
Abstract
The epithelial sodium channel (ENaC) is the major mediator of sodium transport across the apical membranes of the distal nephron, the distal colon, the respiratory tract and the ducts of exocrine glands. It is subject to feedback inhibition by increased intracellular Na+, a regulatory system wherein the ubiquitin protein ligases, Nedd4 and Nedd4-2, bind to conserved PY motifs in the C-termini of ENaC and inactivate the channel. It has been proposed recently that the kinase Sgk activates the channel as a consequence of phosphorylating Nedd4-2, thus preventing it from inhibiting the channels. This proposal predicts that Sgk should interfere with Na+ feedback regulation of ENaC. We have tested this prediction in Xenopus laevis oocytes and in mouse salivary duct cells and found that in neither system did increased activity of Sgk interrupt Na+ feedback inhibition of ENaC. We found, however, that Sgk stimulation was largely abolished in oocytes expressing ENaC channels with C-terminal truncations or mutated PY motifs. We were also unable to confirm that Sgk directly interacts with Nedd4-2 in vitro. We conclude that the stimulatory effect of Sgk on ENaC requires the presence of the channel’s PY motifs, but it is not due to the interruption of Na+ feedback regulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epithelial sodium channels (ENaC) are expressed in the distal nephron, the distal colon, the lungs and the excretory ducts of salivary and sweat glands [15]. They are the major route for Na+ absorption across the apical membranes of these epithelia and play a critical role in the regulation of normal extracellular fluid volume and blood pressure [36]. Their activity is increased by hormones such as aldosterone, insulin and vasopressin (ADH) [18]. Recent studies have implicated the serum- and glucocorticoid-inducible kinase (Sgk), a member of the PKB-Akt family of serine-threonine kinases thought to play a role in cell volume control [34], as a critical mediator of the action of these hormones [2, 16, 31, 43].
The mechanism by which Sgk1 increases Na+ channel activity, however, is unclear. In principle, Sgk could act by increasing the rate of insertion of channels into the plasma membrane, increasing the open probability of the channels that are already in the plasma membrane, or reducing the rate of removal of the channels from the plasma membrane. Data supporting each of these mechanisms have been published [2, 4, 9]. Of particular interest, however, have been the recent reports that Sgk1 may reduce the rate of retrieval of the channels from the plasma membrane as a consequence of phosphorylating the ubiquitin protein ligase, Nedd4-2, so as to render it unable to interact with the Na+ channels [8, 38, 39].
Nedd4-2 and its isoform Nedd4 mediate a negative feedback system wherein increases in the intracellular concentration of Na+ inhibit epithelial Na+ channels [1, 13]. Nedd4-2 and Nedd4 do this by binding to the PY motifs in the carboxyl termini of the β- and γ-subunits of the Na+ channels [21, 24, 40], and then ubiquitinating the N-termini of the channel α- and γ-subunits [41] so as to trigger the endocytosis [37] and degradation of the channels by proteasomes and lysosomes [32, 41].
The concept that both Sgk activation and Na+ feedback inhibition of ENaC are due to alterations in the activity of Nedd4/Nedd4-2-mediated ENaC retrieval suggests that Sgk activation and Na+ feedback inhibition are functionally interrelated. In particular, the model predicts that the stimulatory effect of Sgk should be most prominent when the Nedd4/Nedd4-2 pathway is activated by Na+ feedback inhibition. In contrast, when Na+ feedback inhibition is absent, the stimulatory effect of Sgk should be rather small, because under these conditions the Nedd4/Nedd4-2 pathway is thought to be inactive. The model also predicts that Sgk may lead to a reduction or even loss of the normal inhibitory regulation of epithelial Na+ channels by increased intracellular Na+, depending on the relative strength of the two opposing effects. The present studies resulted from the wish to test these predictions in two experimental systems, the Xenopus laevis oocyte expression system and mouse mandibular duct cells, because Na+ feedback inhibition is known to be mediated by ubiquitin protein ligases of the Nedd4 family in both [1, 13, 17, 21, 25, 29]. In particular, the proposal that Sgk phosphorylates Nedd4-2 so as to prevent it from interacting with epithelial Na+ channels predicts that Sgk should inhibit Na+ feedback in these cell types. Preliminary data from this paper have been reported in abstract form [35].
Materials and methods
Materials
Constitutively active, recombinant Sgk1 (Δ1-60 S422D) was obtained from Upstate Biotechnology (Waltham, MA). Sgktide (KKPNRRLSVA) [33] was synthesised by Mimotopes (Clayton, Vic, Australia). The anti-Nedd4/Nedd4-2 antibody is described in [30].
Expression constructs containing carboxyl regions of mouse α-, β- and γ-ENaC, all three WW domains of mouse Nedd4 and all four WW domains of mouse Nedd4-2 have been described previously [17, 20, 21]. The Sgk1 PY motif glutathione S-transferase (GST) construct was generated by polymerase chain reaction amplification of the PY motif region (amino acids 262–330) from mouse Sgk1 cDNA, followed by cloning into the BamHI/EcoRI sites of pGEX-2TK (Amersham Pharmacia Biotech Australia).
Production of recombinant proteins
GST alone and GST fusion proteins for carboxyl α-, β- and γ-ENaC and WW domain regions of Nedd4 and Nedd4-2 were produced as described in Fotia et al. [17]. The GST–Sgk1 PY motif fusion protein was produced as follows. An overnight culture of E. coli DH5α harbouring the Sgk1 PY motif GST fusion construct was diluted 1:4, grown for 2 h at 22°C, induced with 1 mM isopropylβ-d-thiogalactoside and grown for an additional 5 h at 22°C. The bacterial cell pellet was resuspended in phosphate-buffered saline containing 1 mg/ml lysozyme and then freeze–thawed. The resulting solution was sonicated and then clarified by centrifugation at 13,000×g for 30 min. GST fusion protein was then affinity purified by using glutathione-Sepharose beads (Amersham Pharmacia Biotech) as described previously [20]. Protein concentrations were estimated by electrophoresis alongside bovine serum albumin standards, followed by Coomassie blue staining.
[32P]-labelled protein probes were produced by directly labelling the appropriate GST fusion protein using protein kinase A (New England Biolabs) as previously described [20].
Production of PY motif filters
To prepare PY motif filters, approximately 2 μg of GST alone and each GST fusion protein was resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidine difluoride membranes (Perkin Elmer Life Sciences Biotech, Boston, MA). Membranes were blocked in Hyb75 [23] and then hybridised with Nedd4 or Nedd4-2 WW domain [32P]-labelled protein probes for 4 h at 4°C in Hyb75. Membranes were washed four times in Hyb75, exposed to a phosphorscreen (Molecular Dynamics) and analysed on a Typhoon 9410 variable mode imager (Molecular Dynamics).
Isolation of oocytes and injection of complementary RNA
Adult female Xenopus laevis were anaesthetised in 0.2% MS222 (Sigma, Taufkirchen, Germany), and oocytes were obtained by a partial ovariectomy. The oocytes were isolated from the ovarian lobes by enzymatic digestion at 19°C for 3–4 h with 1 mg/ml type V collagenase from Cl. histolyticum (Sigma) dissolved in OR2 (82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM HEPES, pH 7.4 with Tris). Defolliculated stage V–VI oocytes were injected with 0.1 ng per subunit of α-, β- and γ-ENaC–FLAG complementary RNA (kindly provided by Bernard C. Rossier, Lausanne, Switzerland) and 1 ng of constitutively active Sgk1-SD cRNA (kindly provided by Florian Lang, Tubingen, Germany) or water. In the experiments with truncated and mutated ENaC, 0.02 ng cRNA per subunit of α-, β- and γ-rENaC wild-type, or αP64stop-, β564stop- and γF606stop-rENaC, or αY637A-, βY618A- and γY628A-rENaC and 1 ng of constitutively active Sgk1-SD cRNA or water were injected. The cRNAs were dissolved in RNase-free water, and the total volume injected was 46 nl (Nanoject automatic injector, Drummond, Broomall, PA). Injected oocytes were stored at 19°C in modified Barth’s solution (MBS) with either low Na+ [1 mM NaCl, 40 mM KCl, 60 mM N-methyl-d-glucamine, 0.3 mM Ca(NO3)2, 0.4 mM CaCl2, 0.8 mM MgSO4, 10 mM HEPES, pH 7.4 with HCl] or high Na+ conditions [85 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.3 mM Ca(NO3)2, 0.4 mM CaCl2, 0.8 mM MgSO4, 10 mM HEPES, pH 7.4 with Tris] supplemented with 10 units/ml sodium penicillin and 10 μg/ml streptomycin sulphate to prevent bacterial overgrowth.
Two-electrode voltage clamp
Oocytes were studied 2 days after injection using the two-electrode voltage clamp technique. The oocytes were placed in a small experimental chamber and constantly superfused with ND96 (NaCl 96 mM, KCl 2 mM, CaCl2 1.8 mM, MgCl2 1 mM, HEPES 5 mM, pH 7.4 with Tris) containing 2 μM amiloride (Sigma) at a rate of 3–5 ml/min. Experiments were performed at room temperature. Oocytes were held continuously at a holding potential of −60 mV (OC-725C oocyte clamp, Warner Instruments Corp., Hamden, USA). Current–voltage (I–V) plots were obtained from voltage-step protocols using consecutive 400 ms step changes of the clamp potential from −60 to −120 mV up to +40 mV in 20-mV increments (PULSE and LIH-1600, HEKA, Lambrecht, Germany). The average current values reached during the last 100 ms of the voltage steps were used for the I–V plots. Amiloride-sensitive whole-cell currents (ΔIami) were obtained by washing out amiloride (2 μM) with amiloride-free ND96 and subtracting the whole-cell currents in the presence of amiloride from the corresponding whole-cell currents without amiloride. Amiloride-sensitive whole-cell conductance (Gami) was calculated as chord conductance using the ΔIami values measured at −100 and −80 mV.
Surface labeling of oocytes
Experiments were performed as described recently [28, 45], using mouse anti-FLAG M2 monoclonal antibody (Sigma) as primary antibodies and peroxidase-conjugated sheep anti-mouse IgG (Chemicon, Boronia Victoria, Australia) as secondary antibodies. Chemiluminescence was quantified in a Turner TD-20/20 luminometer (Turner Designs, Sunnyvale, CA) by placing individual oocytes in 50 μl of SuperSignal ELISA Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL), and integrating the signal over a period of 15 s. Results are given in relative light units (RLU).
Isolation of salivary duct cells
Isolated salivary duct cells were prepared by collagenase digestion of mandibular glands from male mice [13].
Patch clamp methods
The standard bath solution, with pH 7.4, contained the following:
Contents of standard bath solution | Amount (mM) |
NaCl | 145 |
KCl | 5.5 |
CaCl2 | 1.0 |
MgCl2 | 1.2 |
NaH2PO4 | 1.2 |
Na-N-[2-hydroxyethyl] piperazine-N,[2-ethanesulfonic acid] (Na-HEPES) | 7.5 |
H-HEPES | 7.5 |
Glucose | 10 |
After establishing the whole-cell configuration in an isolated cell, we replaced the bath solution with a solution containing:
| Amount (mM) |
Na-glutamate (Na-glu) | 145 |
NaCl | 5.0 |
MgCl2 | 1.0 |
H-HEPES | 10 |
Glucose | 10 |
Ethylene-glycol-bis(β-aminoethyl-ether)N,N,N’,N’-tetraacetic acid (EGTA) | 1.0 |
The pH was adjusted to 7.4 with NaOH. The pipettes were filled with pH 7.2 solutions containing
| Amount (mM) |
NMDG-glu and Na-glu (combined) | 150 |
MgCl2 | 1.0 |
H-HEPES | 10 |
Glucose | 10 |
EGTA | 5.0 |
In experiments in which a NO3-rich pipette solution was used, 150 mM NMDG-NO3 was used in place of 150 mM NMDG-glutamate. In experiments in which we used constitutively active Sgk1 (Δ1-60 S422D) and in the corresponding control experiments, we also included in the pipette solution Na-orthovanadate (1 mM), DTT (2 mM) and Na3ATP (1 mM), and we increased the concentration of MgCl2 to 2 mM. We also confirmed that the Sgk1 (Δ1-60 S422D) batch used in the patch clamp solution phosphorylated Sgktide in vitro (data not shown).
Current–voltage relations were obtained by applying 250 ms voltage pulses from a resting potential of 0 mV and were initially measured 4 min after attaining the whole-cell configuration. Steady-state currents are the average currents between 200 and 250 ms after the start of the pulse. Amiloride-sensitive current was calculated by subtracting the current following the addition to the bath of 100 μM amiloride from the current before the addition of amiloride. Chord conductances were calculated at −80 mV (cf. [13]).
Statistical methods
Data are presented as mean±SEM. In patch clamp experiments, n indicates the number of cells studied. In oocyte experiments, N indicates the number of different batches of oocytes, n the number of different oocytes studied in each group. Significance was evaluated by unpaired Student’s t test.
Results
Does Sgk inhibit Na+ feedback in Xenopus oocytes?
Na+ feedback in Xenopus oocytes can be activated by increasing extracellular Na+ [29, 42]. Hence, to test whether the stimulatory effect of Sgk on ENaC channels heterologously expressed in Xenopus oocytes is dependent on the presence of sodium feedback inhibition, we co-injected Sgk1 and ENaC-FLAG cRNA in Xenopus oocytes and incubated them in high and low Na+ MBS.
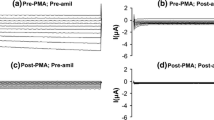
As expected, the reversal potential of ΔIami measured in seven batches of oocytes was more positive in low Na+ oocytes (Fig. 1b right panel) than in high Na+ oocytes (Fig. 1a right panel). The reversal potential of ΔIami in ENaC control oocytes and ENaC + Sgk1 oocytes kept in low Na+ averaged +44 and +38 mV, respectively (Fig. 1b). In contrast, in oocytes kept in high Na+, the reversal potential averaged +18 mV in ENaC control oocytes and +15 mV in ENaC + Sgk1 oocytes (Fig. 1a). This shift of the reversal potential indicates that maintaining the oocytes in high Na+ compared to low Na+ results in an increase of the apparent intracellular Na+ concentration from 17 to 46 mM in ENaC oocytes and from 21 to 54 mM in the ENaC + Sgk1 oocytes. This confirms a similar degree of Na+ loading in the two groups of oocytes due to high Na+ incubation. While using the reversal potential of ΔIami to calculate the apparent intracellular Na+ concentration tends to overestimate the true intracellular Na+ concentration [27], these data indicate that the intracellular Na+ was substantially lower in low Na+ incubated oocytes than in high Na+ incubated oocytes.

The stimulatory effect of Sgk1 on the amiloride-sensitive sodium current (ΔIami) in Xenopus oocytes is preserved in the absence of sodium feedback inhibition. Oocytes were injected with ENaC–FLAG cRNA ± Sgk1-SD and incubated for 2 days in high Na+ (a) or low Na+ (b) MBS. On the left side of panels a and b, representative whole-cell current traces are shown resulting from 400 ms step changes of the clamp potential from −60 to −120 mV up to +40 mV in 20-mV increments in the presence (+ami) and absence (−ami) of 2 μM amiloride. On the right, average current–voltage (I–V) plots are depicted using the amiloride-sensitive whole-cell current (ΔIami; mean±SEM) values obtained by subtracting the current traces recorded in the presence of amiloride from the corresponding traces recorded in its absence. Experiments were performed in seven different batches of oocytes with an equal number of 72 oocytes (N=7, n=72) in each group. Please note the different current scales used in a and b
We found a significant 5.3-fold increase of ΔGami in the low Na+ ENaC group (32.12±2.29 μS, n=72) compared to the high Na+ control group (6.08±0.43 μS, n=72) (Fig. 1). This finding is consistent with previous reports [25, 29, 42] and indicates that sodium feedback inhibition is largely abolished in the low Na+ group. As illustrated by representative current traces and by average I–V plots shown in Fig. 1, co-injection with Sgk1 cRNA led to a significant 52% increase of ΔGami in high Na+ incubated oocytes (9.27±0.65 μS, n=72) and to a significant 64% increase in low Na+ incubated oocytes (52.81±3.39 μS, n=72). Thus, the stimulatory effects of Sgk1 co-expression were not reduced by incubation of the oocytes in low Na+ solution, viz when Na+ feedback inhibition is not operating. Indeed, the absolute stimulatory effect of Sgk was larger in oocytes incubated in low Na+ solution than in oocytes incubated in high Na+ (Fig. 1). We then examined the ratio of ΔGami observed in the oocytes incubated in high Na+ to ΔGami observed in oocytes incubated in low Na+ as a measure of the efficacy of Na+ feedback inhibition. We found no difference in this ratio between oocytes expressing Sgk1 [ΔGami(high Na+)/ΔGami(low Na+)=18%] and oocytes not expressing Sgk1 [ΔGami(high Na+)/ΔGami(low Na+)=19%]. Thus, Sgk co-expression appeared to have no impact on Na+ feedback inhibition in Xenopus oocytes.
In addition, we determined the surface expression level of ENaC with a chemiluminescence assay (Fig. 2). We found a 42% increase of surface expression in low Na+ incubated oocytes when ENaC was co-injected with Sgk1 (68.3±5.3 RLU, n=131) compared to the ENaC control in low Na+ incubated oocytes that had not been injected with Sgk1 (48.1±3.1 RLU, n=131).

The stimulatory effect of Sgk1 on ΔIami can be partially attributed to an increase in ENaC surface expression. In parallel to the two electrode voltage clamp experiments, we determined the surface expression level of ENaC–FLAG with a chemiluminescence assay (N=7, n=131 per group). The results show a significant 42% increase (p<0.0001) of surface expression (measured in Relative light units, RLU) in low Na+ when ENaC was co-injected with Sgk1 (filled bars)
To investigate whether Sgk interferes with acute Na+ feedback inhibition, we also tested the effect of Sgk1 on the response of ΔIami and ENaC surface expression to a sudden increase of the extracellular sodium concentration. Two days after cRNA injection and continuous incubation in low Na+ MBS, oocytes were divided into two groups and either transferred to high Na+ MBS or further maintained in low Na+ MBS. A representative experiment is shown in Fig. 3a. In ENaC and ENaC + Sgk1 oocytes maintained in the low Na+ MBS, ΔIami remained at a relatively constant level. In contrast, exposure to high Na+ MBS rapidly reduced ΔIami in ENaC and in ENaC + Sgk1 oocytes, demonstrating the presence of substantial Na+ feedback inhibition. In similar experiments performed in four different batches of oocytes, we found reductions during acute sodium loading of 69.6±4.2% (3–6 h high Na+ exposure) and 77.7±5.8% (8–10 h high Na+ exposure) of ΔIami in ENaC control oocytes compared to reductions of 54.1±8.8% (3–6 h) and 79.7±3.3% (8–10 h) observed in ENaC + Sgk1 oocytes (Fig. 3b). These findings demonstrate that Sgk does not prevent acute Na+ feedback inhibition of ENaC. This was confirmed by ENaC surface expression measurements (Fig. 3c). Acute exposure to high Na+ reduced ENaC surface expression by 65.2±5.5% in ENaC oocytes and by 73.2±5.7% in ENaC + Sgk1 oocytes. Thus, Na+ feedback inhibition of ENaC surface expression was also preserved in the presence of Sgk.

Sodium feedback inhibition induced by acute Na+ load is not prevented by Sgk1. Oocytes were injected with ENaC-FLAG cRNA + Sgk1-SD or water and incubated for 2 days in low Na+ MBS. On the day of experiment half of the oocytes were transferred into high Na+ MBS. ΔIami was assessed after 0, 3–6 and 8–10 h, surface expression after 3–4 h. a Representative results from one of four similar sets of experiments. After 10 h of acute Na+ loading (open symbols), ΔIami was significantly decreased in the ENaC injected oocytes (circles) and in the ENaC + Sgk1 injected oocytes (squares) compared to corresponding control oocytes maintained in low Na+ MBS (filled symbols). Each data point represents the mean of ten oocytes (N=1, n=10); error bars represent SEM. Average ΔIami reduction (b) and reduction of surface expression (c) in ENaC and ENaC + Sgk1 injected oocytes after acute exposure to high Na+ MBS. Bars represent the mean of four similar sets of experiments in four different batches of oocytes (N=4, n=4); error bars represent SEM. Current measurements were performed as shown in a
Removal or mutation of ENaC PY motifs prevents the stimulatory effect of Sgk in the presence and absence of Na+ feedback inhibition
The concept that Sgk stimulates ENaC by inhibiting its interaction with Nedd4/Nedd4-2 predicts that the removal or mutation of the channel’s PY motifs, which are known to be essential for Nedd4/Nedd4-2 binding, should abolish the stimulatory effect of Sgk. Indeed, it has been reported that mutation of the PY motifs in all three channel subunits abrogates the stimulatory effect of Sgk on ENaC [8, 38]. In contrast, it was reported in an earlier study that the stimulatory effect of Sgk on ENaC was preserved when the PY motifs of all three channel subunits were either mutated or removed by C-terminal truncations [3]. In the present study we re-investigated this important issue in the presence or absence of sodium feedback inhibition, i.e. in oocytes incubated in high (Fig. 4a) or low Na+ (Fig. 4b) MBS, respectively. The finding that wild type ENaC currents were reduced in oocytes incubated in high Na+ compared to those in oocytes incubated in low Na+ confirms that Na+ feedback inhibition is operating (Fig. 4a,b) and is consistent with the results shown in Fig. 1. In oocytes incubated in high Na+, truncation of the C-termini or mutation of the PY motifs resulted in a loss of Na+ feedback inhibition [25] with increased currents compared to wild-type ENaC (Fig. 4a). In contrast, removal or deletion of the PY motifs had no additional stimulatory effect in oocytes incubated in low Na+ to prevent Na+ feedback inhibition (Fig. 4b). Importantly in the two groups of oocytes (high and low Na+), the stimulatory effect of Sgk1 was largely abolished by the removal or deletion of the PY motifs. In oocytes incubated in high Na+, co-expression of Sgk1 had a similar stimulatory effect on wild type ENaC currents as the removal or mutation of the PY motifs. In oocytes incubated in low Na+, co-expression of Sgk1 resulted in ENaC currents that were substantially larger than those observed in oocytes expressing the truncated or mutated ENaC (Fig. 4b). When taken together, these findings indicate that the stimulatory effect of Sgk requires the presence of intact PY motifs, but is largely independent of the Na+ feedback inhibitory mechanism.

In oocytes incubated in high or low Na+ the stimulatory effect of Sgk1 is largely abolished by removal or mutation of ENaC’s PY-motifs. Oocytes were injected with cRNA coding either for ENaC-wt, C-terminal truncated ENaC (ENaC trunc), or ENaC with mutated PY-motifs (ENaC PY-mut), with (+) or without (−) Sgk1 and incubated for 2 days in high Na+ (a) or low Na+ (b) MBS. Bars show mean+SEM of ΔIami obtained at a holding potential of −60 mV (A N=4, n=46, B N=4, n=52)
Does Sgk block Na+ feedback in mouse mandibular duct cells?
As described in the previous sections, we were unable to observe an effect of Sgk on the inhibition of ENaC by increased intracellular Na+ in Xenopus oocytes. We thus decided to attempt to confirm this observation in a native cell system in which intracellular Na+ is known to inhibit epithelial Na+ channels via Nedd4-(2), mouse mandibular salivary duct cells [13, 17].
Mouse mandibular salivary duct cells, when studied in the whole-cell patch clamp configuration with Na-glutamate solution in the bath and NMDG-glutamate solution in the pipette, have an amiloride-sensitive Na+ conductance (Fig. 5a,b) [10]. The magnitude of this conductance is decreased by increasing the intracellular concentration of Na+ [26], as seen here, for example, when we use a patch pipette solution containing 72 mM Na+ (Fig. 5a,c).

The amiloride-sensitive sodium current in mouse mandibular duct cells is inhibited by increased intracellular Na+. a Amiloride-sensitive currents during dialysis with 0 Na and 72 Na solution. b Mean steady-state current–voltage relations of cells (n=9) studied with 0 Na pipette solution (filled circles), and following the addition of 100 μmol/l amiloride to the bath (open circles). The amiloride-sensitive component of the current is also shown (filled triangles). c Mean steady-state current–voltage relations of cells (n=7) studied with 72 Na pipette solution (filled circles), following the addition of 100 μmol/l amiloride to the bath (open circles). The amiloride-sensitive component of the current is also shown (filled triangles)
To determine whether Sgk inhibits Na+ feedback regulation of the amiloride-sensitive Na+ channels in mandibular duct cells, we examined whether inclusion of recombinant constitutively active Sgk1 (Δ1-60 S422D) (400 ng/ml) in the 72 mM Na pipette solution overcomes the inhibitory effect of the increase in Na+ in the pipette solution. We found that it did not (Fig. 6). Given that Sgk1 activates epithelial Na+ channels in macropatches from Xenopus oocytes over a period of 20 min, and that this activation is dependent on the ambient concentration of Ca2+ [9], we also examined the effect including Sgk1 (Δ1-60 S422D) in the 0 Na pipette solution. We found that Sgk1 did not change the amiloride-sensitive current when studied over a period of 20–25 min. Similarly, increasing the free Ca2+ in the pipette solution to 1 μmol/l from <1 nmol/l, also did not reveal any effect of Sgk1 (Δ1-60 S422D) on the amiloride-sensitive current.

Sgk does not prevent the inhibition of the amiloride-sensitive channels in mouse mandibular duct cells by increased intracellular Na+. Amiloride-sensitive conductance during dialysis with 0 Na solution, 72 mM Na solution, 72 mM Na solution + 400 ng/ml recombinant Sgk1 (Δ1-60 S422D), or with NMDG-NO3 solution±400 ng/ml recombinant Sgk1 (Δ1-60 S422D)
The amiloride-sensitive Na+ channels in mouse salivary duct cells are also inhibited by the presence of anions, such as Cl−, Br− and\(NO^{ - }_{3} \), in the cytosol [11]. These anions act via a G protein dependent mechanism [12] that does not involve Nedd4 or Nedd4-2 [13]. We thus examined whether Sgk could overcome the inhibition of the Na+ channels produced by the anion feedback system. We found that inclusion of recombinant active Sgk1 (Δ1-60 S422D) in the pipette solution was unable to overcome the inhibition of the amiloride-sensitive current produced by the\(NO^{ - }_{3} \)-containing pipette solution (Fig. 6).
Does Sgk directly interact with Nedd4-2?
As mentioned above, it has been suggested that phosphorylation of Nedd4-2 by Sgk1 is mediated via interactions between the PY motif of Sgk1 (PPFY) and the WW domains of Nedd4-2 [8, 38]. However, whilst Snyder and colleagues [38] have demonstrated the requirement of the Sgk1 PY motif for an interaction with Nedd4-2, Debonneville et al. [8] were unable to demonstrate binding between these two proteins. To investigate binding between Sgk1 and Nedd4-2, we generated a GST fusion protein of the Sgk1 PY motif-region (Fig. 7a). Equal amounts of this affinity-purified protein, GST alone and GST fusions of the PY motif-containing carboxyl termini of ENaC α-, β- and γ-subunits were immobilised on PVDF membranes after separation by SDS-PAGE, then probed with the [32P]-labelled WW domain regions of mouse Nedd4 and Nedd4-2. As shown in the radiograms, the two probes bound strongly with the carboxyl termini of all three ENaC subunits as previously reported [17, 21], whereas no detectable binding was seen to the PY motif of Sgk1 or GST (Fig. 7b).

a The mSgk1 sequence used as a GST fusion protein. b A far-Western analysis of binding between the 32P-labelled WW domain region of Nedd4(-2) and the PY motif regions of α-, β- and γ-ENaC subunits and Sgk1. Approximately 2 μg of each affinity purified GST fusion protein and GST alone was used in far-Western assays. Coomassie blue-stained gel of GST proteins as indicated on top of the gel (top). Proteins were transferred to PVDF membranes and hybridised to 32P-labelled Nedd4 (middle) or Nedd4-2 (bottom) protein probes. Molecular mass markers in kDa are indicated on the left-hand side of the gels
Discussion
We have shown that ENaC stimulation by Sgk is fully preserved when Na+ feedback inhibition is reduced. Moreover, we have demonstrated that increased activity of Sgk does not lead to inhibition of Na+ feedback regulation of epithelial Na+ channels. In Xenopus oocytes, where Na+ feedback inhibition can be triggered by increasing the extracellular Na+ concentration [25, 29, 42], we found no evidence that co-expression of Sgk leads to a reduced activity of Na+ feedback, or that the stimulatory effect of Sgk is abrogated when the Na+ feedback system is not operating. These results make it unlikely that an inhibitory action of Sgk on Na+ feedback inhibition is responsible for the stimulatory effect of Sgk on ENaC. We have also confirmed previous findings [8, 38] that the stimulatory effect of Sgk is largely abolished by removal or mutation of the channel’s PY motifs, confirming the importance of these motifs for mediating the Sgk effect. Our results, however, are at odds with an earlier study [3] and we have no explanation for this discrepancy. Our Xenopus studies further confirm that the increase in Na+ channel activity produced by co-expression of Sgk1 is accompanied by an increase in channel surface expression.
Similarly, increasing Sgk activity in mouse mandibular duct cells by the inclusion of recombinant constitutively active Sgk in the pipette solution did not prevent the Na+ channels being inhibited by increased intracellular Na+. Because we have shown in vitro that the recombinant Sgk1 (Δ1-60 S422D) remains active in the pipette solution (see “Materials and methods”) and this recombinant Sgk has been successfully used in other patch clamp studies [9], the absence of an effect of Sgk1 is unlikely to be due to a lack of activity of the enzyme. Furthermore, we used the protocol that we have previously used to show that the kinase Grk2 inhibits Na+ feedback regulation of the Na+ channels [14]; hence the failure of Sgk1 to act is not attributable to a technical difficulty such as the failure of the kinase to reach the channels or the ubiquitin protein ligase. Thus, the present studies permit us to conclude that Sgk does not inactivate Na+ feedback in at least one aldosterone-sensitive tissue, the salivary ducts [19].
Our failure to observe a functional interrelation of Sgk action and Na+ feedback inhibition in these two systems and the finding that the stimulation of ENaC by Sgk does not seem to require that Nedd4-2 mediated channel retrieval has been activated by high intracellular sodium suggest the presence of additional pathways through which Sgk stimulates ENaC independently of Nedd4-2. For example, the macropatch studies by Diakov and Korbmacher have found that S621, which is located in a putative Sgk-phosphorylation consensus site in the C-terminus of the α-subunit of the channel, is required for stimulation of ENaC activity by Sgk [9]. An obvious prediction from this is that Sgk exerts its action by directly or indirectly modifying the phosphorylation of S621, although an earlier study did not observe phosphorylation by Sgk of any of the channel subunits [44]. Moreover, Sgk may stimulate channel insertion rather than acting solely through an inhibition of channel retrieval. This is supported by earlier reports that Sgk does not reduce the rate of endocytosis of Na+ channels expressed in Xenopus oocytes [3] and does not increase the half-life of ENaC subunits in A6 cells [7]. Moreover, the stimulatory effects of aldosterone and vasopressin on Na+ channels are not prevented by the deletion of the PY motifs in the channels [6].
We were unable to demonstrate that the PY motif of Sgk1 is a target of the Nedd4-2 WW domains in vitro. This is consistent with the failure of Debonneville et al. [8] to co-immunoprecipitate heterologously expressed Sgk and Nedd4-2 from Xenopus oocytes, and in vitro interaction studies, which found no interaction [22], or only a weak interaction [5], between these two proteins. Snyder et al. [38], on the other hand, were able to demonstrate binding between Sgk and Nedd4 or Nedd4-2 in vitro. The differences in experimental protocol between these studies include: the Nedd4-2 construct used [full-length [8, 38], individual WW domains [5, 22] or the entire WW domain region (present study)], the Sgk1 construct used [full-length [8, 38], or an Sgk peptide 5, 22, (present study)], the type of assay [in vivo [8] or in vitro 5, 22, 38, (present study)] and finally the species of origin of the constructs [human [38], Xenopus [8, 22] or mouse [5] (present study)]. Of these, only the use of constructs of human origin, rather than of mouse or Xenopus origin, appears to be associated with the clear demonstration of an association between Nedd4-2 and Sgk1. This suggests that in non-human species the interaction between Sgk and Nedd4-2 may be too weak or transient to be detected.
In conclusion our study provides evidence that there is no direct functional link between Sgk-mediated ENaC regulation and Na+ feedback inhibition. Our results confirm the importance of the channel’s PY motifs for mediating the stimulatory effect of Sgk but suggest that the stimulation of ENaC by Sgk involves more than the inhibition of Nedd4-2 mediated channel retrieval. This makes sense physiologically because Sgk-mediated ENaC stimulation is likely to occur in sodium depleted states with a high level of plasma aldosterone and a low renal tubular sodium concentration. Under these conditions Sgk and the lack of sodium feedback inhibition will act synergistically to stimulate ENaC mediated sodium absorption to maintain sodium balance.
References
Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O (1999) Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest 103:667–673
Alvarez de la Rosa D, Canessa CM (2003) Role of SGK in hormonal regulation of epithelial sodium channel in A6 cells. Am J Physiol 284:C404–C414
Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM (1999) The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem 274:37834–37839
Alvarez de la Rosa D, Li H, Canessa CM (2002) Effects of aldosterone on biosynthesis, traffic, and functional expression of epithelial sodium channels in A6 cells. J Gen Physiol 119:427–442
Asher C, Sinha I, Garty H (2003) Characterization of the interactions between Nedd4-2, ENaC, and sgk-1 using surface plasmon resonance. Biochim Biophys Acta 1612:59–64
Auberson M, Hoffmann-Pochon N, Vandewalle A, Kellenberger S, Schild L (2003) Epithelial Na+ channel mutants causing Liddle’s syndrome retain ability to respond to aldosterone and vasopressin. Am J Physiol 285:F459–F471
De La Rosa DA, Paunescu TG, Els WJ, Helman SI, Canessa CM (2004) Mechanisms of regulation of epithelial sodium channel by SGK1 in A6 cells. J Gen Physiol 124:395–407
Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O (2001) Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20:7052–7059
Diakov A, Korbmacher C (2004) A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s α-subunit. J Biol Chem 279:38134–38142
Dinudom A, Young JA, Cook DI (1993) Amiloride-sensitive Na+ current in the granular duct cells of mouse mandibular glands. Pflügers Arch 423:164–166
Dinudom A, Young JA, Cook DI (1993) Na+ and Cl− conductances are controlled by cytosolic Cl− concentration in the intralobular duct cells of mouse mandibular glands. J Membr Biol 135:289–295
Dinudom A, Komwatana P, Young JA, Cook DI (1995) Control of the amiloride-sensitive Na+ current in mouse salivary ducts by intracellular anions is mediated by a G protein. J Physiol (Lond) 487:549–555
Dinudom A, Harvey KF, Komwatana P, Young JA, Kumar S, Cook DI (1998) Nedd4 mediates control of an epithelial Na+ channel in salivary duct cells by cytosolic Na+. Proc Natl Acad Sci USA 95:7169–7173
Dinudom A, Fotia AB, Lefkowitz RJ, Young JA, Kumar S, Cook DI (2004) The kinase Grk2 regulates Nedd4/Nedd4-2-dependent control of epithelial Na+ channels. Proc Natl Acad Sci USA 101:11886–11890
Duc C, Farman N, Canessa CM, Bonvalet JP, Rossier BC (1994) Cell-specific expression of epithelial sodium channel α, β, and γ subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol 127:1907–1921
Faletti CJ, Perrotti N, Taylor SI, Blazer-Yost BL (2002) Sgk: an essential convergence point for peptide and steroid hormone regulation of ENaC-mediated Na+ transport. Am J Physiol 282:C494–C500
Fotia AB, Dinudom A, Shearwin KE, Koch JP, Korbmacher C, Cook DI, Kumar S (2003) The role of individual Nedd4-2 (KIAA0439) WW domains in binding and regulating epithelial sodium channels. FASEB J 17:70–72
Garty H, Palmer LG (1997) Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77:359–396
Gruber WD, Knauf H, Frömter E (1973) The action of aldosterone on Na+ and K+ transport in the rat submaxillary main duct. Pflügers Arch 344:33–39
Harvey KF, Dinudom A, Komwatana P, Jolliffe CN, Day ML, Parasivam G, Cook DI, Kumar S (1999) All three WW domains of murine Nedd4 are involved in the regulation of epithelial sodium channels by intracellular Na+. J Biol Chem 274:12525–12530
Harvey KF, Dinudom A, Cook DI, Kumar S (2001) The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem 276:8597–8601
Henry PC, Kanelis V, O’Brien MC, Kim B, Gautschi I, Forman-Kay J, Schild L, Rotin D (2003) Affinity and specificity of interactions between Nedd4 isoforms and the epithelial Na+ channel. J Biol Chem 278:20019–20028
Kaelin Jr WG, Krek W, Sellers WR, DeCaprio JA, Ajchenbaum F, Fuchs CS, Chittenden T, Li Y, Farnham PJ, Blanar MA et al (1992) Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell 70:351–364
Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O (2001) A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15:204–214
Kellenberger S, Gautschi I, Rossier BC, Schild L (1998) Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest 101:2741–2750
Komwatana P, Dinudom A, Young JA, Cook DI (1996) Cytosolic Na+ controls an epithelial Na+ channel via the Go guanine nucleotide-binding regulatory protein. Proc Natl Acad Sci USA 93:8107–8111
Konstas AA, Mavrelos D, Korbmacher C (2000) Conservation of pH sensitivity in the epithelial sodium channel (ENaC) with Liddle’s syndrome mutation. Pflügers Arch 441:341–350
Konstas AA, Bielfeld-Ackermann A, Korbmacher C (2001) Sulfonylurea receptors inhibit the epithelial sodium channel (ENaC) by reducing surface expression. Pflügers Arch 442:752–761
Konstas AA, Shearwin-Whyatt LM, Fotia AB, Degger B, Riccardi D, Cook DI, Korbmacher C, Kumar S (2002) Regulation of the epithelial sodium channel by N4WBP5A, a novel Nedd4/Nedd4-2-interacting protein. J Biol Chem 277:29406–29416
Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA (1997) cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics 40:435–443
Lang F, Henke G, Embark H, Waldegger S, Palmada M, Bohmer C, Vallon V (2003) Regulation of channels by the serum and glucocorticoid-inducible kinase—implications for transport, excitability and cell proliferation. Cell Physiol Biochem 13:41–50
Malik B, Schlanger L, Al-Khalili O, Bao HF, Yue G, Price SR, Mitch WE, Eaton DC (2001) ENaC degradation in A6 cells by the ubiquitin-proteosome proteolytic pathway. J Biol Chem 276:12903–12910
Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA (1999) Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J 18:3024–3033
Pearce D (2003) SGK1 regulation of epithelial sodium transport. Cell Physiol Biochem 13:13–20
Rauh R, Paulides M, Diakov A, Korbmacher C (2004) Stimulation of ENaC by SGK1 does not require sodium feedback inhibition. Pflügers Arch 447:S124
Rossier BC, Pradervand S, Schild L, Hummler E (2002) Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol 64:877–897
Shimkets RA, Lifton RP, Canessa CM (1997) The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem 272:25537–25541
Snyder PM, Olson DR, Thomas BC (2002) Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277:5–8
Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC (2004) cAMP and SGK regulate ENaC through convergent phosphorylation of Nedd4-2. J Biol Chem 279:45753–45758
Staub O, Dho S, Henry PC, Correa J, Ishikawa T, McGlade J, Rotin D (1996) WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J 15:2371–2380
Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciecanover A, Schild L, Rotin D (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquination. EMBO J 16:6325–6336
Volk T, Konstas AA, Bassalay P, Ehmke H, Korbmacher C (2004) Extracellular Na+ removal attenuates rundown of the epithelial Na+ channel (ENaC) by reducing the rate of channel retrieval. Pflügers Arch 447:884–894
Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC (2002) Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus Oocytes. J Gen Physiol 120:191–201
Wang J, Barbry P, Maiyar AC, Rozansky DJ, Bhargava A, Leong M, Firestone GL, Pearce D (2001) SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol 280:F303–F313
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22:537–548
Acknowledgements
This project was supported by the National Health and Medical Research Council of Australia, the Australian Kidney Foundation, the Deutsche Forschungsgemeinschaft (SFB423) and the Johannes and Frieda Marohn Stiftung. The expert technical assistance of Ralf Rinke is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Robert Rauh and Anuwat Dinudom cotributed equally to this work.
Rights and permissions
About this article
Cite this article
Rauh, R., Dinudom, A., Fotia, A.B. et al. Stimulation of the epithelial sodium channel (ENaC) by the serum- and glucocorticoid-inducible kinase (Sgk) involves the PY motifs of the channel but is independent of sodium feedback inhibition. Pflugers Arch - Eur J Physiol 452, 290–299 (2006). https://doi.org/10.1007/s00424-005-0026-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-005-0026-5