
Abstract
The aim of this study was to examine the effects of reduced glycogen concentration on sarcoplasmic reticulum (SR) Ca2+-ATPase activity in rat fast-twitch muscles. In the first experiment, the gastrocnemius (GAS) muscle from one leg was removed, followed by starvation for 24–72 h, after which the remaining GAS was removed. Intra-animal comparisons revealed that starvation caused a 25% reduction (P<0.05) in the glycogen concentration but no change in SR Ca2+-ATPase activity in the GAS. In the second experiment, the SR was purified from a mixture of the GAS and vastus lateralis muscles. In half of the samples obtained from each animal, glycogen was extracted from the SR by treatment with glucoamylase. Treatment resulted in a 94.1 and 70.2% decrease (P<0.01) in glycogen and glycogen phosphorylase, respectively, and a 41.5% increase (P<0.05) in a fluorescein isothiocyanate (FITC) binding to SR Ca2+-ATPase. On the other hand, SR Ca2+-ATPase activity and the affinity of the enzyme for ATP were unaltered. These results do not implicate depletion of muscle glycogen as a contributor to impaired SR Ca2+-ATPase activity as measured in vitro. Therefore, it is concluded that muscle glycogen does not influence exercise tolerance and work productivity in working muscles by modulating the structure of protein involved in Ca2+ sequestering. Furthermore, it is suggested that the FITC binding assay may be inappropriate as a method for examining the mechanisms for the altered activity of SR Ca2+-ATPase.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Skeletal muscles induced to contract repeatedly respond with a progressive loss in their ability to generate a target force or power. This decline in function, referred to as muscle fatigue, has a complex etiology that can involve various metabolic and ionic factors [2, 28, 33]. Among the numerous factors implicated in fatigue, none have been more evident or universally agreed upon than the need for a reserve of glycogen in the working muscle cell. Evidence linking muscle glycogen to muscle fatigue has been most apparent during large muscle group activity, e.g., running and cycling, at intensities ranging from 60 to 85% of maximal aerobic power [12, 15]. Despite the well-accepted view of a close correlation between glycogen and fatigue, the precise mechanisms by which glycogen depletion disrupts cellular function remain incompletely understood. During repeated contraction at moderate intensities, glycolysis functions to provide essential precursors for the generation of citric acid cycle intermediates. Therefore, it is assumed that fatigue associated with glycogen depletion would be due to the loss of an energy source. This energy crisis hypothesis remains the widely accepted explanation to account for fatigue during low-glycogen states. The possibility, however, seems remote since either little or no reduction in the ATP concentration occurs during prolonged exercise [15].
Skeletal muscle sarcoplasmic reticulum (SR) regulates intracellular free calcium concentration and plays a central role in muscle contraction and relaxation. Accumulating evidence has implicated altered intracellular Ca2+ regulation as a major contributor to fatigue. Studies utilizing muscles from human, horse, and rat have demonstrated that exercise that results in muscle fatigue disturbs SR Ca2+ handling properties in working muscle [6, 10, 17]. The Ca2+ handling properties that are attenuated with exercise include both Ca2+ uptake and Ca2+ release. The major protein responsible for Ca2+ uptake is the 110-kDa ATPase (SR Ca2+-ATPase) that translocates 2 mol of Ca2+ across the SR bilayer membrane upon the hydrolysis of 1 mol of ATP, and the exercise-induced depression in Ca2+ uptake results primarily from the impaired catalytic function of this enzyme [17, 29]. There is evidence to suggest that the lowered SR Ca2+-ATPase activity induced by vigorous contractile activity may be ascribed to structural changes in its nucleotide-binding domain, i.e., the ATP binding site [25, 27]. A fraction of muscle glycogen appears to be specifically associated with the SR membrane [13, 23]. Glycogen phosphorylase (GP) is also associated with the SR via its binding to the glycogen particles [23]. Thus, glycolysis of SR glycogen accompanied by muscle contraction elicits the release of GP from the SR membrane [23]. In vitro study of Cuenda et al. [8] revealed that the addition of exogenous GP to SR vesicles shifted the ATP binging site of SR Ca2+-ATPase towards an ATP binding conformation, suggesting that endogenous GP associated with the SR may exert the effects on the structure of the ATP binding site.
These findings raise the plausible hypotheses that depressions in SR Ca2+-ATPase activity and structural alterations in the ATP binging site with contractile activity might occur secondarily to the decreased glycogen and GP contents and that deteriorations in exercise tolerance and work productivity of muscles resulting from glycogen depletion might be mediated through such mechanisms. However, few data were available concerning the effects of glycogen status on SR function. No published study presently exists, moreover, that examines whether a decrease in the glycogen content that occurs in vivo might influence SR Ca2+-ATPase activity.
These findings prompted us to investigate changes in SR Ca2+-ATPase activity in muscles where the glycogen content was repressed. In the first experiment (starvation experiment), we subjected rats to starvation, which resulted in the varied degrees of reductions in muscle glycogen. The results from the starvation experiment suggest that, for conditions in which the muscle glycogen content is reduced by up to 50%, glycogen status does not alter SR Ca2+-ATPase activity. However, it was envisaged that the magnitude of the decline in glycogen observed in the starvation experiment might be too small to affect the activity. Therefore, we performed the second experiment (glycogen-extraction experiment) in which glycogen was extracted from the SR membrane by treatment with glucoamylase. Considering the findings reported by Cuenda et al. [8], the glycogen-extraction experiment focused on changes not only in SR Ca2+-ATPase activity but also in the structure of the ATP binding site and the affinity of the enzyme for ATP.
Methods
Starvation experiment
The Animal Care Committee of Hiroshima University approved all procedures for this study. Experiments were performed on the gastrocnemius (GAS) muscles from male Wistar strain rats ranging in body weight from 190 to 220 g. Rats had free access to standard rodent chow and water until the time of experiment. On the day of experiment, while animals were anesthetized using an intraperitoneal injection of pentobarbital sodium (40 mg kg−1), the GAS (control muscle) from all rats was surgically removed from one leg according to Ianuzzo et al. [16]. Then, animals were arbitrarily assigned to a control and a starvation group. After surgery, control rats were given chow and water ad libitum, whereas starved rats were given water only. The GAS (experimental muscle) was removed from the other leg under anesthesia 1, 2, or 3 days later for starved rats and 3 days later for control rats (n=7 per group).
Immediately after ablation, muscle pieces of ∼100 mg wet weight were diluted with 9 volumes (mass vol−1) of ice-cold homogenizing buffer composed of (in mM) 300 sucrose, 20 3-(N-morpholino)propanesulfonic acid (MOPS)/KOH, pH 7.4, 0.0014 pepstatin, 0.83 benzamidine, 0.0022 leupeptin, and 0.2 phenylmethanesulfonyl fluoride (PMSF) and homogenized for 3×30 s at 5,000 rpm. The homogenates were centrifuged at 2,000×g for 15 min. The resulting supernatant was used for measurement of SR Ca2+-ATPase activity. The remaining portion of the muscle was stored at −80°C for later analysis.
SR Ca2+-ATPase activity was spectrophotometrically determined in homogenates at 37°C according to Simonides and van Hardeveld [31]. The analytical procedure for measurement has been described in detail elsewhere [36]. Briefly, the assay mixture consisted of 20 mM N-2-hydroxyethyl-piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.5, 1 mM ethyleneglycotetraacetic acid (EGTA), 200 mM KCl, 15 mM MgCl2, 0.8 mM CaCl2, 10 mM sodium azide, 0.4 mM nicotinamide adenine dinucleotide (NADH), 10 mM phosphoenolpyruvate, 18 U ml−1 pyruvate kinase, 18 U ml−1 lactate dehydrogenase, and 1 μg ml−1 Ca2+ ionophore A23187. The reaction was started by adding MgATP to give a final concentration of 4 mM.
Muscle samples were freeze-dried at −70°C for 24 h, and glycogen concentration was measured in perchloric-acid-denatured protein pellets. To hydrolyze glycogen, pellets were boiled in 2 N HCl for 2 h. The concentration was determined fluorometrically using the production of nicotinamide adenine dinucleotide phosphate (NADPH) [24].
Glycogen-extraction experiment
Experiments were performed on male Wistar strain rats ranging in body weight from 250 to 260 g (n=6 per group). Each animal was deeply anaesthetized using an intraperitoneal injection of pentobarbital sodium (40 mg kg−1). The GAS and vastus lateralis muscles were exposed, dissected free from surrounding muscle, and excised from both legs. These two muscles were combined for microsomal preparations.
Microsomes were prepared according to Lees and Williams [22]. The muscle was placed in 5 volumes (mass vol−1) of ice-cold homogenizing buffer consisting of 20 mM HEPES, pH 7.0, 0.2% (mass vol−1) sodium azide, 0.2 mM PMSF, and 1 mM ethylenediaminetetraacetic acid (EDTA) and homogenized for 3×30 s at 5,000 rpm. Homogenates were centrifuged at 8,000×g for 15 min at 4°C. After filtration through four layers of gauze, the supernatants obtained from each muscle were divided into a glycogen-extracted (GE) and a control (CON) group, and KCl was added to give a final concentration of 600 mM. In order to extract SR glycogen, the GE group buffer was supplemented with 17.4 U ml−1 glucoamylase. Samples were gently stirred on ice for 1 h and then centrifuged at 50,000×g for 120 min at 4°C. The resulting pellet was resuspended in the storage buffer (homogenizing buffer containing 300 mM sucrose and 150 mM KCl). The storage buffer used for SR glycogen measurement did not contain sucrose [22]. Protein concentrations were determined by the Bradford assay [5] using bovine serum albumin as a standard.
The SR Ca2+-ATPase and GP contents in microsomal proteins were determined by densitometric analysis of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Ten micrograms microsomal protein was applied to gels. Electrophoresis was carried out using a 10% acrylamide separating gel and a 4% acrylamide stacking gel and lasted 15 h at constant voltage (60 V) in a cold room. The gels were stained with Coomassie blue R in 45% (vol vol−1) methanol and densitometrically evaluated using NIH Image software. In addition to the CON and GE samples, each gel always contained microsomal proteins obtained from the plantaris muscle. This was utilized as a standard. Densitometrically evaluated amounts of SR Ca2+-ATPase and GP were normalized by reference to those in a standard.
Thirty micrograms microsomal protein was incubated in a solution containing (in mM) 50 Tris–HCl, pH 8.8, 250 sucrose, 0.1 CaCl2, 5 MgCl2, and 0.05 fluorescein isothiocyanate (FITC) for 60 min at room temperature. The reaction was stopped by adding a solution consisting of (in mM) 100 Tris–HCl, pH 6.8, 250 sucrose, and 5 Na2ATP [21]. Proteins were separated by electrophoresis as mentioned above and then fixed by incubating the gel in 25% (vol vol−1) isopropanol. The gel was photographed with ultraviolet illumination. The FITC binding to SR Ca2+-ATPase was densitometrically evaluated using the negative of the photograph and the NIH Image software.
With the exception of the use of microsomes and the method used for hydrolysis of glycogen, glycogen concentration and SR Ca2+-ATPase activity were measured using the same methods as described in the starvation experiment. Five and ten micrograms microsomal proteins were used for measurements of the glycogen content and enzyme activity, respectively. To hydrolyze glycogen, proteins were incubated with gentle shaking in the assay mixture composed of 174 mM acetic acid, pH 4.8 and 8.7 U ml−1 glucoamylase for 120 min at 40°C. Determinations of ATP dependence, namely, the ATP concentration needed to obtain half-maximum activity, were performed by varying the ATP concentration in the assay mixture. On the basis of a double reciprocal plot for the ATP concentration and SR Ca2+-ATPase activity, the ATP dependence was calculated.
Statistical analyses
All data were expressed as means±SD. Significances of the effects of starvation on muscles from the left and right legs (starvation experiment) and of glycogen extraction (glycogen-extraction experiment) were assessed using Student’s t test for paired samples. A two-way variance analysis was performed to evaluate the influence of starvation and duration of starvation (starvation experiment). If an overall F-value was obtained, a Scheffé post hoc analysis was used to isolate the significance different means. All comparisons were performed at the 95% confidence level.
Results
Starvation experiment
No differences existed in the glycogen contents of control muscles among four groups studied. As shown in control rats, ablation per se caused no changes in the glycogen content (Table 1). Intra-animal comparisons revealed that starvation for up to 3 days evoked significant reductions (P<0.05) in the glycogen content. The reductions in muscles of rats starved for 1, 2, and 3 days averaged 24, 23, and 25%, respectively. Large differences were observed between animals with regard to the extent of these changes; the range of decreases was from 0 to 50%. For rats starved for 3 days, significant decreases (P<0.05) were also observed when compared to experimental muscles from control rats. For all groups, there were no differences in SR Ca2+-ATPase activities between control and experimental muscles, suggesting no causal relationship between glycogen status and the enzyme activity. In order to more unequivocally elucidate this, SR Ca2+-ATPase activity was plotted against differences in the glycogen concentration for all samples (including control and experimental muscles from each rat). As expected, no significant correlation was observed (Fig. 1).

Plot of muscle glycogen concentration vs sarcoplasmic reticulum (SR) Ca2+-ATPase activity for gastrocnemius. The gastrocnemius muscle (control) was removed from one leg, followed by starvation for 24–72 h, after which the remaining gastrocnemius (experimental) was removed. On the average, starvation elicited a 25% decrease in the glycogen concentration. The values for all samples (including control and experimental muscles from each animal) were plotted. Glycogen concentration is expressed as micromole glucosyl units per gram dry weight. There was no significant correlation between glycogen concentration and enzyme activity (r=0.05)
Glycogen-extraction experiment
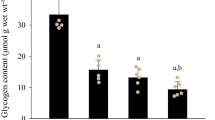
Treatment with glucoamylase resulted in pronounced decreases (P<0.01) in the glycogen and GP contents in microsomes, which amounted to 5.9 and 29.8% of control levels, respectively (Figs. 2 and 3). It was possible that the reduction in the GP content would lead to a decrease in the relative concentration of SR Ca2+-ATPase in microsomes. However, its content was unchanged. The unexpected result was that extraction evoked significant increases in an FITC binding to SR Ca2+-ATPase (Fig. 4). Densitometry of the SR Ca2+-ATPase band revealed a 41.5% increase (P<0.05) as compared to control. As shown in previous studies [21, 25], FITC is a fluorescein molecule that binds specifically to a lysine residue located in the ATP binding site of the SR. Thus, alterations in the FITC binding reflect structural changes in the ATP binding site [21, 25]. From the increased FITC binding observed in the glycogen extraction experiment, it was expected that glycogen-extraction might be responsible for increases in maximal SR Ca2+-ATPase activity or in the affinity of the enzyme for ATP [8, 23]. However, both parameters were unaltered (Fig. 5).

Glycogen concentration associated with sarcoplasmic reticulum (SR) for control (CON) and glycogen-extracted (GE) conditions. Glycogen associated with the SR was extracted by incubating muscle homogenates in a solution containing glucoamylase. Values are means±SD. **P<0.01, compared with control

Electrophoretic separation of sarcoplasmic reticulum (SR) Ca2+-ATPase and glycogen phosphorylase (GP) (a) and their contents (b) in microsomal fractions for control (solid bars) and glycogen-extracted (open bars) conditions. For treatment of glycogen-extracted condition, see legend to Fig. 2. Microsomal proteins were separated on a 10% polyacrylamide gel. Only Ca2+-ATPase and GP regions are shown (a). The contents of SR Ca2+-ATPase and GP are expressed as a percentage of control (b). Values are means±SD. **P<0.01, compared with control

Electrophoresis of FITC-labeled sarcoplasmic reticulum (a) and densitometric evaluation of FITC binding (b) for control (CON) and glycogen-extracted (GE) conditions. For treatment of glycogen-extracted condition, see legend to Fig. 2. The FITC labeling was visualized by photography under ultraviolet light. Values are means±SD. *P<0.05, compared with control

Sarcoplasmic reticulum (SR) Ca2+-ATPase activity and ATP dependence of SR Ca2+-ATPase for control (solid bars) and glycogen-extracted (open bars) conditions. For treatment of glycogen-extracted condition, see legend to Fig. 2. The ATP dependence represents the ATP concentration needed to obtain half-maximum activity. Values are means±SD
Discussion
The fact that muscle fatigue is highly correlated with glycogen depletion at work loads ranging from 60 to 85% of maximal aerobic power was clearly established by a series of reports published in the late 1960s and early 1970s [1, 4, 18]. Despite years of investigation, the precise mechanisms underlying glycogen-depletion-induced fatigue remain incompletely understood. The possible causality of SR glycogen in changes of SR Ca2+-ATPase as assessed in vitro stems from the following previous observations: (1) repressions in SR Ca2+-ATPase activity due to vigorous muscle contraction are always accompanied by reductions in the FITC binding to the enzyme [21, 25]; (2) the reduced SR glycogen content leads to the release of GP from the SR membrane [23]; and (3) the increased GP content in SR vesicles results in the enhancement in the FITC binding to SR Ca2+-ATPase [8]. Taking these findings into account, we hypothesized that decreases in the glycogen content associated with the SR might induce the inactivation of SR Ca2+-ATPase that is mediated through structural alterations in the ATP binding site. However, this assumption is not supported by our results demonstrating that in vivo and in vitro decreases in the glycogen content fail to influence SR Ca2+-ATPase activity.
Glycolytic enzymes as well as glycogen particles are associated with the SR membrane [20]. The rate of utilization of glycogen during intense muscle contraction is several-fold higher for the SR vesicles than for whole muscles [23], suggesting that locally generated ATP supplied by glycolytic enzymes bound to the SR membrane may be utilized to sequester Ca2+ from the cytoplasm to the SR lumen [13, 23]. It has been shown that these metabolic systems are necessary for optimal SR Ca2+ sequestering and in some cases are more efficient than exogenous ATP [35]. It appears likely, therefore, that the decreased glycogen located on the SR membrane may adversely affect SR Ca2+ uptake capacity in vivo.
Pronounced decreases in muscle glycogen can be achieved by exposing muscles to prolonged contractile activities. In most cases, such activities cause appreciable reductions not only in the glycogen content, but also in SR Ca2+-ATPase activity. The depression of SR Ca2+-ATPase activity observed in these muscles seems to result to some extent from protein oxidation [19, 26]. It is obvious that, using muscles undergoing repetitive contraction, the question cannot be assessed as to whether the reduced content of muscle glycogen might by itself account for changes in SR Ca2+-ATPase activity because the effects of glycogen are indistinguishable from those of oxidation. Therefore, the first experiment utilized starvation, which allows examination of the impact of the reduced glycogen on its own. Although not measured in the starvation experiment because of restriction on the amount of tissue available, the GP content associated with the SR has been reported to reduce by 50 to 75% in muscles from rabbits that were starved for 48 h [9]. According to the aforementioned findings observed by Cuenda et al. [8], it is conceivable that the loss of GP would be responsible for structural changes in the ATP binding site, leading to the decreased SR Ca2+-ATPase activity. However, the results obtained in the starvation experiment demonstrated that similar treatment, i.e., starvation for 24–72 h, was not capable of bringing about the decay of enzyme activity (Table 1).
Historical studies on humans have demonstrated that only when glycogen levels in working muscles are nearly depleted does muscular fatigue become manifest [14, 18]. The possibility exists that a maximum of 50% reduction in the glycogen content in the starvation experiment may be too small to result in a decrease in SR Ca2+-ATPase activity. On the basis of this assumption, in the second experiment, glycogen was extracted from the SR membrane by incubating a solution containing glucoamylase. The glycogen extraction experiment confirms observations of Lees and Williams [22] who found marked decreases in the GP content associated with the SR to result from glycogen extraction. On the other hand, we found the enhancement of the FITC binding to the SR to be without concomitant changes in SR Ca2+ATPase activity and in the affinity of the enzyme for ATP (Fig. 5), whereas they observed an increase in the enzyme activity with no alterations in the FITC binding [22]. Although the reason for this controversy is unclear, an important implication of observations by Lees and Williams [22] and in the glycogen-extraction experiment is that it could negate the use of the FITC binding assay as a method for examining the mechanisms for the altered catalytic activity of SR Ca2+-ATPase.
Glycogen-depletion-mediated muscular fatigue could be explained by the failure of the excitation–contraction coupling processes other than SR Ca2+-sequestering ability. Both dihydropyridine receptor, voltage sensor of transverse (T)-tubule, and SR Ca2+-release channel have several phosphorylation sites [11]. It is purported that phosphorylation of some of these sites, which is an ATP-requiring process, is necessary for normal transmission of signals within the triad [34]. Similar to the SR membrane, the triads comprise glycogen and a complex of glycolytic enzymes that can synthesize ATP. Single-fiber studies by Allen and colleagues [3, 7] suggest that the repressed force output that occurs with repetitive contraction may be ascribed to reductions in the ATP concentration within the narrow space of the triad gap, which result from compartmentalized depletion of glycogen and lead to the altered phosphorylation status in the triad. In contrast, a more recent study by Stephenson et al. [32] has indicated that even when the ATP concentration is maintained, glycogen depletion is associated with loss of fiber ability to respond to T-system depolarization. Their observations lead to the implication that glycogen might exert the effects on the structure of the T-tubular SR Ca2+-release site complex. It is also pointed out that glycogen status may be involved in plasma membrane excitability. The Na+-K+-ATPase is an integral membrane protein and can maintain low internal Na+ and high internal K+ concentrations by the vectorical transport of three Na+ and two K+ per each molecule of ATP utilized. ATP synthesized through glycolysis appears to be the predominant source of the fuel for the Na+-K+-ATPase [30]. Therefore, energy homeostasis for this enzyme would be jeopardized if glycogen is depleted.
Although it appears likely that not only does muscle glycogen provide the primary energy source during exercise, but that glycogen affects the structure and function of organs in skeletal muscles [8], the results reported here do not implicate depletion of muscle glycogen as a contributor to impaired SR Ca2+-ATPase activity as measured in vitro. Therefore, it is concluded that muscle glycogen does not influence exercise tolerance and work productivity in working muscles by modulating the structure of proteins involved in Ca2+ sequestering. Furthermore, it is suggested that the FITC binding assay may be inappropriate as a method for examining the mechanisms for the altered activity of SR Ca2+-ATPase.
References
Ahlborg BJ, Bergström J, Ekelund L-G, Hultman E (1967) Muscle glycogen and muscle electrolytes during prolonged physical exercise. Acta Physiol Scand 70:129–142
Allen DG (2004) Skeletal muscle function: role of ionic changes in fatigue, damage and disease. Clin Exp Pharmacol Physiol 31:485–493
Allen DG, Lännergren, Westerblad H (1997) The role of ATP in the regulation of intracellular Ca2+ release in single fibres of mouse skeletal muscle. J Physiol (Lond) 498:587–600
Bergström J, Hermanson L, Hultman E, Saltin B (1967) Diet, muscle glycogen and physical performance. Acta Physiol Scand 71:140–150
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Byrd SK, McCutcheon LJ, Hodgson DR, Gollnick PD (1989) Altered sarcoplasmic reticulum function after high-intensity exercise. J Appl Physiol 67:2072–2077
Chin ER, Allen DG (1997) Effects of reduced muscle glycogen concentration on force, Ca2+ release and contractile protein function in intact mouse skeletal muscle. J Physiol (Lond) 497:17–29
Cuenda A, Centeno F, Gutierrez-Merino C (1991) Modulation by phosphorylation of glycogen phosphotylase–sarcoplasmic reticulum interaction. FEBS Lett 283:273–276
Cuenda A, Henao F, Nogues M, Gutierrez-Merino C (1994) Quantification and removal of glycogen phosphorylase and other enzymes associated with sarcoplasmic reticulum membrane preparations. Biochim Biophys Acta 24:35–43
Duhamel TA, Green HJ, Perco JG, Sandiford SD, Ouyang J (2004) Human muscle sarcoplasmic reticulum during submaximal exercise in normoxia and hypoxia. J Appl Physiol 97:180–187
Favero TG (1999) Sarcoplasmic reticulum Ca2+ release and muscle fatigue. J Appl Physiol 87:471–483
Fitts RH (1994) Cellular mechanisms of muscle fatigue. Physiol Rev 74:49–94
Friden J, Seger J, Ekblom B (1989) Topographical localization of muscle glycogen: an ultrahistochemical study in human vastus lateralis. Acta Physiol Scand 135:381–391
Gollnick PD, Armstrong RB, Saubert IV CW, Sembrowich WL, Shepherd RE, Saltin B (1973) Glycogen depletion patterns in human skeletal muscle fibers during prolonged work. Pflügers Arch 344:1–12
Green HJ (1991) How important is endogenous muscle glycogen to fatigue in prolonged exercise? Can J Physiol Pharmacol 69:290–297
Ianuzzo C, Gollnick P, Armstrong R (1976) Compensatory adaptations of skeletal muscle fiber types to long term functional overload. Life Sci 19:1517–1524
Inashima S, Matsunaga S, Yasuda T, Wada M (2003) Effect of endurance training and acute exercise on sarcoplasmic reticulum function in rat fast- and slow-twitch muscles. Eur J Appl Physiol 89:142–149
Karlsson J, Saltin B (1971) Diet, muscle glycogen, and endurance performance. J Appl Physiol 31:203–206
Klebl BM, Ayoub AT, Pette D (1998) Protein oxidation, tyrosine nitration, and inactivation of sarcoplasmic reticulum Ca2+-ATPase in low-frequency stimulated rabbit muscle. FEBS Lett 422:381–384
Korge P, Campbell KB (1994) Local ATP regeneration is important for sarcoplasmic reticulum Ca2+ pump function. Am J Physiol 267: C357–C366
Leberer E, Härtner K-T, Pette D (1987) Reversible inhibition of sarcoplasmic reticulum Ca-ATPase by altered neuromuscular activity in rabbit fast-twitch muscle. Eur J Biochem 162:555–561
Lees SJ, Williams JH (2004) Skeletal muscle sarcoplasmic reticulum glycogen status influences Ca2+ uptake supported by endogenously synthesized ATP. Am J Physiol 286:C97–C104
Lees SJ, Franks PD, Spangenburg EE, Williams JH (2001) Glycogen and glycogen phosphorylase associated with sarcoplasmic reticulum: effects of fatiguing activity. J Appl Physiol 91:1638–1644
Lowry OH, Passonneau JV (1972) A flexible system of enzymatic analysis. Academic, New York
Luckin KA, Favero TG, Klug GA (1991) Prolonged exercise induces structural changes in SR Ca2+-ATPase of rat muscle. Biochem Med Metab Biol 46:391–405
Matsunaga S, Inashima S, Yamada T, Watanabe H, Hazama T, Wada M (2003) Oxidation of sarcoplasmic reticulum Ca2+-ATPase induced by high-intensity exercise. Pflügers Arch 446:394–399
Matsushita S, Pette D (1992) Inactivation of sarcoplasmic-reticulum Ca2+-ATPase in low-frequency-stimulated muscle results from a modification of the active site. Biochem J 285:303–309
McCully KK, Authier B, Olive J, Clark BJ III (2002) Muscle fatigue: the role of metabolism. Can J Appl Physiol 27:70–82
Mishima T, Yamada T, Matsunaga S, Wada M (2005) N-acetylcysteine fails to modulate the in vitro function of sarcoplasmic reticulum of diaphragm in the final phase of fatigue. Acta Physiol Scand 184:195–202
Okamoto K, Wang W, Rounds J, Chambers EA, Jacobs DO (2001) ATP from glycolysis is required for normal sodium homeostasis in resting fast-twitch rodent skeletal muscle. Am J Physiol 281:E479–E488
Simonides WS, van Hardeveld C (1990) An assay for sarcoplasmic reticulum Ca2+-ATPase activity in muscle homogenate. Anal Biochem 191:321–331
Stephenson DG, Nguyen LT, Stephenson GMM (1999) Glycogen content and excitation–contraction coupling in mechanically skinned muscle fibres of the cane toad. J Physiol (Lond) 519:177–187
Tupling AR (2004) The sarcoplasmic reticulum in muscle fatigue and disease: role of the sarco(endo)plasmic reticulum Ca2+-ATPase. Can J Appl Physiol 29:308–329
Westerblad H, Allen DG, Bruton JD, Andrade FH, Lännergren J (1998) Mechanisms underlying the reduction of isometric force in skeletal muscle fatigue. Acta Physiol Scand 162:253–260
Xu K, Zweier J, Becker L (1995) Functional coupling between glycolysis enzyme and sarcoplasmic reticulum Ca2+ transport. Circ Res 77:88–97
Yamada T, Inashima S, Matsunaga S, Nara I, Kajihara H, Wada M (2004) Different time course of changes in sarcoplasmic reticulum and myosin isoforms in rat soleus muscle at early stage of hyperthyroidism. Acta Physiol Scand 180:79–87
Acknowledgement
This study was supported by Grants-in-Aid for Scientific Research of Japan 17300209.
Author information
Authors and Affiliations
Corresponding author
Additional information
The work was done at the Faculty of Integrated Arts and Sciences, Hiroshima University, Hiroshima, Japan.
Rights and permissions
About this article
Cite this article
Mishima, T., Sugiyama, M., Yamada, T. et al. Effects of reduced glycogen on structure and in vitro function of rat sarcoplasmic reticulum Ca2+-ATPase. Pflugers Arch - Eur J Physiol 452, 117–123 (2006). https://doi.org/10.1007/s00424-005-0018-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-005-0018-5