
Abstract
Light scattering is an empirical technique employed to measure rapid changes in cell volume. This study describes a new configuration for the method of light scattering and its corroboration by measurements of cell height (as a measure of cell volume). Corneal endothelial cells cultured on glass cover-slips were mounted in a perfusion chamber on the stage of an inverted microscope. A beam of light was focused on the cells from above the stage at an angle of 40° to the plane of the stage. The scattered light intensity (SLI), captured by the objective and referred to as forward light scatter (FLS), increased and decreased in response to hyposmotic and hyperosmotic shocks, respectively. The rapid increase and decrease in SLI corresponded to cell swelling and shrinkage, respectively. Subsequently, SLI decreased and increased as expected for a regulatory volume decrease (RVD) and increase (RVI), respectively. These data are in agreement with measurements of cell height, demonstrating that the method of light scatter in FLS mode is useful for monitoring rapid changes in cell volume of cultured cells. Changes in SLI caused by gramicidin were consistent with cell volume changes induced by equilibration of NaCl and KCl concentrations across the cell membrane. Similarly, an additional decrease in SLI was recorded during RVD upon increasing K+ conductance by valinomycin. Decreasing K+ conductance of the cell membrane with Ba2+ changed the time course of SLI consistent with the effect of the K+ channel blocker on RVD. Bumetanide and dihydro-ouabain inhibited increases in SLI during RVI. In conclusion, FLS is a valid method for qualitative analysis of cell volume changes with a high time resolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Changes in the volume of animal cells following an osmotic shock are characterized by two dynamic components [16, 19]. The first component is the approach towards osmotic equilibrium by either the influx or efflux of water across the plasma membrane through water channels and lasts only a few seconds. The second component is a volume regulatory phenomenon, that, through either regulatory volume decrease (RVD) or increase (RVI), leads to restoration of the original volume. Unlike the first component, RVD and RVI are coupled to the net movement of electrolytes and are realized through a number of active and passive volume-sensitive ion transport mechanisms [16, 19, 22]. As a result, characterization of mechanisms underlying RVD and RVI provide an alternative approach for investigating factors regulating various mechanisms of ion transport in epithelia.
A direct approach for characterization of RVD and RVI is to follow changes in cell volume in response to acute osmotic shocks. A number of methods have been developed to measure rapid changes in cell volume during RVD/RVI, including those based on the principles of dye dilution [1, 11, 26], light scattering [6, 9, 12, 13, 42], dynamic fluorescence quenching (DFQ) [35, 37], fluorescence self-quenching [15], cell height [5, 17, 33, 39, 40], morphometry [10, 25], atomic force microscopy [14, 27, 28, 31, 32], laser scanning reflection microscopy [21], scanning conductance microscopy [18], ion-sensitive electrodes [8] and electrical impedance [30]. Although the dye dilution method is suitable for real-time measurements, this technique is limited by poor volume sensitivity (especially in thin cells), inevitable dye loss, photobleaching and the requirement of a confocal microscope for optical sectioning [1, 26]. The principle of DFQ was demonstrated previously using the Cl−-sensitive fluorescent dye 6-methoxy-N-(3-sulphopropyl)quinolinium (SPQ) [35, 37]. The volume-sensitive fluorescence of SPQ was obtained through substitution of Cl− in the Ringers' with NO3 −. This method has high sensitivity and temporal resolution. However, quencher-dye pairs insensitive to Cl− have not been discovered, limiting the broad use of this approach. The light scattering method is an empirical approach and requires complementary cell volume measurements of comparable temporal and volume sensitivity for corroboration. Correlating relative changes in cell height to those in cell volume is a direct method applicable to confluent cell monolayers [5, 17, 33, 40, 43]. Such measurements have been made in real-time by determining cell height as the distance between bright fluorescent microspheres attached to the apical and basal surfaces of a cell. This method relies on the location of the microspheres rather than on intensity of fluorescence [40]. As a consequence, the method of cell height is less vulnerable to optical artefacts than other fluorescence/scatter-based methods.
The objective of this study was to demonstrate the implementation of a new optical configuration to measure cell volume through light scattering. Our results show that changes in the intensity of the scattered light along the forward path of the incident beam, called forward light scatter (FLS), are related to changes in cell volume. We confirmed the volume sensitivity of FLS by measurements of cell height and by using certain well-established pharmacological manoeuvres that yield predictable volume changes. The FLS configuration became necessary as the scattered light collected at acute angles in the direction opposite to that the illumination beam, as described earlier [9], gave poor volume sensitivity with confluent corneal endothelial cells.
Materials and methods
Cell culture
As described earlier [35, 37], second or third passage cultures of bovine corneal endothelial cells (BCEC) were grown to confluence on glass cover-slips (25 mm diameter, 150 µm thick) or permeable Anopore filter supports (pore size 0.2 µm, Nunc Intermed, Roskilde, Denmark). Primary cultures from fresh cow eyes were established in DMEM and supplemented with 10% fetal calf serum and an antibiotic-antimycotic mixture (penicillin 100 U/ml, streptomycin 100 mg/ml and fungizone 0.25 mg/ml). The cells were maintained at 37 °C in a CO2 (5%)/air (95%) incubator and fed every 2–3 days.
Solutions
All experiments were conducted using \( {\text{HCO}}^{ - }_{3} \)-free Ringer's solutions equilibrated with air. Isosmotic solutions for cell height experiments consisted of (mM): 153.5 Na+, 4 K+, 0.6 Mg2+, 1.4 Ca2+, 118.2 Cl−, 1 \( {\text{HPO}}^{{2 - }}_{4} \), 10 HEPES, 31.3 gluconate, 6 glucose, pH 7.4, adjusted to 320 mOsm/(kg H2O) with sucrose. The hyposmotic solutions were obtained by removing 63.5 Na+, 35 Cl− and 28.5 gluconate, and adjusted to 230 mOsm/(kg H2O) by sucrose.
Light scattering experiments were performed using two types of solutions: type 1 was similar to the solutions in cell height experiments, but adjusted to 297 mOsm/(kg H2O). Hyposmotic solutions were made by dilution with de-ionized water. Type-2 Ringer's was similar to type 1 with the exception of 85 Na+ and 85 Cl− and more sucrose to adjust to 297 mOsm/(kg H2O). Hyposmotic solutions were here obtained by including less sucrose.
Low-Na+ Ringer's contained 8 Na+, 4 K+, 1.4 Ca2+, 0.6 Mg2+, 28.5 N-methyl-d-glucamine (NMDG+), 10 TRIS, 1 \( {\text{HPO}}^{{2 - }}_{4} \), 10 HEPES, 116.2 Cl−, 2.8 gluconate, 107 choline, 14.25 \( {\text{SO}}^{{2 - }}_{4} \) and 6 glucose. pH was adjusted to 7.4. Hyposmotic solutions were made by removing 27 choline, 27 Cl−.
Perfusion system for light scattering
The cell-covered cover-slip was sealed at the bottom of an aluminium perfusion chamber (volume 80 µl). A plain cover-slip served as the top cover. The chamber was held in a brass thermostatic collar maintained at 37 °C and placed on the stage of an inverted microscope (Nikon Diaphot 200). The solutions were perfused via gravity flow in excess of 4–6 chamber volumes/min.
Perfusion system for cell height measurement
We used an Ussing-type chamber (volume 0.3 ml), made of Plexiglass and designed for cell height measurement with an upright microscope [40]. The bottom of the chamber was sealed with a standard glass cover-slip. Cell-coated glass cover-slips were placed upside down. This eliminated disturbances in image quality caused by changes in the refractive index of the perfusate between the objective and the cells (especially during hyperosmotic shocks). Uniform perfusion of the cells was obtained via gravity flow at a rate of 8 ml/min. A custom-built bubble trap prevented the formation of air bubbles. Pressure changes in the chamber caused by switching solutions were avoided by using non-interrupting valves. The temperature of the solutions was maintained at ~34 °C. A few experiments were conducted with cells grown on Anopore filters by placing them between the two halves of the perfusion chamber.
Measurement of light scattering
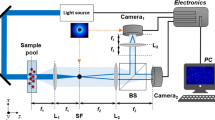
Light from a halogen lamp (20 W) was filtered (>420 nm) and focused onto the cells through an achromatic lens from above the stage (Fig. 1) at an adjustable angle of incidence (θi). In most experiments, the beam subtended θi=40° to the plane of the cover-slip and had a spot size of approximately 1 mm. Light scattered in the forward direction (Fig. 1) was collected by a long-working-distance objective (Olympus PL2; 20×; working distance 1.2 cm; NA=0.4) with a field of view of approximately 600–800 cells. The scattered light intensity (SLI) was then directed to a photomultiplier tube (HC125-01, Hamamatsu, Bridgewater, N.J., USA) by a dichroic mirror centred at 520 nm (made for fluorescein isothiocyanate fluorescence by Nikon). Several variations of this configuration included in the experiments are as follows:

Schematic diagram of the set-up for forward light scattering (FLS). Light from a halogen lamp was filtered (>420 nm) and focused on a monolayer of cells grown to confluence on glass cover-slips and perfused continuously with Ringer's at 37 °C. The scattered light collected by the objective was detected by a photomultiplier (PMT) and demodulated by a lock-in amplifier. Other variations of this configuration were also employed (see text). The apparatus shown here achieved the highest volume sensitivity
-
1.
The illumination beam was mechanically chopped (400 Hz; SR800 Optical Chopper; Stanford Research Systems, Sunnyvale, Calif., USA) and corresponding photomultiplier responses were demodulated using a lock-in amplifier (SRS 830; Stanford Research Systems). The inclusion of the lock-in amplifier in the setup enabled sensitive detection of changes in SLI, especially when the signal at the photomultiplier tube was low under isosmotic conditions.
-
2.
The halogen lamp output was filtered using interference filters (530±10 nm) to determine the wavelength sensitivity of scatter responses. Although extensive experiments were not conducted, the general pattern of SLI in response to cell swelling or shrinkage was not affected by choosing an illumination beam of narrow spectrum.
-
3.
The halogen lamp, chopper assembly and light-guide assembly were replaced by blue- (LNG992CF9; Panasonic; peak λ 470 nm) or red-light-emitting diodes (LEDs) and were sine- or square-wave modulated by an electronic function generator (model CFG253; Tectronix) at 1–4 kHz. Scattered light was detected using a photomultiplier, while the output was demodulated with the lock-in amplifier. The use of LEDs enabled the detection of FLS by modulating the illumination at higher frequencies enabling additional noise rejection.
-
4.
The illumination beam from the halogen lamp was directed from below the microscope stage to yield a configuration similar to that in [9] (Fig. 4B). This was the first configuration tested and yielded a very low volume sensitivity after more than 150 trials (also see Results).
Measurement of cell height
Cell height was measured as the distance between fluorescent microspheres (1 µm diameter), which served as high-contrast landmarks for top and bottom surfaces of an individual cell (Fig. 2B). Details of the experimental setup, along with the depth-scanning algorithm employed in the study, have been discussed earlier [40, 41]. Microspheres were localized in real-time through depth scanning with a fluorescence imaging microscope. The focal plane was placed ~30 positions across the endothelial monolayer with a piezoelectric nanopositioner (PIFOC Model 721; Physik Instrumente, Waldbronn, Germany; see Fig. 2A) coupled to the microscope objective (Achroplan, 40×, NA=0.75, water-immersion, working distance 1.9 mm). At each focal plane, an image of the microspheres was acquired in the field of view (Fig. 2B, left of the panel). The position of the microsphere along the z-axis was ascertained using a computer algorithm that identified the maximum fluorescent intensity corresponding to each microsphere in the field of view (Fig. 2B; right of the panel). The distance between the focal planes corresponding to maximum intensity of any top microsphere to that of a specified reference bottom microsphere is taken as a measure of cell height (Fig. 2B; right of the panel). The computer program permitted simultaneous tracking of up to 20 top microspheres in real-time.

Diagram showing the set-up for measuring cell height. A Monolayers of bovine corneal endothelial cells (BCEC) were coated at the apical and basolateral surfaces with fluorescent microspheres and excited by blue light. The distance between the top fluorescent beads on any given cell was measured relative to a common bottom microsphere designated as the reference. The relative locations of the top microspheres along the z-axis were determined by depth scanning accomplished by the piezoelectric drive attached to the objective. Cells cultured on permeable supports (Anopore filters) as well as grown on cover-slips can be used. In this study, we mostly used monolayers cultured on cover-slips that were mounted upside-down in the chamber. This avoided solution changes between the objective and the cells as well as disturbances in image quality caused by changes in the refractive index of the perfusate. B The focal plane of the objective was stationed every 150 nm and an image of the entire field of view was acquired using an image acquisition program (left). The same program also controlled the piezoelectric drive and determined the peak intensity of each microsphere in the field of view. The distance between the peak intensity of a top microsphere on any given cell and that of the reference microsphere represents the cell height (T c) of the cell to which the top microsphere adheres (right)
Attachment of microspheres
For bottom microspheres, a suspension of fluorescent latex microspheres (F-8852, Molecular Probes, Eugene, Ore., USA; ~1 µm diameter) was prepared in ethyl alcohol (4 µl microspheres suspension in 10 ml ethanol) and plated on cover-slips. The cells were seeded after the ethanol evaporated. For porous filters, a suspension of the same microspheres in 1% gelatine solution (4 µl microspheres suspension in 10 ml gelatine) was applied. Microspheres at the apical surface of the cells, referred to as top microspheres, were biotin-coated (F-8768, Molecular Probes) or avidin-coated (F-8776, Molecular Probes). These were suspended in an isosmotic Ringer's solution (5 µl/ml), applied to the monolayer of cells, and allowed to adhere for approximately 45–60 min. Non-adherent microspheres were removed by repeated replacement of the Ringer's before measurements.
Measurement of cell height by confocal microscopy
In addition to real-time measurement of cell height, microspheres on the cell surface were visualized using a confocal laser scanning microscope (LSM 510; Zeiss; equipped with a Plan-Neofluar 40×/1.3 oil-immersion objective). Cells with attached microspheres were loaded with the pH-sensitive fluorescent dye 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF), by exposure to the dye's acetoxymethyl ester (BCECF-AM; Molecular Probes), as previously described [3]. BCECF and microspheres were excited at 488 nm. Fluorescence images from cells and microspheres were collected through a band-pass filter centred at 530 nm at intervals of 0.21 µm to construct a z-stack under computer control.
Chemicals
Cell culture supplies were obtained from Gibco BRL (Grand Island, N.Y., USA). All other chemicals were obtained from Sigma (St. Louis, Mo., USA). Stock solutions of gramicidin D (10 mM) and valinomycin (10 mM) in dimethylsulphoxide were stored desiccated at −20 °C.
Results
Typical light scattering responses to osmotic shocks
Cells were focused with a 20× objective using phase-contrast illumination and the illumination beam for light scattering measurements switched on. With the incident beam at an angle (θi) of ~40° in the FLS mode (Fig. 1), the measured SLI increased with cell swelling and decreased with cell shrinkage. Typical SLI responses with type-1 solutions, shown in Fig. 3A and B, were obtained in response to hyposmotic and hyperosmotic shocks, respectively. The characteristics of these responses did not change as the wavelength of the incident beam was altered, or when anisosmotic shocks were induced using type-2 Ringer's (data not shown). Due to the high plasma membrane water permeability, the first component of the response corresponding to osmotic equilibrium across the plasma membrane was complete within a few seconds. In typical responses shown in Fig. 3A and B, a 50 mOsm/(kg H2O) anisosmotic shock resulted in a change of SLI equivalent to ~10% of that observed under isosmotic conditions. The magnitude of this change is the measure of volume sensitivity of the scatter response, and varied among cover-slips. This variability can be attributed partly to small differences in the angle of incidence from cover-slip to cover-slip and to the thickness of the basement membrane. Although a detailed analysis of this angle sensitivity was not undertaken, very high sensitivity to volume change was observed in most trials with the lock-in amplifier, with which small signals could be detected reliably. A typical response to an osmotic shock of 10 mOsm/(kg H2O) is shown in Fig. 4A. In contrast to the high sensitivity achieved in the FLS mode, volume sensitivity was relatively low with the illumination in the backward scattering mode. Figure 4B illustrates a typical backward scatter response to a 150 mOsm/(kg H2O) shock. The apparent volume-sensitive scatter is less than 0.1% of the isosmotic scatter. All experiments discussed below were therefore conducted in the FLS configuration with an θi of ~40°. Despite variable scatter responses between cover-slips, the change in SLI was inversely proportional to the change in osmolarity (±50 mOsm/(kg H2O)) for any given cover-slip and configuration of the illumination beam, as expected from Van't Hoff's law (Fig. 5A and B).

Characteristics of the scattered light responses caused by anisosmotic conditions. Osmolality of the perfusate in isosmotic conditions was 295 mOsm/(kg H2O). Scattered light intensity (SLI) is given in arbitrary units (A.U.) A FLS response to a reduction of the osmolality by 50 mOsm/(kg H2O) (HYPO). Concomitant with the fast approach to osmotic equilibrium, increased light scatter (cell swelling) ensued immediately after the shock. The subsequent slow decrease in SLI towards its original value is the regulatory volume decrease (RVD). B SLI response to an increase of the osmolality by 50 mOsm/(kg H2O) (HYPER). The Ringer's was made hyperosmotic by adding sucrose. The subsequent slow increase in SLI towards its original value is the regulatory volume increase (RVI). The results shown in A and B are typical of more than 20 independent experiments

Sensitivity of the scattered light responses to volume changes in the forward and the backward light scattering configurations. Initially, cells were perfused with isosmotic Ringer's and subsequently exposed to hyposmotic Ringer's. A FLS response to a 10 mOsm/(kg H2O) hypotonic shock. Inset display optical configuration for FLS. B Typical backward light scatter (BLS) response to a 150 mOsm/(kg H2O) hyposmotic shock showing RVD. Inset demonstrate optical configuration for BLS. θ i is the angle of incidence of the light beam

Linearity of the scattered light response. A SLI response during hyperosmotic treatment and hyposmotic challenge. Under isosmotic conditions SLI was 3.36 A.U. Hypertonicity decreased SLI to 3.24 A.U. Hypotonicity increased SLI to 3.50 A.U. B Normalized SLI as a function of the reciprocal of osmolality (π). SLI was normalized by reference to SLI under isosmotic conditions. The data were obtained from two osmotic shocks on the same cover-slip. SLI values at the peak of the response (i.e. immediately after the osmotic shock) were chosen to represent the cell volume in response to immediate osmotic equilibrium resulting from a shock
Typical cell height responses to osmotic shocks
The bottom microspheres remained un-endocytosed by endothelial cells after 3–4 days of cell culture. The bottom microspheres applied to the gelatine layer and plain glass surface were used as reference for determining cell height. One bottom microsphere served as the reference for measuring the displacement of top microspheres upon changes in cell height (see Fig. 2B). Figure 6A shows an optical section (confocal image) of the bottom microspheres in focus. Figure 6B shows an optical section located below the nuclear dome of the cell, while Fig. 6C is a section near the top of the dome. Note that the endothelial cells are relatively flat (2–4 µm) at the periphery and have a dome-shaped morphology, being tallest over the nucleus. This dome-shaped morphology is also evident in the reconstruction of the z-stacks shown in the side panels of Fig. 6A–C. Due to the dome-shaped morphology, many microspheres were found downhill from the domes. However, in general, a sufficient number of microspheres remained attached close to the top of the dome. Under isosmotic conditions, the average distance between bottom and top microspheres (centre-to-centre) was 2.96±0.6 µm (n=79). Since the microspheres attach frequently on the slope of the dome, this average only represents an "apparent cell height". In fact, based on z-stacks from confocal observations, peak cell height was 4.5±0.4 µm (n=11) (Figs. 6A–C).

Confocal images of the fluorescent microspheres. Cells were grown on glass cover-slips that had been coated with microspheres (bottom microspheres). The cells were loaded with 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF) and the upper surface also labelled with microspheres (top microspheres). Next, depth scans were conducted under computer control. z-Stacks were viewed with Zeiss LSM viewing software. A Optical section at the centre of the reference microsphere that was chosen among many bottom microspheres. Top (T) and bottom (B) microspheres are indicated by filled arrows. B Optical section at the centre of a top microsphere. C Optical section taken at a point downhill on the cell surface. D Apparent cell height (T a ) is the distance between a top and a bottom microsphere. The true cell height T c is given by the position of the top microsphere at the top of the dome. ΔT is the actual change in cell height, ΔT a the apparent change in cell height. A single cell may have many adherent bottom beads but only one (B r ) was chosen as reference. Volume increase is proportional to ΔT a
Figure 7 illustrates typical cell height responses to hyposmotic (Fig. 7A and D) and hyperosmotic (Fig. 7B) shocks. Exposure to hyposmotic shock resulted in a cell height profile consistent with the predicted cell volume response. Thus, the initial rise in apparent cell height (33.3±3.9%; n=64; eight independent cover-slips) corresponds to an immediate rise in cell volume, due to osmotic water equilibrium, similar to that of the light scattering response (Fig. 3). The return to isosmotic Ringer's led to a precipitous decrease in apparent cell height concomitant with cell shrinkage. Responses shown in Fig. 7B to hyperosmotic shocks were collected with a water-immersion objective mounted on an upright microscope where the bottom microspheres are closer to the objective. This manoeuvre circumvents disturbances in image quality caused by changes in the refractive index of the perfusate due to the addition of sucrose. Exposure to hyperosmotic Ringer's decreased apparent cell height by 24.3±1.16% (n=81; N=10 independent cover-slips), followed by a slow and slight increase (over 20 min) (Fig. 7B). A typical response to anisosmotic shocks with cells grown on permeable filters is shown in Fig. 7D. The hyposmotic and hyperosmotic responses from various experiments were consolidated to assess the linearity of response. It was assumed that the change in apparent cell height is proportional to the change in cell volume. The resulting Van't Hoff's plot is illustrated in Fig. 7C. The linear profile over a 200-mOsm/kg H2O osmotic range indicates that apparent cell height is a valid indicator of cell volume.

Changes in cell height (shown as cell height T c , as a percentage of the cell height prior to the first osmotic perturbation) in response to anisosmotic Ringer's. In A, B and D, the solid line is the average of n beads from N experiments. The dotted lines are ±SEM. A Response to hyposmotic Ringer's; n=16, N=1. T c under isosmotic conditions prior to the hyposmotic challenge was 2.72±0.31 µm. Osmolality was reduced from 320 to 230 mOsm/(kg H2O). B Response to hyperosmotic Ringer's; n=58, N=6. T c under isosmotic conditions prior to the hyperosmotic challenge was 3.39±0.22 µm. Osmolality was increased from 320 to 430 mOsm/(kg H2O). C Van't Hoff plot constructed from experiments similar to those shown in A and B. The maximal initial change in T c (expressed as a percentage of isosmotic values) recorded in hypo- or hyperosmotic conditions is shown as a function of the reciprocal of the osmolality (π). D As in A, but with the cells grown on a permeable support. Both basolateral and apical surfaces were exposed simultaneously to hyposmotic Ringer's; n=9, N=1. T c under isosmotic conditions prior to the first hyposmotic challenge was 1.94±0.40 µm. Osmolality was reduced from 320 to 230 mOsm/(kg H2O)
Changes in light scattering in response to pharmacologically induced cell volume changes
In many cell types, RVD is induced partially by K+ and Cl− loss through conductive pathways, the latter often activated by cell swelling [16, 19]. Gramicidin increases permeability to K+/Na+ and valinomycin, a K+ ionophore, can be used to alter whole-cell cation (Na+, K+) conductance, and induce predictable patterns of cell volume changes. This paradigm has been demonstrated through the DFQ technique [35, 37]. Specifically, BCEC undergo secondary cell swelling when cells are exposed to gramicidin a few minutes after hyposmotic shock. Similar experiments were conducted while measuring SLI or changes in cell height. Accordingly, exposure to gramicidin (2 µM) led to swelling when cells were perfused with Na+-rich Ringer's. Typical responses are shown in Fig. 8A and B with the light scattering and cell height methods, respectively. Conversely, Fig. 9 shows volume decrease (indicated by a decrease in light scatter) upon exposure to gramicidin when cells were perfused with low-Na+ Ringer's. This additional volume decrease in low-Na+ Ringer's indicates the whole-cell cation conductance is limiting for RVD in BCEC. Consistent with these findings, Fig. 10A shows that exposure to valinomycin (0.3 µM) accelerated the decrease in SLI, indicating a rapid RVD when cells were perfused with Na+-rich Ringer's. In further confirmation of the role of K+ conductance, Fig. 10B shows that RVD decelerated when cells were perfused with Ringer's containing Ba2+ (5 mM) as a K+ channel blocker.

Secondary cell swelling in response to gramicidin. Cells were perfused initially with isosmotic Ringer's and then exposed to a hyposmotic shock and subsequently to gramicidin (GC; 2 µM). A Response recorded with the FLS method (N=6). B Time course of T c. The solid line is the mean from five cells from one experiment. The dotted lines are ±SEM. T c under isosmotic conditions prior to the hyposmotic challenge was 5.98±0.21 µm

RVD is limited by K+ conductance. Swollen cells were exposed to gramicidin (2 µM) in low-Na+ Ringer's (extracellular [Na+] 8 mM, the remainder of Na+ was replaced by equimolar N-methyl-d-glucamine (NMDG +), to which the gramicidin-formed cation channels are impermeable. The rapid decrease in SLI indicates cell shrinkage

RVD is limited by K+ conductance. A Exposure to valinomycin (Val; 0.3 µM) during RVD led to an accelerated volume loss indicating potentiation of RVD. B Fractional recovery (FR) of SLI, calculated as [100 (SLIMAX−SLI)]/[SLIMAX−SLIISO], where SLIMAX is the peak SLI after imposing the osmotic challenge and SLIISO the SLI under isosmotic conditions prior to the hyposmotic shock. Extent of RVD was reduced by Ba2+ (5 mM). In the same cover-slip, valinomycin showed increased RVD after Ba2+ removal. The results shown in each panel are typical of at least three independent experiments
Involvement of Na+-K+-2Cl− in RVI
In the absence of \( {\text{HCO}}^{ - }_{3} \) RVI in BCEC is brought about by activated Na+-K+-2Cl− cotransport [2, 7, 16], as in other cell types. Cells were exposed to bumetanide and ouabain to test whether or not FLS was capable of detecting altered RVI. Figure 11A illustrates a control RVI response followed by an RVI response in the presence of 100 µM bumetanide, a specific inhibitor of the cotransporter. Bumetanide inhibited the apparent RVI significantly, whilst the apparent RVI response returned when bumetanide was removed. Similarly, reducing the activity of Na+-K+ATPase disrupts the Na+/K+ gradients for the cotransporter, and can therefore be expected to inhibit RVI. Slowing of the Na+-K+ATPase can be expected to cause cell swelling. This swelling is due to the net influx of Na+ through leakage pathways, and Cl− through resting anion channels in the endothelium [29]. Under isosmotic conditions, SLI increased slowly after a few minutes of exposure to 100 µM dihydro-ouabain (Fig. 11B). The immediate response was a small decrease in SLI that indicated a volume decrease. The latter could be secondary to immediate depolarization in the membrane potential and a consequent loss of K+. This response was not investigated further. During the slow swelling phase, cells were exposed to hyperosmotic shocks in order to determine the apparent RVI response. As expected, the RVI was very small. However, the cells continued to swell at a rate matching swelling even before osmotic shock. Upon removal of dihydro-ouabain, SLI decreased slowly towards the baseline under isosmotic conditions.

RVI response is inhibited by bumetanide. Cells perfused with isosmotic Ringer's were exposed to hyperosmotic Ringer's and the cell volume response recorded by light scattering in the FLS mode. A Control RVI response is followed by RVI in the presence of bumetanide (100 µM). Note that the presence of bumetanide completely inhibits RVI. However, when bumetanide is removed, the RVI response returns. B Exposure to dihydro-ouabain (100 µM) leads to an immediate decrease in SLI followed by a slow increase. In the presence of dihydro-ouabain and consequent swelling, RVI in response to hyperosmotic shock is not apparent in two subsequent shocks. Removal of dihydro-ouabain resulted in a slow return of SLI towards the baseline. The results shown in each panel are typical of at least three independent experiments
Discussion
Forward light scattering (FLS) vs. backward light scattering (BLS)
In this study, we examined a modified optical configuration for a microscope-based implementation of the method of light scattering to measure rapid changes in cell volume. Although Echevarria and Verkman [9] also used an inverted microscope the illumination beam in their set up made an acute angle with the optical axis of the objective (depicted in Fig. 4B, inset). Since the direction of propagation of the collected scattered light is not along the illumination beam, but rather back towards the source itself, we refer to Echevarria and Verkman's configuration as backward light scattering. Conversely, we refer to the configuration demonstrated in Fig. 1A (also depicted in the inset of Fig. 4A) as forward light scattering since the scattered light captured by the objective is in the direction of propagation of the illumination beam. Our references to configurations shown in the insets of Fig. 4A and B as FLS and BLS, respectively, are consistent with reports on flow cytometry in which FLS is also employed for the measurement of cell size [4, 34].
Angle sensitivity of the scatter response
In attempts to duplicate Echevarria and Verkman's configuration [9], initial experiments were conducted in the BLS mode. Although we succeeded in recording volume-sensitive responses after a considerable number of trials, the volume sensitivity could not be improved (Fig. 4B). This is presumably due to the limited range of the incidence angle (θi) of the illumination beam. Mechanical limitations imposed by the size of the illumination optics beneath the microscope stage precluded a detailed investigation into the dependence of the volume sensitivity on θi. However, the FLS scheme permitted the same illumination optics to be held from above the stage and to be manoeuvred readily to yield a wider range of incidence angles (θi=30–50°; Fig. 1). In this configuration, we obtained consistently higher volume sensitivity with a θi of approximately 40°. This is evident from results illustrated in Figs. 3A and B and 4A, which demonstrate that the apparent volume sensitive scatter is much larger than with the BLS approach (Fig. 4B). Furthermore, although the SLI intensity varied with large changes in θi, small changes around θi=40° (within ±4°) did not result in a significant loss of volume sensitivity. This lack of local sensitivity is evident in measurements with consistently higher volume sensitivity, despite inevitable repositioning of the illumination optics from experiment to experiment.
Taking the above observations on FLS and BLS together, it seems that the volume sensitivity of the scatter is affected significantly by large changes in θi. Specifically, the large changes affect even the sign of the slope of the relationship between Δ[scatter intensity] and Δ[cell volume]. Thus, scatter intensity increased with cell swelling and vice versa (Fig. 3A and B) in FLS mode, but decreased with cell swelling and vice versa (Fig. 4B) in BLS mode. These findings suggest that at illumination angles between those comprising the FLS and BLS modes, scatter sensitivity to cell volume becomes negligible and an inflection point is reached. Consistent with this argument is the fact that scatter intensity varied inversely with cell volume in the BLS mode as in [9], as well as in this study with BLS configuration (Fig. 4B). Reduced sensitivity observed in the BLS mode could suggest that the θi chosen for experiments similar to those shown in Fig. 4B may have been near an inflection point indicated above. It may be noted that several non-microscopic studies have shown both an inverse proportionality between cell volume and light scatter [12, 13, 24, 38] as well as direct proportionality between light scatter and cell volume [6].
Linearity of the scatter response
It is apparent from previous studies that scattered light contains information on size, shape and state of the aggregation of the cells [9, 20, 23]. However, in the absence of changes in cell shape and state of aggregation, changes in the intensity of light scattering have been shown to vary linearly with changes in cell volume (Fig. 5B). Consistent with a rapid approach towards osmotic equilibrium and slow volume changes in response to regulatory volume control, our results (Fig. 3A and B) show that changes in SLI follow two distinct dynamic modes in response to acute osmotic shocks: (1) immediate cell swelling or shrinkage due to osmotically induced water movement and (2) regulatory volume control mechanisms (i.e. RVD or RVI). If the FLS method were corrupted by changes in the refractive index between isosmotic and hyper/hyposmotic Ringer's, RVD and RVI would not have necessarily shown consistent dynamic profiles. Whatever their origin, scattered light responses during RVD and RVI are sensitive to changes in cell volume. This is evident in the slow responses of changes in SLI consistent with the longer time constant (approximately minutes) of the RVD/RVI phenomenon. These slow changes in SLI are not corrupted by extracellular changes in the refractive index, which has a time constant of the order of seconds due to the high flow through the small (80 µl) chamber. In addition, peak changes in SLI (a true measure of the osmometric change in cell volume) after osmotic shocks were related inversely to changes in predicted cell volume, according to Van't Hoff's law (Fig. 5B). Taken together, these findings suggest the scatter signal is volume sensitive.
SLI changes in response to gramicidin or valinomycin are consistent with predicted changes in cell volume and with previous findings by the DFQ technique and intracellular [Na+] measurements [35, 36, 37]. Thus, upon exposure to gramicidin at normal levels of extracellular Na+ (Fig. 8A and B, light scattering and cell height measurements, respectively), the secondary increase in SLI (indicating increase in cell volume) is due to a net Na+ influx through gramicidin channels facilitated by an inwards gradient for Na+, as well as parallel Cl− influx through volume-regulated anion channels [35, 36, 37]. After reversing the gradient for Na+, exposure to gramicidin resulted in a precipitous decrease in SLI (indicating rapid RVD; shown in Fig. 9). On the other hand, the K+-selective ionophore valinomycin favours K+ loss and induces a decrease in SLI (indicating enhanced RVD) in the presence of normal extracellular [Na+] (Fig. 10A). Reduced FLS during RVD in response to Ba2+ (Fig. 10B) is also consistent with reduced RVD by a block of K+ channels.
To validate the response during cell shrinkage, we recorded SLI during the RVI response in the presence of bumetanide and, prior to the exposure of cells, to dihydro-ouabain. It is clear from Fig. 11A that bumetanide, a known inhibitor of the Na+-K+-2Cl− cotransporter, reduced the rate of increase in SLI during RVI in a reversible manner. Activation of the Na+-K+-2Cl− cotransporter has been demonstrated in BCEC as a response to hyperosmotic conditions [7]. Accordingly, the reduced increase in SLI during RVI (Fig. 11A) is consistent with reduced RVI by inhibition of Na+-K+-2Cl− cotransport. Finally, ouabain, which induced a constant increase in SLI, inhibited RVI by disrupting the gradients for Na+ and K+ through inhibition of the Na+-K+-ATPase.
Cell height as a measure of cell volume and verification of key FLS methodology
The principle of using cell height as a measure of cell volume is applicable to cells in confluent monolayers [5, 17, 33, 40]. Since measurement of cell height is direct and accomplished in real-time, we used this technique to corroborate FLS measurements. The setup we have chosen to measure cell height has been utilized extensively to measure changes in volume of A6 cells (a polarized and tight epithelial cell line from the distal tubule of Xenopus laevis kidney) grown on permeable supports [40, 41]. Although the specific type of labelling microspheres (i.e. avidin-coated) was suitable for A6 cells [40, 41], current experiments showed that they adhere less avidly to BCEC. This lower affinity could be attributed to the dome shape of the BCEC cells, leading to the microspheres' slipping downhill. Although small lateral movements were handled adequately by a dynamic selection of the region of interest in the image acquisition program, the loss of microspheres from the dome region made it difficult to conduct a large number of successful experiments. No specific problems were noted from the bottom microspheres. Results presented in this study provide the first evidence to suggest that although BCEC are dome-shaped, apparent cell height is indicative of cell volume. For this study, the most important point is that cell height measurements corroborated measurements by FLS (Fig. 8A vs. B) and the DFQ method [35, 36, 37].
In conclusion, we have modified the configuration of the light-scattering method for measuring changes in cell volume [9]. This new FLS configuration was corroborated through cell height measurements. It yielded a higher volume sensitivity for measurements of rapid changes in cell volume of BCEC cells grown to confluence on glass cover-slips compared with the BLS configuration. We have demonstrated volume sensitivity of SLI using the principles of RVD/RVI and predictable volume changes using gramicidin, valinomycin, and ouabain. Many of the results shown in this study have also been shown in BCEC cells using DFQ, offering comparable temporal resolution [35, 36, 37].
References
Altamirano J, Brodwick MS, Alvarez-Leefmans FJ (1998) Regulatory volume decrease and intracellular Ca2+ in murine neuroblastoma cells studied with fluorescent probes. J Gen Physiol 112:145–160
Bildin VN, Yang H, Crook RB, Fischbarg J, Reinach PS (2000) Adaptation by corneal epithelial cells to chronic hypertonic stress depends on upregulation of Na:K:2Cl cotransporter gene and protein expression and ion transport activity. J Membr Biol 177:41–50
Bonanno JA, Yi G, Kang XJ, Srinivas SP (1998) Reevaluation of Cl−/\( {\text{HCO}}^{ - }_{3} \) exchange in cultured bovine corneal endothelial cells. Invest Ophthalmol Vis Sci 39:2713–2722
Bouvier T, Troussellier M, Anzil A, Courties C, Servais P (2001) Using light scatter signal to estimate bacterial biovolume by flow cytometry. Cytometry 44:188–194
Crowe WE, Wills NK (1991) A simple method for monitoring changes in cell height using fluorescent microbeads and an Ussing-type chamber for the inverted microscope. Pflugers Arch 419:349–357
Dho S, Chou S, Chang XB, Rommens JM, Foskett JK (1992) Right-angle light scattering to assay basal and regulated plasma membrane Cl− conductances. Am J Physiol 263:C530–C534
Diecke FP, Zhu Z, Kang F, Kuang K, Fischbarg J (1998) Sodium, potassium, two chloride cotransport in corneal endothelium: characterization and possible role in volume regulation and fluid transport. Invest Ophthalmol Vis Sci 39:104–110
Dierkes PW, Coulon P, Neumann S, Schlue WR (2002) Potentiometric measurement of cell volume changes and intracellular ion concentrations under voltage-clamp conditions in invertebrate nerve cells. Anal Bioanal Chem 373:762–766
Echevarria M, Verkman AS (1992) Optical measurement of osmotic water transport in cultured cells. Role of glucose transporters. J Gen Physiol 99:573–589
Espelt MV, Mut PN, Amodeo G, Krumschnabel G, Schwarzbaum PJ (2003) Volumetric and ionic responses of goldfish hepatocytes to anisotonic exposure and energetic limitation. J Exp Biol 206:513–522
Farinas J, Simanek V, Verkman AS (1995) Cell volume measured by total internal reflection microfluorimetry: application to water and solute transport in cells transfected with water channel homologs. Biophys J 68:1613–1620
Fischbarg J, Kuang KY, Hirsch J, Lecuona S, Rogozinski L, Silverstein SC, Loike J (1989) Evidence that the glucose transporter serves as a water channel in J774 macrophages. Proc Natl Acad Sci USA 86:8397–8401
Fischbarg J, Li J, Kuang K, Echevarria M, Iserovich P (1993) Determination of volume and water permeability of plated cells from measurements of light scattering. Am J Physiol 265:C1412–C1423
Goerge T, Niemeyer A, Rogge P, Ossig R, Oberleithner H, Schneider SW (2002) Secretion pores in human endothelial cells during acute hypoxia. J Membr Biol 187:203–211
Hamann S, Kiilgaard JF, Litman T, Alvarez-Leefmans FJ, Winther BR, Zeuthen T (2002) Measurement of cell volume changes by fluorescence self-quenching. J Fluorescence 12:139–145
Hoffmann EK, Dunham PB (1995) Membrane mechanisms and intracellular signalling in cell volume regulation. Int Rev Cytol 161:173–262
Kawahara K, Onodera M, Fukuda Y (1994) A simple method for continuous measurement of cell height during a volume change in a single A6 cell. Jpn J Physiol 44:411–419
Korchev YE, Gorelik J, Lab MJ, Sviderskaya EV, Johnston CL, Coombes CR, Vodyanoy I, Edwards CR (2000) Cell volume measurement using scanning ion conductance microscopy. Biophys J 78:451–457
Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Häussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physiol Rev 78:247–306
Latimer P (1979) Light scattering vs. microscopy for measuring average cell size and shape. Biophys J 27:117–126
Maric K, Wiesner B, Lorenz D, Klussmann E, Betz T, Rosenthal W (2001) Cell volume kinetics of adherent epithelial cells measured by laser scanning reflection microscopy: determination of water permeability changes of renal principal cells. Biophys J 80:1783–1790
McCarty NA, O'Neil RG (1992) Calcium signaling in cell volume regulation. Physiol Rev 72:1037–1061
McGann LE, Walterson ML, Hogg LM (1988) Light scattering and cell volumes in osmotically stressed and frozen-thawed cells. Cytometry 9:33–38
McManus ML, Strange K (1993) Acute volume regulation of brain cells in response to hypertonic challenge. Anesthesiology 78:1132–1137
Mitchell CH, Fleischhauer JC, Stamer WD, Peterson-Yantorno K, Civan MM (2002) Human trabecular meshwork cell volume regulation. Am J Physiol 283:C315–C326
Muallem S, Zhang BX, Loessberg PA, Star RA (1992) Simultaneous recording of cell volume changes and intracellular pH or Ca2+ concentration in single osteosarcoma cells UMR-106-01. J Biol Chem 267:17658–17664
Nagao E, Dvorak JA (1998) An integrated approach to the study of living cells by atomic force microscopy. J Microsc 191:8–19
Quist AP, Rhee SK, Lin H, Lal R (2000) Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J Cell Biol 148:1063–1074
Rae JL, Watsky MA (1996) Ionic channels in corneal endothelium. Am J Physiol 270:C975–C989
Rutledge EM, Aschner M, Kimelberg HK (1998) Pharmacological characterization of swelling-induced d-[3H]aspartate release from primary astrocyte cultures. Am J Physiol 274:C1511–C1520
Schneider SW, Yano Y, Sumpio BE, Jena BP, Geibel JP, Gekle M, Oberleithner H (1997) Rapid aldosterone-induced cell volume increase of endothelial cells measured by the atomic force microscope. Cell Biol Int 21:759–768
Schneider SW, Pagel P, Rotsch C, Danker T, Oberleithner H, Radmacher M, Schwab A (2000) Volume dynamics in migrating epithelial cells measured with atomic force microscopy. Pflugers Arch 439:297–303
Shiga N, Wangemann P (1995) Ion selectivity of volume regulatory mechanisms present during a hypoosmotic challenge in vestibular dark cells. Biochim Biophys Acta 1240:48–54
Sloot PM, Hoekstra AG, Figdor CG (1988) Osmotic response of lymphocytes measured by means of forward light scattering: theoretical considerations. Cytometry 9:636–641
Srinivas SP, Bonanno JA (1997) Measurement of changes in cell volume based on fluorescence quenching. Am J Physiol 272:C1405–C1414
Srinivas SP, Bonanno JA, Hughes BA (1998) Assessment of swelling-activated Cl− channels using the halide-sensitive fluorescent indicator 6-methoxy-N-(3-sulfopropyl)quinolinium. Biophys J 75:115–123
Srinivas SP, Guan Y, Bonanno JA (1999) Swelling activated chloride channels in cultured bovine corneal endothelial cells. Exp Eye Res 68:165–177
Strange K, Morrison R (1992) Volume regulation during recovery from chronic hypertonicity in brain glial cells. Am J Physiol 263:C412–C419
Su X, Pang T, Wakabayashi S, Shigekawa M (2003) Evidence for involvement of the putative first extracellular loop in differential volume sensitivity of the Na+/H+ exchangers NHE1 and NHE2. Biochemistry 42:1086–1094
Van Driessche W, De Smet P, Raskin G (1993) An automatic monitoring system for epithelial cell height. Pflugers Arch 425:164–171
Van Driessche W, De Smet P, Li J, Allen S, Zizi M, Mountian I (1997) Isovolumetric regulation in a distal nephron cell line (A6). Am J Physiol 272:C1890–C1898
Verkman AS (1995) Optical methods to measure membrane transport processes. J Membr Biol 148:99–110
Wangemann P, Marcus DC (1990) K+-induced swelling of vestibular dark cells is dependent on Na+ and Cl− and inhibited by piretanide. Pflugers Arch 416:262–269
Acknowledgements
Supported by NIH grants E11107 (SP) and EY00834 (JAB) and by grant "Fonds voor Wetenschappelijk Onderzoek Vlaanderen" G.0179.00 (WVD). The authors thank Prof. Bernard Himpens, M.D. and Priya Gomez, Laboratory of Physiology, K. U. Leuven, Campus Gasthuisberg O/N, Leuven (Belgium) for their help in obtaining confocal images shown in Fig. 6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Srinivas, S.P., Bonanno, J.A., Larivière, E. et al. Measurement of rapid changes in cell volume by forward light scattering. Pflugers Arch - Eur J Physiol 447, 97–108 (2003). https://doi.org/10.1007/s00424-003-1145-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-003-1145-5