−) male mice. The generation, breeding, and genotyping of these mice have been described previously [3]. All experiments were performed according to Swiss Animal Welfare laws and approved by the local veterinary authority (Veterinäramt Zürich).
Metabolic studies
To induce metabolic acidosis, male C57BL/6J and NaPi-IIa deficient mice were given 0.28 M NH4Cl/ 2% sucrose in drinking water for 2 or 7 days. The control group received only 2% sucrose in drinking water for 7 days. Each group consisted of five animals for each time point, treatment, and genotype. Forty-eight hours prior to sacrifice, mice were housed in metabolic cages (single mouse metabolic cages, Tecniplast, Buguggiate, Italy) and had free access to standard mouse chow and drinking water ad libitum. Daily chow (standard rodent chow GLP3433, Kliba AG, Switzerland: 0.8 % phosphate, 1.05 % calcium, 0.2 % magnesium), water intake, and body weights were measured, and urine was collected under mineral oil. For the acute induction of metabolic acidosis, mice were trained over 5 days to receive food only for 6 h in the morning. Two days before the actual experiment, mice were adapted to metabolic cages. On the day of the experiment, mice received either standard chow or 2 g NH4Cl/100g standard chow over a period of 6 h with free access to water. Urine was collected over 6h. Before and after the collection period, urinary bladders were emptied by abdominal massage. At the end of all metabolic cage experiments, mice were anesthetized with ketamine–xylazine, and heparinized mixed arterial-venous blood was collected and analyzed immediately for pH, blood gases, and electrolytes on a Radiometer ABL 800 (Radiometer, Copenhagen, Denmark) blood gas analyzer. Plasma was collected and frozen until further analysis. Both kidneys were harvested and rapidly frozen in liquid nitrogen and stored at −80°C.
Urinary pH was measured using a pH microelectrode (691 pH-meter, Metroholm). Urinary creatinine was measured by the Jaffe method [29]. Ammonium in urine was measured by the method of Berthelot [5]. Urine and plasma phosphate were measured using a commercial kit (Sigma Diagnostics, Munich, Germany). Urinary electrolytes (Na+, K+, Ca2+, Mg2+, Cl−) were measured by ion chromatography (Metrohm ion chromatograph, Herisau, Switzerland).
RNA extraction and reverse transcription
Snap-frozen kidneys (five kidneys for each condition) were homogenized in RLT-Buffer (Qiagen, Basel, Switzerland) supplemented with β-mercaptoethanol to a final concentration of 1%. Total RNA was extracted from 200 μl aliquots of each homogenized sample using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Quality and concentration of the isolated RNA preparations were analyzed by the ND-1000 spectrophotometer (NanoDrop Technologies). Total RNA samples were stored at −80°C. Each RNA sample was diluted to 100 ng/μl and 3 μl used as a template for reverse transcription using the TaqMan Reverse Transcription Kit (Applied Biosystems, Forster City, CA, USA). For reverse transcription, 300 ng of RNA template were diluted in a 40-μl reaction mix that contained (final concentrations) RT buffer (1×), MgCl2 (5.5mM), random hexamers (2.5 μM), RNase inhibitor (0.4U/μl), the multiscribe reverse transcriptase enzyme (1.25U/μl), dNTP mix (500 μM each), and RNase-free water.
Real-time quantitative PCR
Quantitative real-time qRT-PCR was performed on the ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Primers for all genes of interest (Table 1) were designed using Primer Express software from Applied Biosystems. Primers were chosen to result in amplicons no longer than 150bp spanning intron-exon boundaries to exclude genomic DNA contamination. The specificity of all primers was first tested on mRNA derived from kidney and always resulted in a single product of the expected size (data not shown). Probes were labeled with the reporter dye FAM at the 5’ end and the quencher dye TAMRA at the 3’ end (Microsynth, Balgach, Switzerland). Real-Time PCR reactions were performed using TaqMan Universal PCR Master Mix (Applied Biosystems). Briefly, 3.5 μl cDNA, 1 μl of each primer (25 μM), 0.5 μl labeled probe (5 μM), 6.5 μl RNAse-free water, and 12.5 μl TaqMan Universal PCR Master Mix reached 25 μl of final reaction volume. Reaction conditions were denaturation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15s and annealing/elongation at 60°C for 60s with auto ramp time. All reactions were run in duplicate. For analyzing the data, the threshold was set to 0.06 as this value had been determined to be in the linear range of the amplification curves for all mRNAs in all experimental runs. The expression of gene of interest was calculated in relation to hypoxanthine guanine phosphoribosyl transferase (HPRT). Relative expression ratios were calculated as R = 2(Ct(HPRT) − Ct(test gene), where Ct represents the cycle number at the threshold 0.06.
BBM vesicles preparation
NH4Cl-treated WT and KO mice were anesthetized with ketamine–xylazine intraperitoneally, and kidneys were removed and rapidly frozen in liquid nitrogen. Brush border membranes vesicles (BBMV) were prepared as described previously using the Mg2+-precipitation technique [6, 7] and further used for western blotting and transport studies.
Transport studies
The transport rate of phosphate into renal BBMV was determined at 30s and 120min (equilibrium value) as described [14] at 25°C in the presence of inward gradients of 100mM NaCl or 100mM KCl and 0.1mM K2HPO4. All measurements were performed in triplicates.
Western blot analysis
After measurement of the protein concentration (Bio-Rad, Hercules, CA, USA), 10 μg of brush border membrane proteins were solubilized in Laemmli sample buffer, (2 % 2-mercaptoethanol), and SDS-PAGE was performed on a 10 % polyacrylamide gel. For immunoblotting, the proteins were transferred electrophoretically to polyvinylidene fluoride membranes (Immobilon-P, Millipore, Bedford, MA, USA). After blocking with 5% milk powder in Tris-buffered saline/0.1% Tween-20 for 60 min, the blots were incubated with the primary antibodies: rabbit polyclonal anti-NaPi-IIa (1:6,000) [10], rabbit affinity-purified anti-NaPi-IIc (1:1,000) raised against the C-terminal amino acids 588–601 (NH2–CYENPQVIASQQL–COOH; Pineda Antibody Services, Berlin, Germany) and mouse monoclonal anti-β-actin antibody (42kDa; Sigma, St. Louis, MO; 1:5,000) either for 2 h at room temperature or overnight at 4°C. The specificity of the NaPi-IIc antibody was tested by western blotting and immunohistochemistry (see supplementary Figs. 1 and 2). The membranes were then washed three times, blocked for 1 h, and again incubated for 1 h at room temperature with the secondary goat anti-rabbit or donkey anti-mouse antibodies 1:5,000 linked to alkaline phosphatase (Promega, USA). The protein signal was detected with the CDP Star chemiluminescence system (Roche Diagnostics, Basel, Switzerland) using the DIANA III-chemiluminescence detection system (Raytest, Straubenhardt, Germany). All images were analyzed using appropriate software (Advanced Image Data Analyser AIDA, Raytest) to calculate the protein of interest/β-actin ratio.
Immunohistochemistry
Mouse kidneys were perfusion-fixed through the right ventricle with a fixative solution, and subsequent immunohistochemistry was performed as described previously [11]. Serial cryosections (5 μm) were taken and incubated at 4°C overnight with a rabbit anti NaPi-IIa 1:1,000 as described previously [10] or immunopurified rabbit anti NaPi-IIc 1:1,500. The specificity of the immunopurified anti-NaPi-IIc antibody was characterized as shown in the supplementary figures (supplementary Fig. 2). Binding sites of the primary antibodies were detected using Alexa 555-conjugated goat/anti-rabbit antibodies (Invitrogen, Basel, Switzerland). F-actin was visualized using fluorescein-coupled phalloidin (Invitrogen, Basel, Switzerland). Sections were studied by epifluorescence with a Polyvar microscope (Reichert Jung, Vienna, Austria), and digital images were acquired with a charged coupled device camera.
Statistical analysis
Results are expressed as mean ± SEM. All data were tested for significance using the ANOVA and unpaired Student’s test where appropriate. Only values with p < 0.05 were considered as significant.
Results
Induction of metabolic acidosis
NH4Cl-induced metabolic acidosis was achieved after 48h and 7 days in both WT and NaPi-IIa KO mice, as evident from the reduction in blood pH and bicarbonate concentration and the increase in serum chloride concentration (Table 2). Urinary pH was decreased only in NH4Cl-loaded WT mice. In NaPi-IIa-deficient animals, urine pH, which was significantly lower than that of WT mice under control conditions (5.27 ± 0.07 vs. 6.85 ± 0.30, respectively), was not changed by NH4Cl treatment (Table 2). Urinary ammonium excretion was increased in WT and KO acid-loaded animals. As expected, the plasma phosphate concentration was lower in NaPi-IIa KO mice compared to WT mice under all conditions examined; however, metabolic acidosis did not significantly affect blood Pi concentration either in WT or in KO animals (Table 2). Urinary Pi excretion was higher in KO mice compared to WT mice under all conditions examined and was further increased only in WT animals after 2 days of NH4Cl loading (Table 2; Fig. 1). In KO animals, acid loading reduced renal phosphate excretion non-significantly. Metabolic acidosis was more pronounced in animals receiving NH4Cl for 2 days, as evident from blood pH and plasma bicarbonate concentrations, indicating partial adaptation of these animals to acid-base disturbances after 7 days of acid loading (Table 2).

Renal phosphate excretion during metabolic acidosis. Urine was collected over 24 h in metabolic cages from wild-type (Slc34a1+/+) control mice and mice receiving NH4Cl for 2 or 7 days. Mice deficient for NaPi-IIa (Slc34a1−/−) were treated in parallel. Acid loading increased 24 h urinary phosphate excretion in wild-type mice after 2 days, but no effect was observed after 7 days. Slc34a1−/− mice had higher 24-h phosphate excretion, which was non-significantly reduced by acid loading for 2 or 7 days (n = 5 animals/group). All data are presented as mean ± SEM (n = 5). *p < 0.05
Acidosis alters mRNA expression of renal Na+/phosphate cotransporters
To investigate the effect of metabolic acidosis on mRNA expression of renal phosphate transporters, real-time quantitative RT-PCR was performed for NaPi-IIa, NaPi-IIc, Pit1, and Pit2 and the housekeeping gene HPRT. Induction of metabolic acidosis with NH4Cl resulted in a decrease in renal NaPi-IIa mRNA abundance in WT mice after 2 days but not after 7 days (Fig. 2a). In contrast, a strong decrease in the relative mRNA abundance of NaPi-IIc was evident in both WT and KO mice after 2 and 7 days (Fig. 2b). Under control conditions, NaPi-IIc mRNA expression was 2-fold higher in KO mice compared to WT animals. Metabolic acidosis resulted in increased mRNA expression of Pit1, which reached significance in WT and KO mice only after 7 days of acid loading (Fig. 2c). mRNA abundance of Pit2 was not affected by NH4Cl treatment in WT mice and increased after 7 days of metabolic acidosis in NaPi-IIa KO mice (Fig. 2d).

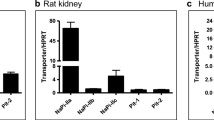
Expression of genes encoding renal phosphate transporters. Real-time RT-PCR was used to assess NaPi-IIa, NaPi-IIc, Pit1, and Pit2 mRNA levels in kidneys from control and NH4Cl-loaded animals (n = 5/group). a NaPi-IIa mRNA expression significantly decreased after 2 days acid loading and returned to normal values after 7 days. b NaPi-IIc mRNA expression decreased in normal and NaPi-IIa-deficient mice during MA. Under control conditions, NaPi-IIc expression was 2-fold higher in NaPi-IIa-deficient mice compared to wild-type animals. Expression of Pit1 was higher in the absence of NaPi-IIa and increased in both wild-type and Slc34a1−/−mice only after 7 days NH4Cl loading (c), whereas Pit2 expression was not affected by acidosis in wild-type mice but increased after 7 days NH4Cl loading in NaPi-IIa deficient mice (d). All data are presented as mean ± SEM (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001
Metabolic acidosis increases protein expression of renal Na+/phosphate cotransporters
Western blotting of brush border membranes prepared from kidneys of control and acid-loaded mice demonstrated that in contrast to mRNA expression, NH4Cl loading resulted in a greater abundance of both NaPi-IIa and NaPi-IIc, immunoreactive protein in WT mice after 2 and 7 days of treatment (Fig. 3). In NaPi-IIa KO mice expression of NaPi-IIc was elevated only after 2 days acid loading and returned to normal values after 7 days (Fig. 3). Effects of NH4Cl loading on Pit1 and Pit2 protein abundance could not be investigated due to the lack of specific antibodies against these two transporters.

Expression of NaPi-IIa and NaPi-IIc proteins during acid loading. a Immunoblotting for the Na+/phosphate cotransporters NaPi-IIa and NaPi-IIc in brush border membranes from control and acid-loaded mice. All membranes were reprobed for β-actin. b Bar graphs summarizing data from immunoblotting. All data were normalized against β-actin, mean ± SEM (n = 5). Protein abundance of NaPi-IIa and NaPi-IIc was higher in wild-type mice after acid loading for 2 and 7 days. c In NaPi-IIa-deficient mice, NaPi-IIc expression increased only after 2 days NH4Cl loading and normalized after 7 days. d Relative NaPi-Iic expression data in Slc34a3−/− mice. All data were normalized against β-actin, mean ± SEM (n = 5). *p < 0.05, **p < 0.01
Metabolic acidosis affects brush border membrane Na/Pi cotransport activity
Sodium-dependent phosphate uptake was increased after 2 days of H4Cl loading in BBMV from wild-type mice with metabolic acidosis and returned to normal values after 7 days (Fig. 4a). In contrast, in NaPi-IIa-deficient mice, sodium-dependent phosphate transport was progressively and significantly decreased in both acid-loaded KO mouse groups (Fig. 4b).

BBM sodium-dependent phosphate (Na/Pi) transport activity in wild-type and NaPi-IIa-deficient mice. Na/Pi cotransport was determined by sodium-dependent 32P-uptake in BBMV. a NH4Cl loading reduced Na/Pi transport activity in BBMV from normal mice after 2 days but not after 7 days. b In NaPi-IIa-deficient mice, MA resulted in a significant decrease in Na/Pi cotransport activity after 2 and 7 days. n = 5 per group. *p < 0.05, **p < 0.01, ***p < 0.001
Metabolic acidosis does not alter distribution of renal Na+/phosphate cotransporters
Immunohistochemistry was used to determine whether metabolic acidosis altered segmental and/or subcellular distribution of renal Na+-dependent phosphate cotransporters NaPi-IIa and NaPi-IIc [20, 26, 30, 32]. Under standard dietary conditions, NaPi-IIa is expressed in convoluted (S1 + S2) and straight (S3) proximal tubules of superficial and juxtamedullary nephrons, whereas the expression of NaPi-IIc is restricted to the renal cortex in the S1 segment of deep and superficial proximal tubule (Fig. 5). The apparent renal distribution of NaPi-IIa and NaPi-IIc was not changed during metabolic acidosis in WT animals treated for 2 or 7 days. We also were not able to detect major signal intensity changes during acid loading in the kidneys of these animals (Fig. 5a and b). In NaPi-IIa −/− mice, NaPi-IIc was localized to apical brush border membranes of proximal S1 segments (Fig. 5c). Furthermore, the signal intensity of NaPi-IIc staining was stronger in superficial S1 proximal tubules than in wild-type animals (Fig. 5a vs. c). In addition, few S2 proximal tubules are expressed NaPi-IIc in NaPi-IIa −/− mice. Acidosis treatment for 2 or 7 days in these animals did not induce any change in NaPi-IIc signal intensity or its cortical distribution (Fig. 5c).

Immunolocalization of type II Na/Pi cotransporters in kidneys from wild-type and NaPi-IIa-deficient mice after 2 and 7 days acid loading. NaPi-IIa (a) or NaPi-IIc (b and c) stainings on renal cortical overviews and single S1 proximal tubules (higher magnification) from wild-type (a and b) and NaPi-IIa-deficient mice (c) during acid loading. No difference in the localization of either NaPi-IIa or NaPi-IIc were detected in WT and NaPi-IIa-deficient mice after 2 and 7 days of acid loading
Effect of acute metabolic acidosis
We examined the effect of acute metabolic acidosis to determine whether direct interactions between protons and the phosphate transporters could reduce renal phosphate reabsorption and thereby contribute to phosphaturia. The induction of acute metabolic acidosis was confirmed by the presence of lower blood pH and bicarbonate concentration, lower urine pH, and higher urinary ammonium excretion. Acute acidosis was also associated with significantly elevated urinary phosphate excretion (Table 3, Fig. 6a). However, NaPi-IIa and NaPi-IIc protein abundance in the brush border membrane fraction remained unchanged (Fig. 6b).

Effects of acute metabolic acidosis. a Urinary phosphate excretion was increased during acute metabolic acidosis. b Western blotting for NaPi-IIa and NaPi-IIc abundance in brush border membranes from control and acidotic mice. All membranes were reprobed for β-actin. c Quantification of NaPi-IIa/β-actin and NaPi-IIc/β-actin ratios, n = 5 for each group. All data are presented as mean ± SEM (n = 5). **p < 0.01
Discussion
Metabolic acidosis is associated with and causes increased renal acid elimination that requires adaptive changes in metabolic and transport pathways. Phosphate, working as a buffer in blood and as titratable acid in urine, greatly contributes to increased renal acid secretion [18], and its excretion into urine is elevated during MA. Previous transcriptome analysis of kidneys from acid-loaded mice revealed a robust downregulation of the type IIc Na+/Pi cotransporter NaPi-IIc mRNA [25], suggesting that it may be responsible for the phosphaturia observed.
In the present study, we analyzed the regulation of all known renal sodium-dependent phosphate cotransporters, namely, the type II NaPi-IIa and NaPi-IIc and the type III Pit1 and Pit2. NaPi-IIa-deficient mice served as an additional tool to distinguish between type IIa-dependent and IIa-independent transport activities in vivo and ex vivo and to test the contribution of renal mechanisms to acidosis-induced phosphaturia. Our findings demonstrate that during NH4Cl-induced metabolic acidosis in mouse, (1) phosphaturia is only transient in wild-type mice and no acid-induced phosphaturia was observed in NaPi-IIa-deficient mice, (2) sodium-dependent phosphate transport activity is transiently increased in BBMV from wild-type mice and decreased in BBMV from NaPi-IIa deficient mice, (3) NaPi-IIa and NaPi-IIc protein are upregulated in wild-type mice and NaPi-IIc protein in KO mice, (4) NaPi-IIc mRNA is strongly reduced, (5) renal localization of NaPi-IIa and NaPi-IIc is not affected, (6) phosphaturia in mice following induction of acute metabolic acidosis was not associated with changes in the abundance of the type IIa and IIc phosphate cotransporters in the brush border membrane, and (7) Pit1 and Pit2 mRNA are increased after 7 days.
Phosphaturia during metabolic acidosis has been attributed mainly to the release of phosphate from bone and subsequent renal excretion [17]. However, several causes may underlie phosphaturia independent from renal phosphate transporter protein abundance. The present demonstration that acidosis-induced phosphaturia is not observed in NaPi-IIa-deficient mice may suggest that phosphaturia in normal mice can be ascribed to decreased renal phosphate reabsorption via NaPi-IIa rather than originating from bone release. However, NaPi-IIa is also expressed in osteoclasts [13], and it has been suggested that NaPi-IIa-deficient osteoclasts are less active in bone resorption, which may also contribute to the reduced phosphaturia in acid-loaded KO mice. A third mechanism that may also contribute to the acidosis-induced phosphaturia is the direct interaction of protons with the phosphate transporter(s) in the brush border membrane of the proximal tubule. Amstutz et al. have demonstrated in rat isolated brush border membrane vesicles that a reduction in extracellular pH causes a decrease in sodium-dependent phosphate transport [2]. The function of the cloned and heterologously expressed type NaPi-IIa cotransporter is decreased by increasing extracellular proton concentrations, which interfere at multiple stages of the transport cycle [9, 36]. Similarly, transport activity of mouse NaPi-IIc has been shown to be decreased by increasing extracellular proton concentrations from pH 7.5 to pH 6.5 [26]. The finding that acutely induced metabolic acidosis elicits phosphaturia without altering the abundance of brush border membrane NaPi-IIa and NaPi-IIc proteins supports the hypothesis that direct inhibition of transport activity contributes to the development of phosphaturia.
In contrast to the present findings, a previous study linked metabolic acidosis-induced phosphaturia to decreased activity and abundance of the renal Na+/Pi cotransporter NaPi-IIa in rat kidney [1]. However, a subsequent study performed both in mouse and rat kidney could not confirm these observations and reported additionally elevated expression of the intestinal Na+/Pi cotransporter NaPi-IIb [33]. Our data are in good agreement with the latter study, demonstrating that both transport activity and NaPi-IIa and NaPi-IIc protein abundance in the brush border membrane are increased during metabolic acidosis. Interestingly, mRNA levels of NaPi-IIa are reduced slightly after 2 days of acidosis, and NaPi-IIc mRNA expression is strongly decreased during metabolic acidosis. Similar discrepancies between NaPi-IIa mRNA expression, protein abundance, and transport activity have been observed in potassium-depleted rats [37]. There, changes in BBM lipid composition and fluidity were found to be linked to altered transport activity [37]. Also. Tenenhouse and colleagues observed the unexpected downregulation of NaPi-IIc mRNA together with elevation of protein abundance in NaPi-IIa KO mice [34]. Thus, further investigation is required to delineate the underlying mechanism for the lack of concordance between cotransporter mRNA and protein abundance. Interestingly, we also observed the regulation of two additional sodium-dependent phosphate cotransporters, Pit1 and Pit2, at the mRNA level following the induction of metabolic acidosis. Pit1 and Pit2 mRNA were elevated after 7 days of metabolic acidosis, suggesting that both transporters could contribute to renal phosphate reabsorption. However, the exact localization of both transporters in kidney is not well studied. In situ hybridization for Pit1 (previously known as Glvr-1) described localization of Pit1 in most mouse kidney structures [35]. Interestingly, the transport activity of both Pit-1 and Pit-2 appears to be increased by extracellular acidification [15, 27].
In summary, we reanalyzed the regulation of renal sodium-phosphate cotransporters in mouse kidney during metabolic acidosis. Our data indicate that phosphaturia during metabolic acidosis cannot be explained by downregulation of protein expression of luminal phosphate cotransporters. In contrast, we found evidence for increased renal expression of NaPi-IIa and NaPi-IIc proteins and Pit1 and Pit2 mRNAs. Thus, phosphaturia may be induced rather by direct interactions between protons and phosphate cotransporters IIa and IIc, thereby reducing transport activity. The role of skeletal phosphate release and increased delivery to the kidney may be less important than previously suggested.
References
Ambuhl PM, Zajicek HK, Wang H, Puttaparthi K, Levi M (1998) Regulation of renal phosphate transport by acute and chronic metabolic acidosis in the rat. Kidney Int 53:1288–1298
Amstutz M, Mohrmann M, Gmaj P, Murer H (1985) Effect of pH on phosphate transport in rat renal brush border membrane vesicles. Am J Physiol 248:F705–F710
Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS (1998) Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U SA 95:5372–5377
Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H (2006) SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78:179–192
Berthelot M (1859) Violet d’aniline. Rep Chim App 1:284
Biber J, Stieger B, Haase W, Murer H (1981) A high yield preparation for rat kidney brush border membranes. Different behaviour of lysosomal markers. Biochim Biophys Acta 647:169–176
Biber J, Stieger B, Stange G, Murer H (2007) Isolation of renal proximal tubular brush-border membranes. Nature protocols 2:1356–1359
Brown AJ, Finch J, Slatopolsky E (2002) Differential effects of 19-nor-1,25-dihydroxyvitamin D(2) and 1,25-dihydroxyvitamin D(3) on intestinal calcium and phosphate transport. J Lab Clin Med 139:279–284
Busch AE, Wagner CA, Schuster A, Waldegger S, Biber J, Murer H, Lang F (1995) Properties of electrogenic Pi transport by a human renal brush border Na+/Pi transporter. J Am Soc Nephrol 6:1547–1551
Custer M, Lotscher M, Biber J, Murer H, Kaissling B (1994) Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol 266:F767–F774
Dawson TP, Gandhi R, Le Hir M, Kaissling B (1989) Ecto-5′-nucleotidase: localization in rat kidney by light microscopic histochemical and immunohistochemical methods. J Histochem Cytochem 37:39–47
Guntupalli J, Eby B, Lau K (1982) Mechanism for the phosphaturia of NH4Cl: dependence on acidemia but not on diet PO4 or PTH. Am J Physiol 242:F552–F560
Gupta A, Tenenhouse HS, Hoag HM, Wang D, Khadeer MA, Namba N, Feng X, Hruska KA (2001) Identification of the type II Na(+)-Pi cotransporter (Npt2) in the osteoclast and the skeletal phenotype of Npt2−/− mice. Bone 29:467–476
Hattenhauer O, Traebert M, Murer H, Biber J (1999) Regulation of small intestinal Na–P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol 277:G756–G762
Kavanaugh MP, Kabat D (1996) Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int 49:959–963
Kavanaugh MP, Miller DG, Zhang W, Law W, Kozak SL, Kabat D, Miller AD (1994) Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci U S A 91:7071–7075
Lemann J Jr., Bushinsky DA, Hamm LL (2003) Bone buffering of acid and base in humans. Am J Physiol Renal Physiol 285:F811–F832
Levi M, Lotscher M, Sorribas V, Custer M, Arar M, Kaissling B, Murer H, Biber J (1994) Cellular mechanisms of acute and chronic adaptation of rat renal P(i) transporter to alterations in dietary P(i). Am J Physiol 267:F900–F908
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM (2006) Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78:193–201
Madjdpour C, Bacic D, Kaissling B, Murer H, Biber J (2004) Segment specific expression of sodium-phosphate cotransporters NaPi-IIa and -IIc and interacting proteins in mouse renal proximal tubules. Pflugers Arch 448:402–410
Miyamoto K, Ito M, Kuwahata M, Kato S, Segawa H (2005) Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther Apher Dial 9:331–335
Murer H, Forster I, Biber J (2004) The sodium phosphate cotransporter family SLC34. Pflugers Arch 447:763–767
Murer H, Hernando N, Forster I, Biber J (2003) Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol 65:531–542
Murer H, Hernando N, Forster I, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80:1373–1409
Nowik M, Lecca MR, Velic A, Rehrauer H, Brandli AW, Wagner CA (2008) Genome-wide gene expression profiling reveals renal genes regulated during metabolic acidosis. Physiol Genomics 32:322–334
Ohkido I, Segawa H, Yanagida R, Nakamura M, Miyamoto K (2003) Cloning, gene structure and dietary regulation of the type-IIc Na/Pi cotransporter in the mouse kidney. Pflugers Arch 446:106–115
Olah Z, Lehel C, Anderson WB, Eiden MV, Wilson CA (1994) The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J Biol Chem 269:25426–25431
Radanovic T, Wagner CA, Murer H, Biber J (2005) Regulation of intestinal phosphate transport. I. Segmental expression and adaptation to low-P(i) diet of the type IIb Na(+)-P(i) cotransporter in mouse small intestine. Am J Physiol Gastrointest Liver Physiol 288:G496–G500
Seaton B, Ali A (1984) Simplified manual high performance clinical chemistry methods for developing countries. Med Lab Sci 41:327–336
Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277:19665–19672
Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, Saito H, Fukushima N, Miyamoto K (2003) Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch 446:585–592
Segawa H, Yamanaka S, Ito M, Kuwahata M, Shono M, Yamamoto T, Miyamoto K (2005) Internalization of renal type IIc Na–Pi cotransporter in response to a high-phosphate diet. Am J Physiol Renal Physiol 288:F587–F596
Stauber A, Radanovic T, Stange G, Murer H, Wagner CA, Biber J (2005) Regulation of intestinal phosphate transport. II. Metabolic acidosis stimulates Na(+)-dependent phosphate absorption and expression of the Na(+)-P(i) cotransporter NaPi-IIb in small intestine. Am J Physiol Gastrointest Liver Physiol 288:G501–G506
Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K (2003) Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol 285:F1271–F1278
Tenenhouse HS, Roy S, Martel J, Gauthier C (1998) Differential expression, abundance, and regulation of Na+-phosphate cotransporter genes in murine kidney. Am J Physiol 275:F527–F534
Virkki LV, Forster IC, Biber J, Murer H (2005) Substrate interactions in the human type IIa sodium-phosphate cotransporter (NaPi-IIa). Am J Physiol Renal Physiol 288:F969–F981
Zajicek HK, Wang H, Puttaparthi K, Halaihel N, Markovich D, Shayman J, Beliveau R, Wilson P, Rogers T, Levi M (2001) Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int 60:694–704
Acknowledgments
M. Nowik is the recipient of a PhD student fellowship from the Zurich Center for Integrative Human Physiology. This study was supported by grants from the Swiss National Science Research Foundation to J. Biber and H. Murer (31-65397.01) and C. A. Wagner (31-109677/1) and the 6th EU Frame work project EuReGene to J. Biber, H. Murer, and C. A. Wagner. The use of the ZIHP Core Facility for Rodent Physiology is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Characterization of the anti-NaPi-IIc antibody by immunoblotting. Brush border membrane vesicles were prepared from kidneys of Slc34a1+/− and Slc34a1−/− mice to test the specificity and cross-reactivity of new antibodies against NaPi-IIc. The antibody against mouse NaPi-IIc was raised in rabbit, affinity-purified, and recognized a single major band of approximately 75kDa (stripe B). This band was prevented by preincubation with the immunizing peptide (stripe C). The preimmune serum from the rabbit used to obtain the anti-NaPi-IIc antibody did not show any reactivity (stripe D). Stripe A shows NaPi-IIa staining in the same samples (PDF 50 KB).
Supplementary Fig. 2
Characterization of anti-NaPi-IIc antibody by immunohistochemistry. Serial kidney sections of normal mice were stained with antibodies against NaPi-IIa (left panel), NaPi-IIc (middle panel), and NaPi-IIc after preincubation with the immunizing peptide (right panel). No staining was detected with the anti-NaPi-IIc antibody after preincubation with the immunizing peptide. Please note: The background level was enhanced in the right panel to detect possible unspecific staining (PDF 342 KB).
Supplementary Fig. 3
Increased abundance of NaPi-IIc protein in the brush border membrane of kidneys from NaPi-IIa-deficient mice. Blots were probed for NaPi-IIc and β-actin, and data are summarized by normalizing NaPi-IIc abundance against β-actin (mean ± SEM). These data are in agreement with previous work demonstrating that renal NaPi-IIc protein expression is significantly increased in NaPi-Iia-deficient mice when compared with normal littermates [34] (PDF 29 KB).
Rights and permissions
About this article
Cite this article
Nowik, M., Picard, N., Stange, G. et al. Renal phosphaturia during metabolic acidosis revisited: molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch - Eur J Physiol 457, 539–549 (2008). https://doi.org/10.1007/s00424-008-0530-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0530-5