
Abstract
Arginase-II (Arg-II) reciprocally regulates nitric oxide synthase (NOS) and offsets basal myocardial contractility. Furthermore, decreased or absent myocardial NOS activity is associated with a depression in myocardial contractile reserve. We therefore hypothesized that upregulation of Arg-II might in part be responsible for depressed myocardial contractility associated with age. We studied arginase activity/expression, NOS expression, NO production in the presence and absence of the arginase inhibitor S-(2-boronoethyl)-l-cysteine (BEC) in old (22 months) and young (3 months) rat hearts and myocytes. The spatial confinement of Arg-II and NOS was determined with immuno-electron-miocrographic (IEM) and immuno-histochemical studies. We tested the effect of BEC on the force frequency response (FFR) in myocytes, as well as NOS abundance and activity. Arginase activity and Arg-II expression was increased in old hearts (2.27 ± 0.542 vs. 0.439 ± 0.058 nmol urea/mg protein, p = 0.02). This was associated with a decrease in NO production, which was restored with BEC (4.54 ± 0.582 vs. 12.88 ± 0.432 μmol/mg, p < 0.01). IEM illustrates increased mitochondrial density in old myocytes (51.7 ± 1.8 vs. 69 ± 2.2 × 106/cm2, p < 0.01), potentially contributing to increased Arg-II abundance and activity. Immunohistochemistry revealed an organized pattern of mitochondria and Arg-II that appears disrupted in old myocytes. The FFR was significantly depressed in old myocytes (61.42 ± 16.04 vs. −5.15 ± 5.65%), while inhibition of Arg-II restored the FFR (−5.15 ± 5.65 vs. 70.98 ± 6.10%). NOS-2 is upregulated sixfold in old hearts contributing to increased production of reactive oxygen species which is attenuated with NOS-2 inhibition by 1400 W (4,735 ± 427 vs. 4,014 ± 314 RFU/min/mg protein, p = 0.005). Arg-II upregulation in aging rat hearts contributes to age-related decreased contractile function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The aging cardiovascular system is characterized by molecular and cellular alterations that lead to clinically relevant cardiac pathophysiologies, including increased myocardial and vascular stiffness, heart valve calcification, coronary atherosclerosis, increased blood pressure, and decreased cardiac contractility (Lee and Oh 2010). Over time, these individual changes culminate in the onset of overall diminished cardiac function, and lead to heart failure (Wessells and Bodmer 2006).
One of the key molecular players in cardiac contractility is nitric oxide (NO). NO can either positively or negatively modulate cardiac contractility depending on its spatial confinement in the cell (Barouch et al. 2002). NO activates soluble guanylyl cyclase and increases intracellular cyclic guanosine monophosphate (cGMP) levels. Evidence suggests that an NO-mediated increase in cGMP contributes to decreased β-adrenergic responsiveness, a key mechanism underlying diminished cardiac function in heart failure (Balligand 1999; Hare et al. 1998). However, NO also modulates a spectrum of proteins via cGMP-independent mechanisms (Stamler et al. 2001). NO-mediated protein modification is thought to involve S-nitrosylation, the coupling of a NO moiety to a reactive cysteine thiol residue on the target protein (Hess et al. 2005). Finally NO mediates the maintenance of the redox balance (Hare 2004). More specifically, NO alters cardiac contractility by modulating the various calcium channels that are integral to excitation–contraction coupling (Khan and Hare 2003). The influence of NO on cardiac contractility depends on the specific isoform of nitric oxide synthase (NOS), its location or domain within the myocyte and the co-localized proteins modulated by NO (Xu et al. 1999). Specifically, NO produced in cardiac myocytes in proximity to the L-type calcium channels activates the cGMP-dependent pathways and inhibits cardiac contractility (Steppan et al. 2006a). Conversely, NO-mediated S-nitrosylation of the ryanodine receptor (RyR) induces activation and therefore increases cardiac contractility (Khan et al. 2003). Furthermore, NO influences cardiac contractility through its role in maintaining superoxide balance in cells. For example, xanthine oxidoreductase (XOR) an enzyme located in close proximity to NOS on the sarcoplasmatic reticulum (SR) produces superoxide anions by purine metabolism (Khan et al. 2004). When superoxide diffuses out of the SR it decreases calcium sensitivity of the contractile machinery, thereby diminishing contractility. NO directly reacts with the superoxide anion and inhibits its release from the SR, thereby reducing superoxide inhibition of cardiac contractility (Khan et al. 2004). Overall, NO modulates cardiac contractility directly by modulating calcium channels, which are important in excitation–contraction coupling, and indirectly by maintaining superoxide balance within the cardiomyocytes.
The concept of spatial confinement of NOS isoforms within the myocyte is critical to understanding the role of NO in the heart (Hare 2003; Khan et al. 2003). NOS-1 localized with the RyR in the SR of myocytes (Jung et al. 2006) is particularly important for cardiac contractility. The substrate for NOS is the basic amino acid l-arginine. Recent evidence suggests that the enzyme arginase (Arg) competes with NOS for l-arginine (Khan and Hare 2003; Steppan et al. 2006b). Substrate (l-arginine) bioavailability is an important determinant of NOS activity. Therefore, an increase in Arg expression in the cell could result in a concurrent decrease in NO production by NOS via competition for a common substrate.
We therefore hypothesized that an increase in Arg activity in aging cardiomyocytes contributes to diminished contractility by decreasing the activity of NOS through substrate depletion.
Materials and methods
Isolation of myocytes
Myocytes were isolated from 3- and 22-month-old Wistar rats. All protocols were conformed to the National Institutes of Health and American Physiological Society Guidelines for the Use and Care of Laboratory Animals. Following perfusion of the rat heart with digestion solution, the atria were removed from the ventricle and thus only ventricular tissue is represented in the myocytes, as described previously (Khan et al. 2003).
Sarcomere shortening
As described in detail by Khan et al. (2003), the isolated myocytes were transferred to a Lucite chamber on the stage of an inverted microscope (Nikon TE 200). The myocytes were continuously perfused in Tyrode solution containing 1.0 mM Ca2+. Next, they were stimulated at 1, 2 and 4 Hz and the resulting sarcomere length (SL) was measured using IonOptix software and iCCD camera. Fast Fourier transform of the Z-line density trace was used to determine the change in average SL and the SL shortening in the presence and absence of BEC (S-(2-boronoethyl)-l-cysteine, 10 μmol/l). Sarcomere shortening (SS) at 1 Hz was taken to be the baseline contractile length.
Western blot
Heart tissue homogenates were added to SDS loading buffer and the samples were loaded onto TrisHCl gels (Biorad) at 200 V for 1 h. The proteins were then transferred to a nitrocellulose membrane and blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS). After washing with TBS-T, the blots were incubated with primary antibodies, including rabbit antibodies against Arg-II (1:1,000, Affinity Bioreagents Inc.). After three washes with TBS-T buffer, the membranes were incubated for 1 h with peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG antibodies (Transduction Laboratories) in a 1:5,000 dilution (Khan et al. 2003). Finally, the resulting bands were visualized using chemiluminescence (Pierce).
Arginase activity assay
Rat hearts or myocytes were homogenized, the suspension was subsequently centrifuged and the supernatant used for the assay. Arg activity was determined from urea production using a method adapted from Zhang et al. (2001). The urea concentration was determined spectrophotometrically by the absorbance at 550 nm measured with a microplate reader. The amount of urea production, after normalization with the protein quantity in the samples, was used as an index for Arg activity (Zhang et al. 2001).
NOS activity, NO and ROS production
Nitrite and nitrate levels were measured to determine NO production (Calbiochem). The assay was performed in PBS (pH 7.4), with and without pre-incubation of the heart homogenates in BEC (10 μmol/l). Levels of ROS (reactive oxygen species) were measured via lucigenin chemiluminescence (Pierce) in old and young rat hearts in the presence and absence of the specific NOS-2 inhibitor 1400 W (N-[[3-(aminomethyl)phenyl]methyl]-ethanimidamide, dihydrochloride) (Sigma).
Histology
Samples of old and young rat myocardium were fixed in 4% paraformaldehyde overnight and re-hydrated in 30% sucrose. The tissue samples were then embedded in OCT, snap frozen and stored at −80º. After blocking the tissue sections with 10% normal goat serum, the primary antibody was applied overnight. After washing the secondary antibody was applied 1:500 for 1 h at room temperature. The slides were viewed on a Zeiss Axiovert 200 confocal microscope with a 510-Meta confocal laser scanning. NOS was visualized with FITC, while caeolin-3 was detected with Texas Red.
Immuno-electron microscopy
Immuno-electron microscopy (IEM) was performed by standard procedures. Rat hearts were perfused with 4% paraformaldehyde, 0.05% glutaraldehyde in PBS and postfixed overnight. 100-μm-thick vibratome sections were cut and collected in PBS, followed by incubation with the primary Ab (rabbit anti-Arg-II; 1:50 dilution) for 24 h at 4°C. After washing, the secondary Ab labeled with 6-nm gold particles was applied. The slides were washed again and the primary NOS antibody was added. For visualization, secondary 12-nm gold antibody particles were used. The tissue sections were examined with a transmission electron microscope (Hitachi 7600TEM) and digital images were acquired. Mitochondria were counted in randomly chosen sections at 20,000× magnification.
Data analysis and statistics
All data are presented as mean ± SEM, with n indicated for each experimental protocol. For dose responses, data were fitted by using the software program PRISM 4 (Graphpad, San Diego). Statistical analysis was performed by using an unpaired Student’s t test or one-way ANOVA with posttest, as appropriate.
Results
Arginase expression and activity in aging cardiac tissue
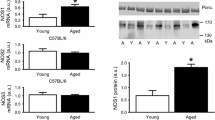
Arg activity is significantly increased in old myocytes, compared to young (Fig. 1a; 2.27 ± 0.542 vs. 0.439 ± 0.058 nmol urea/mg protein, old vs. young, p = 0.02, n = 6 per group). A similar trend is seen for Arg activity in whole heart homogenates from young and old rats (Fig. 1b; 5.1 ± 0.67 vs. 2.0 ± 0.13 pmol urea/mg protein, old vs. young, p = 0.01, n = 3 per group). Figure 1c indicates that Arg-II is the predominant isoform expressed in the rodent heart and there is an increase in Arg-II expression in old rats when compared to young.

a Arg activity is Increased in isolated myocytes from old rats versus young (n = 6 per group, *p < 0.05). b Shows a similar trend in homogenates of whole hearts, i.e., increased Arg activity in old heart homogenates when compared to young (n = 3 per group, *p < 0.05). c Immunoblotting, demonstrating the expression of Arg-II but not Arg-I in young and old heart homogenates. Furthermore, Arg-II is upregulated in old hearts
Mitochondrial ultrastructure, density and spatial confinement of Arg-II and NOS
Immuno-electron microscopy (IEM) shows less mitochondrial staining for Arg-II and NOS in single mitochondria, as well as a disruption and breakdown of the mitochondrial ultrastructure in old (Fig. 2b) versus young hearts (Fig. 2a). Mitochondrial density is, however, greater in old (Fig. 2d) when compared to young (Fig. 2c) myocytes (51.7 ± 1.8 vs. 69 ± 2.2 mitochondria × 106/cm2, *p < 0.01, n = 3 per group Fig. 2e). Immunofluorescence reveals an organized pattern of mitochondria and Arg-II distribution in the myocytes of young rats (Fig. 3a). In contrast, in old rats a disruption of this normal relationship, as well as an increase in the number of mitochondria is observed (Fig. 3b).

a, b Immuno-electron microscopy (IEM, ×120,000) reveals greater levels of Arg-II (white arrows) and NOS (black arrows) in young rats (a) when compared to the old rats (b). Furthermore, there appears to be a breakdown of the mitochondrial ultrastructure in old rats (b) when compared to the young (a). c, d However, there is an increased mitochondrial density in old myocytes (d) in comparison to the young myocytes (c) at ×20,000. e Quantifying this increased mitochondrial density in old rats reveals that the difference is significant (51.7 ± 1.8 vs. 69 ± 2.2 mitochondria × 106/cm2, n = 3 per group, * p < 0.01)

Immuno-histochemistry of old and young rat myocytes [COX IV mitochondria (green), Arg-II (red) and DAPI for nuclei (blue), a merged image is shown lastly]. a Shows organization of Arg-II and the preservation of relationship of mitochondria within the sarcomeric structure within young myocytes. b There appears to be a disruption of this organization in the old myocytes
Regulation of NO production by arginase
The expression and abundance of Arg-II in BEC-treated hearts appears to increase (Fig. 4a), while the activity of Arg-II decreases significantly (Fig. 4b; 2.2 ± 0.65 vs. 5.1 ± 0.67, old treated vs. old untreated, p = 0.04, n = 3 per group). Consistent with this, BEC also results in a significant increase in NO production in the treated old hearts when compared to untreated ones (Fig. 4c; 12.88 ± 0.432 vs. 4.54 ± 0.582 μmol/mg, p < 0.01, n = 2 per group).

a There is increased Arg-II expression in old untreated rats when compared to old BEC-treated rats as shown by immunoblotting. b Treatment with BEC (10 μmol/l) results in significantly decreased Arg activity (n = 3 per group, *p < 0.05) c Concurrently, inhibition of Arg with BEC results in a significant increase in the NO production (n = 2 per group, *p < 0.05) in heart homogenates, suggesting reciprocal regulation of NO production by Arg
Contractility of aging cardiac myocytes
Myocytes from young rats demonstrate an increase in contractile responses with increasing frequency (Fig. 5). In contrast, the response to pacing is markedly attenuated in myocytes from old rats [−5.15 ± 5.65% (old) vs. 61.42 ± 16.04% (young) at 4 Hz, n = 12 myocytes per group from 4 hearts per group (3 myocytes per heart), p = 0.02]. Furthermore, as demonstrated in Fig. 5a the response to pacing in the myocytes that were pre-incubated with BEC was significantly enhanced [4 Hz, −5.15 ± 5.65 vs. 70.98 ± 6.10%, old untreated vs. old BEC-treated, n = 12 myocytes per group from 4 hearts per group (3 myocytes per heart), p = 0.0004].

The FFR in myocytes is depressed in aging rats compared to young. Treatment with BEC restores the FFR in old myocytes
NOS isoforms and function in the aging heart
NOS-2 is significantly upregulated in the aging heart (1.00 ± 0.12 vs. 6.33 ± 1.33, n = 4 per group; Fig. 6a). NOS-1 and NOS-3 show no significant difference in old versus young hearts (data not shown). Furthermore, the increase of NOS-2 enhances ROS production in old versus young hearts (3,153 ± 157 vs. 4,735 ± 427 RFU/min/mg protein, n = 5 hearts per group, p = 0.008), as evidenced by significant inhibition by the NOS-2 inhibitor 1400 W (old vs. old plus 1400 W: 4,735 ± 427 RFU/min/mg protein vs. 4,014 ± 314 RFU/min/mg protein, n = 5 hearts per group, p = 0.005, Fig. 6b).

a NOS-2 is significantly upregulated in aging hearts (1.00 ± 0.12 vs. 6.33 ± 1.33, n = 4 per group). b Increased NOS-2 in old hearts contributes to increased ROS production (3,153 ± 157 vs. 4,735 ± 427, n = 5 hearts per group, p = 0.008), which is significantly inhibited by 1400 W (old vs. old plus 1400 W: 4,735 ± 427 vs. 4,014 ± 314, n = 5 hearts per group, p = 0.005)
Discussion
In this study we demonstrate a potential cellular and molecular mechanism that contributes to age-associated decreased cardiac contractility. An increase in Arg-II activity in old myocytes regulates NOS activity and hence NO production. Since the NO produced by NOS-1 positively modulates cardiac contractility (Khan and Hare 2003), a decrease in NO production in aging cardiac myocytes contributes to diminished contractile response to pacing in these cells. Inhibition of Arg-II activity with BEC restores myocardial NO production and restores the force frequency response in aging cardiac myocytes. This supports the hypothesis that an increase in Arg-II activity is responsible for the diminished contractile reserve in aging cardiac myocytes. Overall, the findings from this study explain in part some of the cellular and molecular mechanisms for age-related myocardial dysfunction.
Regulation of NOS by Arg was a concept initially formulated following the description of upregulation of Arg in murine macrophages when endotoxin was administered (Gotoh et al. 1996; Morris et al. 1998). These groups demonstrated that injection of endotoxin resulted in an induction of Arg (both Arg-I and Arg-II) and inducible NOS (iNOS or NOS-2) (Salimuddin et al. 1999). However, increased NOS-2 expression did not result in sustained increased NO production, suggesting that Arg-I and Arg-II were limiting substrate availability to NOS-2 (Pollock et al. 2001). This led to the suggestion of a model of reciprocal regulation of NOS-3 by Arg in endothelial cells (Berkowitz et al. 2003). We have recently shown that there is a close association between Arg-II and NOS in cardiac tissue which helps regulate cardiac contractility (Steppan et al. 2006b). Furthermore, the study also elicited that Arg-II is primarily confined to the mitochondria of cardiac myocytes (Steppan et al. 2006b), and by reciprocally regulating NOS, Arg-II modulates myocardial contractility in young animals (Steppan et al. 2006a).
In the current study, we performed confocal immunofluorescence and IEM on the hearts of old and young rats. Here we demonstrated that there is a significant increase in mitochondrial density in old myocytes. Considering that Arg-II is predominantly localized to the mitochondria, this may help to explain the increased Arg expression observed in old versus young hearts, despite the decrease in of Arg-II and NOS in a single old mitochondrion. Furthermore, immunofluorescence reveals a disruption of the normal association between Arg-II and mitochondria, as shown in Fig. 2. This disruption most likely plays an integral role in the diminished contractility and overall myocardial dysfunction associated with the aging heart.
Previous work has shown that Arg-II is the predominant isoform expressed in the rat heart (Steppan et al. 2006b). This present study confirms those findings and extends this observation to show and upregulation in aging cardiac tissue, more specifically cardiac myocytes. This is consistent with the upregulation and increased expression of Arg-II in old endothelial cells (Krotova et al. 2010) and endothelial cells exposed to and atherogenic stimulus (Ryoo et al. 2008, 2011; Santhanam et al. 2008) and may contribute to NOS uncoupling or the production of peroxinitrate. This in turn will lead to a change in the nitroso-redox milieu of the cell (Berkowitz 2007; Kim et al. 2009a).
There are three isoforms of NOS that have been implicated in regulating cardiac function, namely NOS-1, NOS-2 and NOS-3 (Umar and van der Laarse 2010). While NOS-1 and NOS-3 regulatory role is constitutively Ca2+-dependant, NOS-2 or inducible NOS is Ca2+ independent and appears to be upregulated in the aging and damaged myocardium (Heinzel et al. 2008; Yang et al. 2004). Xanthine oxidoreductase (XOR) is an enzyme located in close proximity to NOS on the SR and produces superoxide by purine metabolism (Khan et al. 2004). Superoxide diffusion out of the SR inhibits sensitivity of the contractile units in cardiac myocytes, thereby diminishing contractility. NO reacts directly with the superoxide and inhibits its release from the SR, thereby reducing superoxide inhibition of cardiac contractility (Khan et al. 2004). Thus compartmentalized NOS helps maintain this redox balance in the heart by balancing the ratio of superoxide/NO production. However, with age, there is greatly increased production of superoxide/reactive oxygen species (ROS). The combined effect of which eventually leads to decreased contractility (Rueckschloss et al. 2010).
Regarding the interpretation of the effect of NOS-2 inhibition on ROS production, it is well established that aging-associated cardiovascular dysfunction is related to alteration in the nitroso-redox milieu (Kim et al. 2009a; Musci et al. 2006). This nitroso-redox stress/imbalance can lead to nitrosative and oxidative modifications of various proteins and lipids leading to cell damage and dysfunction. It has also been recognized that the upregulation of NOS-2 in aging can lead to nitrosative stress through peroxynitrite formation (Drew and Leeuwenburgh 2002). Therefore the decrease in ROS formation by the NOS-2 inhibitor 1400 W strongly suggests that NOS-2 may in part be the source of ROS and thus in the aging heart NOS-2 is uncoupled.
Arg-II shares the substrate l-arginine with NOS. Thus Arg-II reciprocally regulates NOS activity by substrate competition (White et al. 2006). In this study we have shown that an increase in Arg-II in aging cardiac tissue leads to a significant reciprocal decrease in the NO production by NOS. Furthermore, we have demonstrated that inhibition of Arg activity significantly increases NO production in old rat heart homogenates so that it is not significantly different from that of young hearts. These findings confirm the reciprocal regulation of NOS by Arg-II in aging cardiac tissue (Steppan et al. 2006a). Furthermore, the physiologic implication of this reciprocal regulation was observed by monitoring the FFR in aging cardiac myocytes. As expected, there was diminished contractile response to pacing in aging cardiac myocytes. This diminished response was in part most likely due to the increased Arg-II activity and expression in these aged myocytes, since inhibition of Arg-II activity in these myocytes using the arginase inhibitor BEC restored the FFR. These observations have important potential implications for the treatment of age-related myocardial dysfunction.
Our study is limited by the fact that it has been carried out in a rodent model, on a limited amount of animals and cells. Moreover, we used a simplified ex vivo assay of contractility to demonstrate the physiological principal in the aging heart. However, our recent observations (Kim et al. 2009b) suggest that arginase inhibition decreases age-associated vascular stiffness in old rats in vivo. This modulation in ventricular–vascular coupling may also have significant implications for the cardiac structure and function.
Conclusion
In this study we show that Arginase activity and Arg-II expression are significantly increased in hearts of old versus young rats. This increase was associated with a significant decrease in NO production, being restored to that of young with BEC. Furthermore FFR was significantly depressed in old rat myocytes and inhibition of Arg restored the FFR of old myocytes to that of young. We therefore conclude that Arg-II upregulation in aging rat hearts contributes to decreased cardiac contractile function associated with aging. This finding has important implications for therapy in age-related myocardial dysfunction.
References
Balligand JL (1999) Regulation of cardiac beta-adrenergic response by nitric oxide. Cardiovasc Res 43:607–620
Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM (2002) Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416:337–339
Berkowitz DE (2007) Myocyte nitroso-redox imbalance in sepsis: NO simple answer. Circ Res 100:1–4
Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM (2003) Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 108:2000–2006
Drew B, Leeuwenburgh C (2002) Aging and the role of reactive nitrogen species. Ann NY Acad Sci 959:66–81
Gotoh T, Sonoki T, Nagasaki A, Terada K, Takiguchi M, Mori M (1996) Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett 395:119–122
Hare JM (2003) Nitric oxide and excitation–contraction coupling. J Mol Cell Cardiol 35:719–729
Hare JM (2004) Nitroso-redox balance in the cardiovascular system. N Engl J Med 351:2112–2114
Hare JM, Givertz MM, Creager MA, Colucci WS (1998) Increased sensitivity to nitric oxide synthase inhibition in patients with heart failure: potentiation of beta-adrenergic inotropic responsiveness. Circulation 97:161–166
Heinzel FR, Gres P, Boengler K, Duschin A, Konietzka I, Rassaf T, Snedovskaya J, Meyer S, Skyschally A, Kelm M, Heusch G, Schulz R (2008) Inducible nitric oxide synthase expression and cardiomyocyte dysfunction during sustained moderate ischemia in pigs. Circ Res 103:1120–1127
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS (2005) Protein S-nitrosylation: purview and parameters. Natl Rev Mol Cell Biol 6:150–166
Jung DH, Mo SH, Kim DH (2006) Calumenin, a multiple EF-hands Ca2+-binding protein, interacts with ryanodine receptor-1 in rabbit skeletal sarcoplasmic reticulum. Biochem Biophys Res Commun 343:34–42
Khan SA, Hare JM (2003) The role of nitric oxide in the physiological regulation of Ca2+ cycling. Curr Opin Drug Discov Dev 6:658–666
Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM (2003) Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res 92:1322–1329
Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM (2004) Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation–contraction coupling. Proc Natl Acad Sci USA 101:15944–15948
Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE (2009a) Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 107:1249–1257
Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE (2009b) Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 107:1249–1257
Krotova K, Patel JM, Block ER, Zharikov S (2010) Hypoxic upregulation of arginase II in human lung endothelial cells. Am J Physiol Cell Physiol 299:C1541–C1548
Lee HY, Oh BH (2010) Aging and arterial stiffness. Circ J 74:2257–2262
Morris SM Jr, Kepka-Lenhart D, Chen LC (1998) Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am J Physiol 275:E740–E747
Musci G, Persichini T, Casadei M, Mazzone V, Venturini G, Polticelli F, Colasanti M (2006) Nitrosative/oxidative modifications and ageing. Mech Ageing Dev 127:544–551
Pollock JS, Webb W, Callaway D, Sathyanarayana O’BrienW, Howdieshell TR (2001) Nitric oxide synthase isoform expression in a porcine model of granulation tissue formation. Surgery 129:341–350
Rueckschloss U, Villmow M, Klockner U (2010) NADPH oxidase-derived superoxide impairs calcium transients and contraction in aged murine ventricular myocytes. Exp Gerontol 45:788–796
Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, Sohi J, Santhanam L, Soucy K, Tuday E, Baraban E, Ilies M, Gerstenblith G, Nyhan D, Shoukas A, Christianson DW, Alp NJ, Champion HC, Huso D, Berkowitz DE (2008) Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res 102:923–932
Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH (2011) OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis 214:279–287
SalimuddinNagasaki, Nagasaki A, Gotoh T, Isobe H, Mori M (1999) Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. Am J Physiol 277:E110–E117
Santhanam L, Christianson DW, Nyhan D, Berkowitz DE (2008) Arginase and vascular aging. J Appl Physiol 105:1632–1642
Stamler JS, Lamas S, Fang FC (2001) Nitrosylation: the prototypic redox-based signaling mechanism. Cell 106:675–683
Steppan J, Ryoo S, Schuleri KH, Gregg C, Hasan RK, White AR, Bugaj LJ, Khan M, Santhanam L, Nyhan D, Shoukas AA, Hare JM, Berkowitz DE (2006a) Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. Proc Natl Acad Sci USA 103:4759–4764
Steppan J, Ryoo S, Schuleri KH, Gregg C, Hasan RK, White AR, Bugaj LJ, Khan M, Santhanam L, Nyhan D, Shoukas AA, Hare JM, Berkowitz DE (2006b) Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. Proc Natl Acad Sci USA 103:4759–4764
Umar S, van der Laarse A (2010) Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem 333:191–201
Wessells RJ, Bodmer R (2006) Cardiac aging. Semin Cell Dev Biol
White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, Nyhan D, Shoukas AA, Hare JM, Berkowitz DE (2006) Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 47:245–251
Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC (1999) Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA 96:657–662
Yang B, Larson DF, Watson RR (2004) Modulation of iNOS activity in age-related cardiac dysfunction. Life Sci 75:655–667
Zhang C, Hein TW, Wang W, Chang CI, Kuo L (2001) Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J 15:1264–1266
Acknowledgments
The authors wish to thank all members of the Shoukas-Berkowitz lab for their support and assistance in this project. This work was supported by grant R01AG021523 from the NIH, grant CA01301 from the National Space Biomedical Research Institute through NASA, and NNH04ZUU005N from NASA.
Conflict of interest
None of the authors has any conflict of interest to report.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Keith Phillip George.
M. Khan and J. Steppan contributed equally to the manuscript.
Rights and permissions
About this article
Cite this article
Khan, M., Steppan, J., Schuleri, K. et al. Upregulation of arginase-II contributes to decreased age-related myocardial contractile reserve. Eur J Appl Physiol 112, 2933–2941 (2012). https://doi.org/10.1007/s00421-011-2257-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-011-2257-9