
Abstract
Purpose
To assess the influence of a simulated altitude exposure (~2,900 m above sea level) for a 3 h recovery period following intense interval running on post-exercise inflammation, serum iron, ferritin, erythropoietin, and hepcidin response.
Methods
In a cross-over design, ten well-trained male endurance athletes completed two 8 × 3 min interval running sessions at 85 % of their maximal aerobic velocity on a motorized treadmill, before being randomly assigned to either a hypoxic (HYP: F IO2 ~0.1513) or a normoxic (NORM: F IO2 0.2093) 3 h recovery period. Venous blood was collected pre- and immediately post-exercise, and after 3 and 24 h of recovery. Blood was analyzed for interleukin-6, serum iron, ferritin, erythropoietin, and hepcidin.
Results
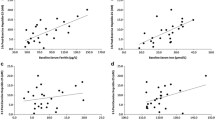
Interleukin-6 was significantly elevated (p < 0.01) immediately post-exercise compared to baseline (NORM: 1.08 ± 0.061 to 3.12 ± 1.80) (HYP: 1.32 ± 0.86 to 2.99 ± 2.02), but was not different between conditions. Hepcidin levels were significantly elevated (p < 0.01) at 3 h post-exercise for both conditions when compared to baseline (NORM: 3.25 ± 1.23 to 7.40 ± 4.00) (HYP: 3.24 ± 1.94 to 5.42 ± 3.20), but were significantly lower (p < 0.05) in the HYP trial compared to NORM. No significant differences existed between HYP and NORM for erythropoietin, serum iron, or ferritin.
Conclusion
Simulated altitude exposure (~2,900 m) for 3 h following intense interval running attenuates the peak hepcidin levels recorded at 3 h post-exercise. Consequently, a hypoxic recovery after exercise may be useful for athletes with compromised iron status to potentially increase acute dietary iron absorption.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adequate iron levels are essential for optimal cognitive and physical performance (Latunde-Dada 2013). However, exercise-induced iron deficiency is a common nutritional disorder diagnosed in athletes (Beard and Tobin 2000). Athletic-induced iron deficiency has been attributed to a number of mechanisms including sweating, gastrointestinal bleeding, hematuria, and hemolysis (Peeling et al. 2008). However, more recent research has focused on the influence of exercise-induced increases in inflammatory cytokines and the iron regulatory hormone known as hepcidin (Peeling et al. 2009a; Sim et al. 2013), in an attempt to further elucidate the mechanisms that underlie iron deficiency in athletes.
Hepcidin is a hepatocyte-derived peptide hormone encoded by the HAMP gene (Krause et al. 2000; Park et al. 2001) which acts as the key regulator of iron metabolism through its interaction with the body’s only known cellular iron exporter, ferroportin (Fpn). These Fpn channels are expressed on a number of different cell types throughout the body, including duodenal enterocytes, macrophages, and hepatocytes (Nemeth et al. 2004a, b). Hepcidin acts to internalize and degrade the Fpn export channels in the small intestine and on the surface of macrophages (Nemeth et al. 2004b; Nemeth and Ganz 2009), thereby inhibiting the absorption of dietary iron in the gut and preventing the release of iron from the macrophages that have collected senescent red blood cells (Nemeth et al. 2004a, b). Clearly, such processes are incongruous to an athlete with compromised iron stores. To date, post-exercise hepcidin regulation has been shown to be driven via the inflammatory response to exercise; and more specifically via the inflammatory cytokine interleukin-6 (IL-6) (Peeling et al. 2009a, b; Sim et al. 2013). Recently, it was reported that peak post-exercise hepcidin activity occurs ~3 h subsequent to the transient increase in IL-6 (Peeling et al. 2009a), which has been demonstrated in both urine (Peeling et al. 2009a, b) and blood (Sim et al. 2013). Factors such as carbohydrate intake and exercise duration and/or modality are proposed to alter the magnitude and association between these responses (Newlin et al. 2012; Robson-Ansley et al. 2011). However, some studies have reported minimal association between exercise-induced inflammatory responses and serum hepcidin levels post-exercise, with Auersperger et al. (2012) reporting no relationship between IL-6 and serum hepcidin concentration following 8 weeks of endurance training in female runners. A similar result was also observed in female soldiers with iron deficiency anemia, but not in those with normal iron stores following 9 weeks of basic combat training (Karl et al. 2010). Of relevance, Peeling et al. (2009a) also observed that hepcidin responses were less pronounced in female participants with ferritin concentrations < 35 μg/L. As a result, it was suggested that the lack of serum hepcidin changes in iron deplete or deficient athletes may be due to the homeostatic regulation of iron metabolism, where low hepatic iron concentrations potentially override inflammatory mediated changes in the hormone, resulting in a decreased hepcidin synthesis, which in turn increases iron absorption from dietary intake (Nicolas et al. 2002). However, it appears that athletes presenting with borderline iron stores may still have an elevated post-exercise hepcidin response, hence suggesting that this hormone is an important mediator in exercise-associated iron deficiency.
In contrast to inflammation, hypoxia is suggested to result in an attenuation of hepcidin activity (Nicolas et al. 2002). Hypoxic inducible factors (HIF-1α and HIF-2α) can be viewed as iron sensors and regulators, which under conditions of sufficient iron and oxygen are virtually undetectable; however, during conditions of iron deficiency and hypoxic exposure these HIF will accumulate (Piperno et al. 2009). Cell cultured animal studies (genetic studies on iron-deficient mice) have demonstrated that HIF activation in hepatocytes results in the suppression of hepcidin expression via the hypoxia response element (HRE)-dependent mechanism, thereby resulting in a reduction in hepcidin levels, which enables the mobilization of iron from sources required for erythropoiesis (Peyssonnaux et al. 2007). However, this model has recently been questioned, with the suggestion that HIF activation does not function as a direct transcriptional suppressor of hepcidin gene expression (Volke et al. 2009). Alternatively, recent research has indicated that erythropoiesis (Lui et al. 2012; Mastrogiannaki et al. 2012; Tanno et al. 2009), EPO itself (Robach et al. 2009), or iron signaling pathways (Babitt et al. 2006; Nemeth and Ganz 2009) may be responsible for triggering the suppression of the hepcidin gene expression. Regardless, the mechanisms seeking to explain the down-regulation of hepcidin under hypoxic stress appear increasingly complex and are currently subject to further investigations (Lui et al. 2012).
At moderate altitudes (2,200–3,500 m) the reduction of the partial pressure of O2 is sufficient to induce EPO production in the kidneys (Huang et al. 1998; Saunders et al. 2009). Continuous exposure at this altitude will see peak EPO levels attained after 24–48 h of exposure (Gore et al. 2001). Hepatic-derived HIF-2α (and not HIF-1α: Mastogiannaki et al. 2009), appears to be the main regulator for increases in erythropoietin (EPO) production (Haase 2010). Based on the findings of Huang et al. (1998), an acute hypoxic stimulus of 2 h may be long enough to extend the half-life of HIF-2α within the liver of mice, allowing for the accumulation of HIF-2α within oxygen-deprived tissue. As a result, it is likely that the stabilization of HIF-2α resulting from an acute hypoxic exposure may result in the up-regulation of erythropoiesis signaling and EPO itself (Knaupp et al. 1992) and a possible corresponding down-regulation of hepcidin levels in vivo. However, whether this occurs in the post-exercise period is yet to be established. Consequently, the aim of the current investigation was to assess the effect of simulated post-exercise altitude exposure for 3 h on the subsequent IL-6 and hepcidin levels in well-trained endurance athletes. It was hypothesized that a simulated post-exercise altitude exposure (~2,900 m above sea level) for 3 h following an intense interval running session (performed in normoxia) would attenuate post-exercise hepcidin levels.
Methods
Participants
Ten well-trained male endurance runners and/or triathletes with no recent exposure to hypoxic stress participated in this study [Age: 26.6 ± 10.7 years; Height: 173.4 ± 21.9 cm; Mass: 72.5 ± 10.5 kg; peak oxygen uptake (VO2peak): 63.3 ± 6.3 ml kg−1 min−1]. Determination of the sample size was attained via a power analysis using customized computer software (GPOWER Version 3.1, Department of Psychology, Bonn University, Bonn Germany) with data from previous investigations that measured similar variables (Peeling et al. 2009a, b). The power analysis suggested a sample size of 10, for an expected power of 0.89 with an alpha level of p ≤ 0.05. The study protocol was approved by the University of Western Australia’s Institutional Review Board, conforming to the Declaration of Helsinki on the use of human subjects. Written informed consent was obtained from participants prior to commencing the investigation.
Experimental overview
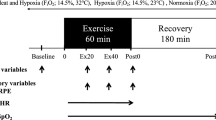
Participants completed three separate running sessions in the exercise physiology laboratory of the Western Australian Institute of Sport. These sessions were separated by a minimum of 7 days, with each conducted at the same time-of-day to minimize the influence of circadian variation. The initial testing session served as a familiarization trial for the participants to become accustomed to the laboratory environment and the equipment to be used during the experimental trials. This session concluded with a graded exercise test (GXT) to exhaustion, to determine each individual’s VO2peak. Subsequently, the velocity corresponding to 85 % of this VO2peak (85 % vVO2peak) was established to determine the pace at which the two subsequent running sessions were performed.
Interval running trials were conducted using a repeated-measure, counterbalanced, crossover design on a motorized treadmill (HP/Cosmos/Venus, Germany). In each session, participants completed a 5-min warm-up at 60 % vVO2peak, followed by a 5-min period of dynamic stretching. Subsequently, the interval session commenced consisting of 8 × 3 min repeats at 85 % vVO2peak with 90 s of active recovery (60 % vVO2peak) between repetitions (i.e., a 2:1 work:rest ratio). This running session was established as a common endurance runner’s training set that would be utilized in the field. In addition, this session has prior precedence from research investigating post-exercise hepcidin responses (Sim et al. 2013). Heart rate (HR) and ratings of perceived exertion (RPE) were recorded at the completion of each 3-min workload. After the final interval repetition, a capillary blood sample for lactate (BLa) analysis was taken, after which a 5-min cool-down run at 60 % vVO2peak was completed. Participants then rested for the next 3 h in the laboratory where they were either seated inside a simulated altitude tent [Colorado Altitude Training (CAT), CAT-430 Walk in Tent] set to ~2,900 m above sea level (F IO2 ~0.1513 ± 0.426; HYP) or in an adjacent room at sea level (FIO2 ~0.2093; NORM). After the 3 h recovery period, participants left the laboratory, but returned 24 h later for a follow-up assessment.
During the HYP and NORM trials, venous blood samples were collected on arrival (baseline), at the conclusion of the interval running (post-exercise) and after 3 and 24 h of post-exercise recovery. In addition, body mass was recorded pre- and post-exercise. Participants were also provided with 400 ml of water to drink ad libitum during each running session to minimize any hemoconcentration effects. During the 3 h post-exercise HYP exposure, the F IO2 was recorded each hour using a hand-held oxygen sensor (Handi®+, Maxtech, UT, USA), and blood oxygenation levels were monitored in the final 15 min of this recovery period by pulse oximetry (SpO2) of the index finger (Fingertip Pulse Oximeter, CMS-50DLP, China).
To ensure that a consistent nutritional intake was obtained before and after the conclusion of each trial, participants completed a 24 h food recall diary leading up to the first interval running session, and then replicated this for the second session. In addition, a standardized snack was provided during the 3 h post-exercise recovery period, which included 1 × 600 mL sports drink (Gatorade©, Schweppes Australia), 750 mL of plain water, 2 × 31.3 g muesli bars (Chewy Muesli bars, Uncle Tobys, Australia), and 1 × 250 mL milk drink (Up and Go, Sanitarium, Australia). The total nutritional intake by participants per session was ~2,523 kJ, comprising of 106 g of carbohydrates, 13 g of protein and 9 g of fat.
Experimental procedures
Graded exercise test (GXT)
The GXT was completed on a motorized treadmill at a constant gradient of 1 % (Jones and Doust 1996) using 3 min exercise and 1 min rest periods. The initial speed was 12 km h−1, with subsequent increments of 2 km h−1 until a speed of 16 km h−1 was reached. Subsequently, the speed increments were increased by 1 km h−1 until voluntary exhaustion. Throughout the GXT, expired air was continuously collected and analyzed for concentrations of O2 and CO2 via a commercial gas analysis system (TrueOne 2,400 Parvo Medics, Utah, USA). The gas analyzers were calibrated pre-test and verified post-test with certified gravimetric gas mixtures. During the test, ventilation was recorded at 15 s intervals via the Parvo Medics pneumotach, which was also calibrated before and verified after exercise using a 3 L syringe in accordance with the manufacturer’s specifications. The data obtained from the GXT was analyzed for VO2peak by summing the four highest consecutive 15 s VO2 values obtained throughout the test.
Blood collection and analysis
Capillary blood samples for BLa analysis were collected from the earlobe (initial drop discarded) into a Lactate Pro Analyzer (Arkray, Japan). In addition, venous blood samples were collected from an antecubital vein in the forearm with the athlete lying supine for 10 min prior to sampling in order to control for any postural-induced shifts in plasma volume. These samples were collected using a 21-gauge needle into two 8.5 mL SST Gel separator tubes, and one 4 ml EDTA collection tube (BD vacutainer, Australia). Immediately after collection, the fresh plasma samples from the EDTA tubes were taken to the Royal Perth Hospital pathology laboratory to be analyzed for hemoglobin concentration and hematocrit. This data was used for the determination of percentage changes in plasma volume as a result of the exercise task (Dill and Costill 1974). The serum samples were allowed to clot for 60 min at room temperature, before subsequently being centrifuged at 10 °C and 3,000 rpm for 10 min. The serum supernatant was divided into 1 mL aliquots and stored at −80 °C until further analysis. Once all samples were collected, the frozen serum was also taken to the Royal Perth Hospital pathology laboratory to be analyzed for circulating levels of IL-6, EPO, serum iron, ferritin, and transferrin. In addition, the remaining frozen serum samples were sent to Radboud University Medical Centre (Nijmegen, The Netherlands) for serum hepcidin-25 analysis.
Serum IL-6 was analyzed using a commercially available ELISA (Quantikine HS, R&D Systems, Minneapolis, USA) with an assay range of 0.38–10 ng L−1. The coefficient of variation (CV) for IL-6 determination at 0.49 and 2.78 ng L−1 was 9.6 and 7.2 %, respectively. Serum iron was measured using the Architect (c1600210) analyzer. Serum iron levels were determined using an iron reagent (Sentinel Diagnostics, Milano, Italy). The CV for iron determination at 12.01 and 43.35 μmol L−1 was 1.7 and 0.6 %, respectively. Serum ferritin levels were determined using an Architect analyzer (1SR06055) and a Ferritin Reagent (Abbott Diagnostics, Illinois, USA). The CV for ferritin determination at 28.62, 223.05, and 497.85 μg L−1 was 4.6, 4.5, and 4.4 %, respectively. Serum EPO was analyzed using a solid phase two-site sequential chemiluminescent enzyme-labeled immunometric assay [Immunlite 2,000 XPi EPO kit (Siemens Medical Solutions Diagnostic, 5210 Pacific Concourse Drive, Los Angeles, CA 90045-6900-USA)], via a Immunite 2,000 XPi Analyzer. The CV for EPO determination at 18.2 mU L−1, 79.5 mU L−1, and 161 mU L−1 was 9.8, 8.7, and 9.6 %, respectively. Serum hepcidin-25 measurements were performed (http://www.hepcidinanalysis.com, Nijmegen, The Netherlands) by a combination of weak cation exchange chromatography and time-of-flight mass spectrometry (WCX-TOF MS) (Kroot et al. 2010). An internal standard (synthetic hepcidin-24; Peptide International Inc.) was used for quantification (Swinkels et al. 2008). Peptide spectra were generated on a Microflex LT matrix-enhanced laser desorption/ionization TOF MS platform (Bruker Daltonics). Serum hepcidin-25 concentrations were expressed as nmol L−1. The lower limit of detection of this method was 0.5 nM; average coefficients of variation were 2.7 % (intra-run) and 6.5 % (inter-run) (Kroot et al. 2010). The median reference level of serum hepcidin-25 is 4.5 nM for men, with a range of 0.5–14.7 nM (Galesloot et al. 2011; http://www.hepcidinanalysis.com Accessed on 2 June 2013).
Measurement of heart rate and perceived exertion
Throughout the experimental trials, HR was measured continuously via a Polar HR monitor (Polar 625X, Finland), and RPE recorded using the Borg perceptual scale (Borg 1982) encompassing the anchor points 6 (very, very light) through 20 (maximal exertion).
Statistical analysis
Results are presented as mean and standard deviation (± SD) unless otherwise stated. Results were analyzed in the IBM Statistical Package for Social Sciences (IBM SPSS version 19.0). A repeated-measure ANOVA was used to analyze the time, trial and time × trial effects of HYP vs. NORM exposures on the subsequent inflammatory, hepcidin, EPO, and iron-related responses. Post-hoc, least significant differences paired samples t tests were used to determine where specific trial differences existed. The alpha level was accepted at p ≤ 0.05. Where appropriate, Cohen’s d effect sizes were calculated to suggest data trends and were interpreted as d < 0.39 (small); d = 0.40–0.69 (moderate); d ≥ 0.7 (large). Only moderate and large effect sizes are presented below.
Results
Physiological responses during running trials and 3 h recovery period post-exercise
The overall mean values of HR, RPE, and BLa recorded during the interval running trials are presented in Table 1. The percentage changes in plasma volume recorded immediately post-exercise were not statistically different between the HYP and NORM conditions (Table 1).
Interleukin-6, EPO, and hepcidin
There were no trial effects for differences in levels of IL-6 between the two conditions (p = 0.86) (Table 2). However, a significant time effect (p = 0.01) showed that IL-6 levels were elevated immediately post-run in both conditions as compared to baseline (HYP: p = 0.01; NORM p = 0.01). The pre-exercise and 3 h post-exercise serum EPO data showed no significant time (p = 0.42) or trial (p = 0.17) effects (Table 2). In addition, no significant time effect was seen for the relative change (p = 0.19) of serum EPO from pre-exercise to 3 h post-exercise between conditions (HYP: 3.4 ± 7.3 mU L−1; NORM 0.94 ± 3.34 mU L−1). Significant time (p = 0.01) and interaction effects (p = 0.049) were recorded between the HYP and NORM hepcidin levels (Table 2). Specifically, the time effect showed hepcidin was elevated in both conditions from baseline to 3 h post-exercise (HYP and NORM, p = 0.01). However, in both conditions at 24 h post-exercise, the levels had returned to baseline (HYP p = 0.46, NORM p = 0.23). No significant differences were evident between HYP and NORM trials at baseline (p = 0.49) or at 24 h post-exercise (p = 0.20); however, a moderate effect (d = 0.45) suggested a trend for lower 24 h post-exercise hepcidin levels in the HYP trial. In addition, the hepcidin levels recorded at 3 h post-exercise were significantly lower in HYP compared to NORM (p = 0.047). The change in hepcidin levels over time (from baseline) was also analyzed, showing a significant difference between the two conditions at 3 h post-exercise (HYP = 2.18 nM; NORM = 4.15 nM, p = 0.02).
Serum iron and serum ferritin
No significant trial (p = 0.14) or time (p = 0.07) effects were recorded for serum iron between the HYP and NORM trials (Table 2). In addition, there was no trial (p = 0.50) effect for differences in serum ferritin levels between the two conditions; however, a significant time effect (p = 0.04) indicated that these levels were elevated immediately post-run in both trials, as compared to baseline (HYP: p = 0.01; NORM: p = 0.02).
Discussion
Previously, it has been suggested that hypoxia may attenuate the activity of the iron regulatory hormone hepcidin; however, a number of suggested in vivo pathways for such an outcome have been proposed, and are still subject to debate (Lui et al. 2012). As a result, this study aimed to determine whether acute hypoxic exposure in the post-exercise recovery period would attenuate the subsequent increases commonly seen in the hepcidin response. Our data demonstrated that hepcidin levels were elevated in both of the tested conditions at the 3 h post-exercise time point; however, these levels were significantly lower in the HYP trial when compared to NORM. This outcome was further supported by a smaller relative change to hepcidin levels in the HYP condition.
The inflammatory response for the current study demonstrated a significant increase in IL-6 immediately post-exercise as compared with baseline; however, no differences were found in the post-exercise responses between the two trials. The IL-6 increases seen here support previous research (Peeling et al. 2009b), where strenuous interval running (10 × 1 km at 90–95 % of VO2max) resulted in a ~five-fold increase in IL-6 levels recorded at the completion of exercise. The IL-6 response in the current study is therefore characteristic of the typical post-exercise acute-phase inflammatory response, demonstrating an exponential rise in IL-6 during exercise, with peak levels obtained at the immediate completion of the work bout (Peeling et al. 2009b). In addition, previous research has also shown that inflammatory cytokines are able to affect ferritin mRNA translation, resulting in an increased ferritin synthesis (Kalantar-Zadeh et al. 2004). Such observations likely explain the increased serum ferritin levels seen within the current study immediately post-exercise compared to baseline, 3 and 24 h post-exercise. As previously suggested, IL-6 has been shown to be one of the primary regulators of hepcidin activity within the body (Nemeth et al. 2004b). However, since the exercise intensity and IL-6 production in both trials were similar here, the subsequent comparison of the hepcidin responses to the intervention is novel and exciting.
The hepcidin levels in the current study were significantly elevated at the 3 h post-exercise time point for both the HYP and NORM trials. This finding is consistent with previous clinical studies demonstrating an increased hepcidin activity 3 h subsequent to the peak activity of IL-6 induced by lipopolysaccharide injection (Kemna et al. 2005). In addition, Peeling et al. (2009a) demonstrated increases in hepcidin levels 3 h post-exercise following a ~seven-fold increase in IL-6 immediately post-exercise, as compared to both baseline and resting control group values. Therefore, it is likely that the IL-6 increases caused by the strain of the interval running session implemented here (~45 min at 85 % vVO2peak) may be the primary mechanism for the increase in hepcidin levels in the 3 h post-exercise period. In accordance with previous research (Peeling et al. 2009a), hepcidin levels at 24 h post-exercise had returned to baseline levels for both conditions.
Regardless of the inflammatory response, the novel data from this investigation is that the hepcidin levels at 3 h post-exercise were significantly lower for HYP compared to NORM. This finding matches that of Nicolas et al. (2002) who first reported that hepcidin expression was reduced during hypoxic exposure. To date, the mechanism explaining hepcidin down-regulation in response to a hypoxic stimulus is not completely established; recent animal studies indicate that the decrease in hepcidin expression following induced anemia was largely dependent on erythropoiesis, and not directly regulated by EPO, anemia, or tissue hypoxia (Pak et al. 2006; Vokurka et al. 2006). However, research in humans is yet to highlight the erythroid protein responsible for the attenuation of hepcidin expression, although recent results have demonstrated a gradual suppression of hepcidin under hypoxic stress, indicating a possible mediation by circulating factors released during erythropoietic activity (Piperno et al. 2011). Regardless, current theories for reduced hepcidin activity under hypoxic stress include the influence of HIF transcription factors. Research that has used a genetic approach to separate the HIF activation under hypoxic stress from EPO synthesis found that HIF-mediated suppression of the hepcidin gene requires erythropoiesis (Lui et al. 2012). When erythropoiesis was pharmacologically inhibited, the hepcidin gene expression was no longer suppressed despite large increases in serum EPO. This final result is similar to that of an associated study (Mastrogiannaki et al. 2012), which found that by ceasing erythropoiesis, hepcidin gene mRNA levels were increased within cell cultured animals. Therefore, it is proposed that under hypoxic stress, hepcidin gene suppression will occur via the stimulation of erythropoiesis as a result of indirect signaling of HIFs, resulting in a reduction of hepcidin levels within the system (Lui et al. 2012; Mastrogiannaki et al. 2012). In addition, GDF-15 has also been identified as a possible erythropoietic regulator of hepcidin activity (Tanno et al. 2007), since it has been proposed to increase during erythropoiesis and may promote the secretion of this erythroblast-derived factor, which has also been associated with the suppression of the hepcidin gene (Tanno et al. 2007). The current study supports the results of this previous research, since no changes were seen in serum EPO itself; however, a reduction in serum hepcidin was recorded under the HYP trial. Potentially, this may indicate that the acute hypoxic stimulus of the HYP condition (3 h at ~2,900 m; F IO2 ~0.1513) was sufficient enough to enable the stabilization of HIF-2α or the accumulation of erythroid proteins that may be released from the stimulus for increased erythropoietic activity, which may have resulted in the suppression of hepcidin. However, the lack of erythroid protein and transcriptional factor measurement remain a limitation of this study, and future research should look to acquire tissue samples for the measurement of HIF-2α and GDF-15 in order to draw definitive conclusions here.
Notwithstanding, it is widely acknowledged that living and training under hypoxic stress may lead to improved athletic performances at sea level (Gore et al. 2001). However, only recently has research focused on the effect that living and training at altitude may have on hepcidin regulation and the subsequent iron metabolism response. At high altitude, the partial pressure of oxygen is reduced, which activates erythropoietic processes resulting in an increased iron demand (Robach et al. 2007). Robach et al. (2009) showed that healthy volunteers treated with recombinant erythropoietin for 1 month efficiently increased erythropoiesis and stimulated bone marrow iron use, which was associated with a prompt and considerable decrease in urinary hepcidin. In addition, research on chronic hypoxic exposure has demonstrated a significant decrease in hepcidin expression, accompanied by a subtle systemic inflammatory response (increase in IL-6) (Goetze et al. 2013; Piperno et al. 2011). Despite these observed increases in IL-6, the elevation of this prominent up-regulator of hepcidin activity did not appear to counteract hepcidin suppression during chronic altitude exposure (Goetze et al. 2013; Piperno et al. 2011). This further suggests that hypoxia-induced erythropoiesis may be an overriding negative regulatory pathway of hepcidin activity as compared to inflammatory driven increases (Robach et al. 2009). During acute hypoxic exposure (as demonstrated with the current study), significantly lower levels of hepcidin would likely mean a more rapid return to baseline levels of this hormone in comparison to the higher levels seen in the NORM trial. Increases in hepcidin levels degrade and internalize the Fpn export channels on the cell surfaces of duodenal enterocytes and macrophages (Nemeth et al. 2004b). During periods of elevated hepcidin activity, the body may be susceptible to reductions in iron absorption and recycling, which over time, is likely to compromise the individuals’ ability to replenish adequate iron for optimal athletic performance. Hence, reduced hepcidin levels in the HYP trial may also attenuate the degradation of Fpn channels throughout the body, potentially reducing the impact on dietary iron absorption in the post-exercise period. For athletes prone to exercise-induced anemia, the strategy of acute hypoxic environment exposure may possibly enable greater iron absorption from meals consumed in the early post-exercise period, providing a novel method for potentially improving iron metabolism in the athletic population. In addition, elite athletes traveling to altitude for a prolonged period of time should continue to be encouraged to use iron supplements during their trip, in order to ensure a maximum absorption of iron while hepcidin levels are reduced, likely enhancing the erythropoietic process. Regardless, further research utilizing similar strategies is required to elucidate the potential benefits suggested here.
Conclusion
The recovery from high-intensity exercise efforts under a hypoxic stimulus of ~2,900 m simulated altitude (F IO2 ~0.1513) for 3 h was effective in attenuating the 3 h post-exercise hepcidin response, possibly via the HIF/HRE pathway of erythropoiesis stimulation and hepcidin gene attenuation. The reduced hepcidin response seen after the hypoxic stimulus might increase an athlete’s ability to absorb iron from any meals consumed subsequent to the training stimulus as a result of a likely reduction to the disruption of the Fpn iron exporters in the gut. Nevertheless, post-exercise hypoxic exposure in the recovery period after training in athletes with compromised iron stores should be further explored as a potential strategy to minimize the impact of elevated hepcidin levels on disruptions to iron absorption.
Abbreviations
- BLa:
-
Blood lactate
- Bpm:
-
Beats per minute
- CO2 :
-
Carbon dioxide
- CV:
-
Coefficient of variation
- EPO:
-
Erythropoietin
- F io2 :
-
Fraction of inspired oxygen
- Fpn:
-
Ferroportin
- GDF-15:
-
Growth differentiation factor-15
- GXT:
-
Graded exercise test
- HAMP gene:
-
Hepcidin encoding gene
- Hb:
-
Hemoglobin
- Hct:
-
Hematocrit
- HIF:
-
Hypoxic inducible transcription factors
- HIF/HRE pathway:
-
Hypoxic inducible transcription factor/Hypoxic response element pathway
- Hp:
-
Haptoglobin
- Hp-Hb:
-
Haptoglobin–Hemoglobin complex
- HR:
-
Heart rate
- HYP:
-
Hypoxic/simulated altitude trial
- IL-6:
-
Interleukin-6
- LSD:
-
Least significant difference
- mRNA:
-
Messenger RNA
- NORM:
-
Control condition
- O2 :
-
Oxygen
- RBC:
-
Red blood cells (erythrocytes)
- RPE:
-
Rating of perceived exertion
- VO2 peak :
-
Peak oxygen consumption
References
Auersperger I, Knap B, Jerin A, Blagus R, Lainscak M, Skitek M, Skof B (2012) The effects of 8 weeks of endurance running on hepcidin concentrations, inflammatory parameters and iron status in female runners. Int J Sports Nutr Exerc Metab 22:55–63
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolfe CJ, Andrews NC, Lin HY (2006) Bone morphogenetic protein signalling by hemojuvelin regulates hepcidin expression. Nat Genet 38:531–539
Beard J, Tobin B (2000) Iron status and exercise. Am J Clin Nutr 72:5945–5975
Borg GAV (1982) Psychophysical bases of perceived exertion. Med Sci Sports Exerc 14:377–381
Dill DB, Costill DL (1974) Calculation of percentage changes in volume of blood, plasma, and red cells in dehydration. J Appl Physiol 37:247–248
Galesloot TE, Vermeulen SH, Geurts-Moespot AJ, Klaver SM, Kroot JJ, van Tienoven D, Wetzels JF, Kiemeney LA, Sweep FC, den Heijer M, Swinkels DW (2011) Serum hepcidin: reference ranges and biochemical correlates in the general population. Blood 117:e218–e225
Goetze O, Schmitt J, Spliethoff K, Theurl I, Weiss G, Swinkels DW, Tjalsma H, Maggiorini M, Krayenbuhl P, Rau M, Fruehauf H, Wojtal KA, Mullhaupt B, Fried M, Gassmann M, Lutz T, Geier A (2013) Adaptation of iron transport and metabolism to acute high-altitude hypoxia in mountaineers. Hepatology 58:2153–2162
Gore CJ, Hahn AG, Aughey J, Martin DT, Ashenden MJ, Clark SA, Garnham AP, Roberts AD, Slater GJ, McKenna MJ (2001) Live high: train low increases muscle buffer capacity and submaximal cycling efficiency. Acta Physiol Scand 173:275–286
Haase VH (2010) Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol 299:F1–F13
Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of hypoxia-inducible factor 1-alpha is mediated by the oxygen-dependent degradation domain via the ubiquitin-proteasome pathway. Pro Natl Acad Sci USA 95:7987–7992
Jones AM, Doust JH (1996) A 1 % treadmill grade most accurately reflects the energetic cost of outdoor running. J Sports Sci 14:321–327
Kalantar-Zadeh K, Rodriguez RA, Humphreys MH (2004) Association between serum ferritin and measures of inflammation, nutrition, and iron in haemodialysis patients. Nephrol Dial Transplant 19:141–149
Karl JP, Lieberman HR, Cable SJ, Williams KW, Young AJ, McClung JP (2010) Randomized double blind, placebo-controlled trial of an iron-fortified food product in female soldiers during military training relations between iron status, serum hepcidin, and inflammation. Am J Clin Nutr 92:93–100
Kemna E, Pickkers P, Nemeth E, van der Hoeven H, Swinkels DW (2005) Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 106:1864–1866
Knaupp S, Khilnani J, Sherwood S, Scharf S, Steinberg H (1992) Erythropoietin response to acute normobaric hypoxia in humans. J Appl Physiol 73:837–840
Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K (2000) LEAP-1, a novel highly disulfide-bonded human peptide exhibits antimicrobial activity. FEBS Lett 480:147–150
Kroot JCC, Laarakkers CM, Geurts-Moespot A, Grebenchtchikov N, Pickkers P, van Ede A, Peters HP, van Dongen-Lases E, Wetzels JFM, Sweep FC, Tjalsma H, Swinkels DW (2010) Immunochemical and mass spectrometry-based serum hepcidin assays for a variety of iron metabolism disorders. Clin Chem 56:1570–1579
Latunde-Dada GO (2013) Iron metabolism in athletes-achieving a gold standard. Euro J Haematol 90:10–15
Lui Q, Davidoff O, Niss K, Haase VH (2012) Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 122:4635–4644
Mastogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C (2009) HIF-2α, but not HIF-1α, promotes iron absorption in mice. J Clin Invest 119:1159–1166
Mastrogiannaki M, Matak P, Mathieu JRR, Delga S, Mayeux P, Vaulont S, Peyssonnaux C (2012) Hepatic hypoxia-inducible factor-2 down regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 97:827–834
Nemeth E, Ganz T (2009) The role of hepcidin in iron metabolism. Acta Haematol 122:78–86
Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T (2004a) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 113:1271–1276
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, McVey-Ward D, Ganz T, Kaplan J (2004b) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2051–2053
Newlin MK, Williams S, McNamara T, Tjalsma H, Swinkels DW, Haymes EM (2012) The effects of acute exercise bouts on hepcidin in women. Int J Sport Nutr Exerc Metab 22:79–88
Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anaemia, hypoxia, and inflammation. J Clin Invest 110:1037–1044
Pak M, Lopez MA, Gabayan V, Gantz T, Rivera S (2006) Suppression of hepcidin during anaemia requires erythropoietic activity. Blood 108:3730–3735
Park CH, Valore EV, Waring AJ, Ganz T (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 276:7806–7810
Peeling P, Dawson B, Goodman C, Landers G, Trinder D (2008) Athletic induced iron deficiency: new insights into the role of inflammation, cytokines and hormones. Eur J Appl Physiol 103:381–391
Peeling P, Dawson B, Goodman C, Landers G, Wiegerink ET, Swinkels DW, Trinder D (2009a) Effects of exercise on hepcidin response and iron metabolism during recovery. Int J Sports Nutr Exerc Metab 19:583–597
Peeling P, Dawson B, Goodman C, Landers G, Wiegerinck ET, Swinkels DW, Trinder D (2009b) Training surface and intensity: inflammation, hemolysis, and hepcidin expression. Med Sci Sports Exerc 41:1138–1145
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS (2007) Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 117:1926–1932
Piperno A, Mariani R, Trombini P, Girelli D (2009) Hepcidin modulation in human diseases: from research to clinic. World J Gastroenterol 15:538–551
Piperno A, Galimberti S, Mariani R, Pelucchi S, Ravasi G, Lombardi C, Bilo G, Revera M, Giuliano A, Faini A, Mainini V, Westerman M, Ganz T, Valsecchi MG, Mancia G, Parati G, HIGHCARE investigators (2011) Modulation of hepcidin production during hypoxia induced erythropoiesis in humans in vivo: data from the HIGHCARE project. Blood 117:2953–2959
Robach P, Cairo G, Gelfi C, Bernuzzi F, Pilegaard H, Viganò A, Santambrogio P, Cerretelli P, Calbet JA, Moutereau S, Lundby C (2007) Strong iron demand during hypoxia-induced erythropoiesis is associated with down-regulation of iron-related proteins and myoglobin in human skeletal muscle. Blood 109:4724–4731
Robach P, Recalcati S, Girelli D, Gelfi C, Aachmann-Andersen NJ, Thomsen JJ, Norgaard AM, Alberghini A, Campostrini N, Castagna A, Vigano A, Santambrogio P, Kempf T, Wollert KC, Moutereau S, Lundby C, Cairo G (2009) Alterations of systemic and muscle iron metabolism in human subjects treated with low dose recombinant erythropoietin. Blood 113:6707–6715
Robson-Ansley P, Walshe I, Ward D (2011) The effect of carbohydrate ingestion on plasma interleukin-6, hepcidin, and iron concentrations following prolonged exercise. Cytokine 53:196–200
Saunders PU, Pyne DB, Gore CJ (2009) Endurance training at altitude. High Alt Med Biol 10:135–148
Sim M, Dawson B, Landers G, Swinkels DW, Tjalsma H, Trinder D, Peeling P (2013) Effect of exercise modality and intensity on post-exercise interleukin-6 and hepcidin levels. Int J Sport Nutr Exerc Metab 23:178–186
Swinkels DW, Girelli D, Laarakkers C, Kroot J, Campostrini N, Kemna EH, Tjalsma H (2008) Advances in quantitative hepcidin measurements by time-of-flight mass spectrometry. PLoS ONE. doi:10.1371/journal.pone.0002706
Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL (2007) High levels of GDF-15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 13:1097–1101
Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, Bhupatiraju A, Lee YT, Goodnough JB, Harandi O, Ganz T, Paulson RF, Miller JL (2009) Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 114:181–186
Vokurka M, Krijt J, Sulc K, Necas E (2006) Hepcidin mRNA levels in mouse liver responds to inhibition of erythropoiesis. Physiol Res 55:667–674
Volke M, Gale DP, Maegdefrau U, Schley G, Klanke B, Bosserhoff AK, Maxwell PH, Eckardt KU, Warnecke C (2009) Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS ONE. doi:10.1371/journal.pone.0007875
Acknowledgments
The authors wish to acknowledge the high performance sports research grant funding received from the Australian Institute of Sport and the Australian Sports Commission.
Conflict of interests
The authors report no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Carsten Lundby.
Rights and permissions
About this article
Cite this article
Badenhorst, C.E., Dawson, B., Goodman, C. et al. Influence of post-exercise hypoxic exposure on hepcidin response in athletes. Eur J Appl Physiol 114, 951–959 (2014). https://doi.org/10.1007/s00421-014-2829-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-014-2829-6