
Abstract
Intercellular junctions play a role in regulating islet cytoarchitecture, insulin biosynthesis and secretion. In this study, we investigated the animal metabolic state as well as islet histology and cellular distribution/expression of CAMs and F-actin in the endocrine pancreas of C57BL/6/JUnib mice fed a high-fat diet (HFd) for a prolonged time period (8 months). Mice fed a HFd became obese and type 2 diabetic, displaying significant peripheral insulin resistance, hyperglycemia and moderate hyperinsulinemia. Isolated islets of HFd-fed mice displayed a significant impairment of glucose-induced insulin secretion associated with a diminished frequency of intracellular calcium oscillations compared with control islets. No marked change in islet morphology and cytoarchitecture was observed; however, HFd-fed mice showed higher beta cell relative area in comparison with controls. As shown by immunohistochemistry, ZO-1, E-, N-cadherins, α- and β-catenins were expressed at the intercellular contact site of endocrine cells, while VE-cadherin, as well as ZO-1, was found at islet vascular compartment. Redistribution of N-, E-cadherins and α-catenin (from the contact region to the cytoplasm in endocrine cells) associated with increased submembranous F-actin cell level as well as increased VE-cadherin islet immunolabeling was observed in diabetic mice. Increased gene expression of VE-cadherin and ZO-1, but no change for the other proteins, was observed in islets of diabetic mice. Only in the case of VE-cadherin, a significant increase in islet content of this CAM was detected by immunoblotting in diabetic mice. In conclusion, CAMs are expressed by endocrine and endothelial cells of pancreatic islets. The distribution/expression of N-, E- and VE-cadherins as well as α-catenin and F-actin is significantly altered in islet cells of obese and diabetic mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The interaction between neighboring cells, which is essential for tissue homeostasis, depends on intercellular junctions (IJs). There are four types of IJs, structurally and functionally distincts, namely tight junction (TJ), adherens junctions (AJ) (zonula or fasciae adherens), desmosomes (or maculae adherens), and gap junctions (GJ). The TJ, AJ and desmosomes are directly involved in intercellular adhesion process and cytoskeleton organization, while GJ mediates cell–cell communication (Ebnet 2008; Harris and Tepass 2010; Mese et al. 2007). Protein complexes associated with IJs have been shown to act as adhesion/signaling molecules involved in several intracellular pathways. They can modulate gene expression and various cellular processes, besides adhesion and communication, such as cell differentiation, proliferation and cell death in various organs, including endocrine pancreas (Benmerah et al. 2003; Conacci-Sorrell et al. 2002; Ebnet 2008; Jain and Lammert 2009; Meda 2013; Spadaro et al. 2012).
The endocrine pancreas is composed of five cell types (α, β, δ, PP and ε cells) that secrete different hormones (i.e., glucagon, insulin, somatostatin, polypeptide P and ghrelin, respectively), all involved direct or indirectly in glycemic homeostasis (Andralojc et al. 2009; Orci 1976). These endocrine cells have a typical species-specific arrangement (cytoarchitecture) within the pancreatic islet (Brissova et al. 2005). In rodents, the beta cells occupy the central region while the other cell types are located at the periphery of the islet (Brissova et al. 2005; Carvalho et al. 2006, 2012; Kim et al. 2009). The islet cells are connected homotypically and heterotypically through tight, adherens, gap junctions and desmosomes, as revealed by electron microscopy (Jain and Lammert 2009; Orci 1976).
The biochemistry and function of IJs within the endocrine pancreas are still not completely known. However, studies have demonstrated that pancreatic endocrine cells do express some junctional proteins such as Cx36 and Cx30.2 (at GJ) (Carvalho et al. 2010; Coronel-Cruz et al. 2013; Nlend et al. 2006), ZO-1 and claudin 8 (at TJ) (Collares-Buzato et al. 2004; Meda 2013; Rieck et al. 2009; Santos-Silva et al. 2012), and N-CAM, E-, N-cadherins and catenins (at AJ) (Carvell et al. 2007; Collares-Buzato et al. 2004; Esni et al. 1999; Meda 2013; Parnaud et al. 2011; Santos-Silva et al. 2012). Some studies have suggested the participation of IJs in the formation/maintenance of islet cytoarchitecture (Esni et al. 1999; Santos-Silva et al. 2012; Yamagata et al. 2002). In addition, IJs seem to be involved in the functional maturation of beta cells in vitro and in vivo (Carvalho et al. 2010; Collares-Buzato et al. 2004; Leite et al. 2005; Santos-Silva et al. 2012), regulate biosynthesis and secretion of insulin (Calabrese et al. 2004; Hauge-Evans et al. 1999; Parnaud et al. 2015; Rogers et al. 2007) as well as mediate beta cell proliferation and death (Meda 2013; Parnaud et al. 2011; Wakae-Takada et al. 2013). However, studies investigating the possible role of these membrane structures in the beta cell dysfunction are scarce. Recently, we demonstrated that prediabetic obese mice, displaying impaired glucose homeostasis and decreased insulin secretory response, show a reduction in gap junctional Cx36 expression. This was associated with a decrease in the number of GJ membrane channels and intercellular coupling between beta cells (Carvalho et al. 2012). However, to the best of our knowledge, the role of other junctional proteins in the pathogenesis of type 2 diabetes mellitus (T2DM) is still unclear.
Type 2 diabetes mellitus is a highly prevalent metabolic-endocrine disease, whose pathogenesis displays strong association with physical inactivity and obesity (Tripathy and Chaves 2010). Pancreatic beta cells play a critical role in the development of T2DM. At early stages, beta cells are able of adaptive response showing increased insulin secretion and/or mass in attempt to offset the peripheral insulin resistance (Ahrén and Pacini 2002; Chang-Chen et al. 2008; Kahn et al. 2006; Sone and Kagawa 2005; Tripathy and Chaves 2010). At advanced stages of T2DM, functional exhaustion of the beta cells occurs, leading to decrease of its number/mass by apoptosis. This state results in reduction or total lack of insulin secreted, making hormone replacement mandatory (Butler et al. 2003; Kahn et al. 2006).
Rodent fed a high-fat/calorie diets have been employed as valuable model to study the pathogenesis of T2DM and other diseases associated with obesity (Ahrén and Pacini 2002; Drolet et al. 2006; Herrera 2009; Oliveira et al. 2015; Shafrir et al. 1999; Surwit et al. 1988; Winzell and Ahrén 2004). Previous studies have focused on the response of peripheral tissues and signaling mechanisms triggered by high-fat/hypercaloric diets over relatively short periods (days to weeks). However, the effect of prolonged exposure (several months) to these diets on the functioning and structure of the endocrine pancreas has been less explored (Ahrén and Pacini 2002; Sone and Kagawa 2005). The objective of the present study was two-fold: 1) to evaluate the effect of prolonged exposure (8 months) to HF diet on the functional and structural aspects of the endocrine pancreas of mice, focusing on beta cell biology, and 2) to investigate the repercussion of this treatment on the cellular distribution and expression of some junctional and cytoskeletal proteins (namely E-, N-, VE-cadherins, ZO-1, β- and α-catenins, and F-actin) in the pancreatic endocrine cells of these animals.
Materials and methods
Animals and diets
Male C57BL/6/JUnib mice were obtained from breeding colonies of the Multidisciplinary Center for Biological Investigation on Laboratory Animal Science (CEMIB) of the University of Campinas (UNICAMP, Brazil). Animals were housed at 22 ± 1 °C in 12-h dark/light cycles with ad libitum access to food and water. Animals (6 to 8 weeks old) were divided into two groups. One group (control) was fed for 240 days (8 months) with a standard rodent diet (Nuvital CR1, Colombo, Paraná, Brazil) (containing 4.5 g % lipid, 53 g % carbohydrates and 23 g % protein). The second group (treated) was fed a high-fat (HF) diet (containing 21 g % lipid, 50 g % carbohydrate and 20 g % protein), with similar content of minerals, vitamins and fibers found in the regular diet. Mice were used in accordance with the guidelines of the National Council for Animal Experimentation (CONCEA-MCTI, Brazil). All experimental protocols used were approved by the Ethics Committee on Animal Use (CEUA) of UNICAMP under protocols # 1885-1 and 3122-1.
Assessment of metabolic parameters
All animals were weighed before and after the diet period; values were expressed as the percentage of body weight gain relative to the initial body weight. Glycemia was determined using a glucometer (Accu-Chek Advantage II,® Roche Diagnostic, Mannheim, Germany) in blood samples collected from the tail of 12-h-fasted or 12-h-fed mice. The insulin tolerance test (ITT) was performed by intraperitoneal injection of insulin (0.5 U/kg body weight of human insulin—Biohulin Biobrás® R, Montes Claros, MG, Brazil) in fed animals (Oliveira et al. 2015). Subsequently, blood glucose was measured in blood samples collected from the tail at 0, 10, 15, 30 and 60 min after insulin administration. The area under the glucose curve was estimated using GraphPad Prism version 5 for Windows (GraphPad Software, La Jolla, CA, USA). For determining insulinemia (at fed state), blood was collected from the cervical veins of euthanized animals in heparinized capillaries and, after centrifugation, plasma samples were stored at −20 °C. The level of plasma insulin was determined by radioimmunoassay (RIA). All blood samples were collected during the period between 09:00 and 11:00 am.
Pancreatic islet isolation and static insulin secretion
Islets were isolated by collagenase digestion (EC 3.4.24.3, Sigma, St. Louis, MO, USA) (Carvalho et al. 2010). After separation with Histopaque 1077 (Sigma), pancreatic islets were individually collected under a dissecting microscope. Fresh islets were used for determining glucose-stimulated insulin secretion, or were homogenized in an anti-protease cocktail for immunoblotting, or added to RNAlater ® (Applied Biosystems, Carlsbad, CA, USA) for real-time PCR (as described later).
To measure the static insulin secretion, pools of 5 islets were placed in microtubes and preincubated at 37 °C for 30 min in Krebs–Ringer bicarbonate buffer (KRBB) containing 115 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 2.5 mM CaCl2.2H2O, 1 mM MgCl2.6H2O and 15 mM HEPES, pH 7.4 plus 5.6 mM glucose and 0.3 % (w/v) BSA for 1 h at 37 °C in an incubator supplied with carbogenic mixture (95 % O2 + 5 % CO2). After careful aspiration of the buffer, islets were incubated for 1 h with the buffer supplemented with 2.8 or 16.7 mM glucose. Aliquots of supernatant (500 μl) were removed and stored at −20 °C for determination of insulin concentration determined by RIA (Carvalho et al. 2010).
Glucose-induced calcium handling
After isolation, islets were pre-incubated in KRBB for 1 h at 37 °C in an incubator supplied with carbogenic mixture (95 O2 + 5 % CO2). Then, the solution was replaced by buffer containing 1 µM Fura-2, AM (Life Technologies, Eugene, Oregon, USA) and islets were incubated for additional 1 h. Then the islets were transferred to a glass coverslip coated with polylysine, which was placed in a thermostatically regulated open chamber (at 37 °C), adapted on the stage of an inverted microscope (Nikon UK, Kingston, UK). The islets were perfused with fresh KRBB (without BSA) at a flow rate of 1 ml/min. Images were acquired every 5 s with a camera (Hammamatsu Photonics Ibérica, Barcelona, Spain) in conjunction with a dual-filter wheel with a 340 and 380 nm bandpass filters (Sutter Instrument Company, Novato, CA). Data were collected using Image Master 5 software (PTI Systems, Birmingham, NJ). The frequency of oscillations was calculated during glucose 11.1 mM perfusion and after the first peak. It was considered oscillation increments 10 times bigger than the variation in fluorescence observed during basal glucose concentration (at 2.8 mM glucose). The area under the curve was calculated during the period with medium at 11.1 mM glucose, after subtracting the basal values.
Histology and cytoarchitecture of pancreatic islets
After animal euthanasia, the pancreas was removed and fixed in 4 % paraformaldehyde (diluted in phosphate buffered saline) for 24 h. The pancreas was sectioned into 5 fragments (1–5 corresponding to the head and the tail region, respectively). Each fragment was processed by routine techniques for embedding in paraffin (Histosec tablets, Merck, Darmstadt, Germany). Semi-serial sections of 5 µm were cut (with 100 µm spacing between sections) until complete exhaustion of the block. The selected sections were stained with hematoxylin–eosin or processed for indirect immunoperoxidase for insulin (Carvalho et al. 2006).
For morphometric analysis, all the islets and the pancreas sections were photographed and the images analyzed using stereology as previously described (Oliveira et al. 2015). The relative area of beta cells (as an estimate of beta cell mass) was determined by the sum of all insulin immunostained-islet areas divided by the respective pancreas section area using the software ImageJ (http://rsbweb.nih.gov/ij/download.html). The rate of beta cell proliferation was determined by counting the number of insulin-positive cell nuclei within the islet. The beta cell size (µm2) was determined by dividing the absolute islet area by the beta cell number (Oliveira et al. 2015).
For the analysis of islet cytoarchitecture, the pancreas sections were processed for dual immunofluorescence for insulin and glucagon using a standard protocol (Oliveira et al. 2015). Alteration to the typical islet cytoarchitecture was evaluated by measuring the number of displaced or non-contiguous alpha cells in the peripheral mantle (around a core of beta cells in the islet). For that, a score system was used as previously described (Oliveira et al. 2015). Each islet was given a score and averaged across experimental groups.
The relative area occupied by alpha (glucagon-positive) and beta cells (insulin-positive) within the islet was also measured in the images of double-labeled islets using the ImageJ free software (http://rsbweb.nih.gov/ij/download.html) and expressed as a percentage of the total islet area (Oliveira et al. 2015).
Localization of junctional proteins and F-actin in the endocrine pancreas
Cellular location of junctional proteins (E-, N-, and VE-cadherins, α- and β-catenins and ZO-1) in the pancreatic islets was determined by indirect immunofluorescence (Collares-Buzato et al. 2004; Santos-Silva et al. 2012). Briefly, pancreas sections were incubated overnight at 4 °C with the following primary antibodies (in 3 % dry skimmed milk in TBS): anti-E-cadherin (dilution 1:50, Abcam, Cambridge, UK, cat no. 11512), anti-N-cadherin (dilution 1:50, Abcam, cat no 18203), anti-VE-cadherin (dilution 1:75, Abcam, cat no 33168), anti-ZO-1 (dilution 1:300; Zymed/Invitrogen, Carlsbad, CA, USA; cat no 617300), anti-α-catenin (dilution 1:600 Sigma, cat no c2081) or anti-β-catenin (dilution 1:500, Zymed/Invitrogen, cat no. 138400). Tissues were then incubated with appropriate secondary antibodies conjugated with FITC (Sigma). When necessary, co-localization of these proteins and insulin or glucagon was performed by further incubating the pancreas sections for 1 h and 30 min with the polyclonal anti-insulin (dilution 1:100, Dako) or anti-glucagon antibody (dilution 1:500, Sigma) followed by its specific polyclonal secondary antibody conjugated with TRITC (Sigma). In addition, co-localization of ZO-1 and CD31, an endothelial marker, was done in some pancreas sections using a primary antibody anti-CD31 (1:50 dilution, Life Technologies, cat no RM5200) and specific secondary antibody (Sigma). The cytochemistry for F-actin was done in 3.7 % formaldehyde-fixed and 0.1 %Triton-permeabilized pancreas cryosections, by incubating them with TRITC-labeled phalloidin (concentration 1 μg/mL; Sigma) for 2 h in PBS at 37 °C, in the dark. Finally, slides were mounted using a commercial antifading agent (Vectashield, Vector) and confocal laser scanning microscope was used for co-localization studies (510 CLSM, Zeiss, Hamburg, Germany) and/or a fluorescence microscope coupled to a conventional system for capturing images (Axio Observer-D1, Zeiss).
To evaluate fluorescence intensity and compare experimental groups, pancreas sections from both groups were processed for immunofluorescence/histochemistry at the same time and analyzed at the same observation session, using identical confocal parameters for image capture. All images were analyzed using ImageJ free software (http://rsbweb.nih.gov/ij/). For ZO-1 and β-catenin immunolocalization, total fluorescence values were expressed as islet integrated pixel density normalized against the area of the islet. For E-cadherin, N-cadherin, α-catenin, and F-actin, the integrated density of 50 points per islet placed at the intercellular region of the beta cells (co-immunolabeled insulin) was measured in all captured images (a total of 1680–4200 points sampled per experimental group). For VE-cadherin, the linear area of the immunostaining for this protein was measured in pixels (using the GIMP free software, http://www.gimp.org/) and normalized against the area of the islet (Image J) and expressed as pixels/islet area. For N-cadherin, the “Grid” ImageJ tool was used to determine the relative islet area containing cells with diffuse staining of the cytoplasm (instead of the usual intercellular location). This area value was normalized against the total area of the islet (expressed as arbitrary units), which was taken as an index of cell redistribution.
Western blot
Cellular content of junctional proteins was analyzed by Western blot on homogenates of pancreatic islets isolated as previously described (Santos-Silva et al. 2012). Approximately 300–400 islets isolated per group were sonicated in an anti-protease cocktail (composition: 10 mmol/L imidazole, pH 7.4, 4 mmol/L EDTA, 1 mmol/L EGTA, 0.5 μg pepstatin A/mL, 200 KIU aprotinin/mL, 2.5 μg leupeptin/mL, 30 μg trypsin inhibitor/mL, 200 μmol/L DL-dithiothreitol, DTT, and 200 μmol/L phenylmethylsulfonylfluoride (PMSF); Sigma). Aliquots of homogenates (containing 50 μg total protein) were incubated for 1 h at 37 °C in 30 % 5X concentrated Laemmli buffer (1 mol/L sodium phosphate, pH 7.8, 0.1 % bromophenol blue, 50 % glycerol, 10 % sodium dodecyl sulfate (SDS), and 2 % β-mercaptoethanol, Sigma) and then applied to an 8 or 10 % SDS-PAGE gel. Proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Membranes were stained with Ponceau S solution (Bio-Rad) to confirm loading and transfer efficiency. Membranes showing comparable lane staining for both groups were incubated with primary antibodies (the same used in the immunofluorescence method), followed by incubation with appropriate secondary antibodies conjugated with peroxidase (Sigma). Immunoreactive bands were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent kit Substrate, Pierce, Thermo Fisher Scientific, Waltham, MA, USA) and autoradiography. Membranes were reincubated with anti-β-actin antibody (Zymed/Invitrogen, 1:1500 dilution) and subsequently with the respective peroxidase- conjugated secondary antibody, as an internal total protein control. The optical density of the bands was analyzed using ImageJ free software (http://rsbweb.nih.gov/ij/) and expressed as ratio of junctional protein/β-actin.
Real-time PCR
Extraction of total RNA
Total RNA from biological triplicates per group (each one containing 300–400 isolated islets from 2 to 3 animals) was extracted with RNAqueous®-Micro Kit (Ambion®, WAustin, Texas, USA), following the manufacturer’s guidelines. RNA purity and concentration were confirmed with a Nanodrop ND-1000 UV–Vis (Nanodrop Technologies, Wilmington, USA) reading at 260 nm and calculating the 260/280 nm ratio. RNA integrity was analyzed by electrophoresis in a 0.8 % agarose gel in TBE buffer (90 mm Tris, 90 mM boric acid, 50 mM EDTA, pH 8.0) in the presence of ethidium bromide and then stored at −80 °C for later use.
cDNA synthesis was performed using the ImProm Kit ™ (Promega, Madison, USA) from 1 µg total RNA, according to manufacturer’s instructions. The integrity of cDNA was verified as described above for RNA. cDNA was stored at −20 °C.
Primers
Primers were designed using Primer-BLAST from NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov) according to primer parameters (Table I; Supplementary material). Primers were tested for specificity of amplification by PCR. Primer amplification efficiency was tested using cDNA dilutions (10−1, 10−2, 10−3, 10−4, 10−5) in triplicate. Amplification efficiency (E) for each gene was estimated by the equation E = 10 (−1/slope) (Ramakers et al. 2003), using SDS Software 1.3.1 ™ (Applied Biosystems®, California, USA). Efficiencies of 90 and 110 % (−3.6 > slope > −3.1) were considered ideal for the calculation of relative expression.
For this assay, we used RPS29 as an endogenous control, which maintained the lowest variability among 3 genes tested for constitutive expression (rps29, actb, gapdh)(data not shown).
Analysis of relative gene expression by RT-qPCR
RT-qPCR amplifications were performed in triplicate, using 20 ng cDNA per reaction. Reactions were prepared with SYBR® Green PCR Master Mix (Applied Biosystems) primers (Forward and Reverse) and H2O DEPC Reaction conditions were 50 °C for 2 min, 95 °C for 10 min, 40 repetitions of 95 °C for 15 s, 61.5 °C for 1 min followed by a final step of 95 °C for 15 s, 60 °C for 1 min and 95 °C for 30 s and 60 °C for 15 s on a 7500 Fast Real-Time PCR System7500 (Applied Biosystems, USA). Analysis was performed by Sequence Detection Software (SDS) v1.3 (Applied Biosystems). The relative quantity (RQ) was calculated using the 2-ΔΔCt (Livak and Schmittgen 2001) comparative Ct method using the following formula, QR = E − ΔΔCt (where 2 is reaction efficiency, ΔΔCt = ΔCt treated − ΔCt control (Livak and Schmittgen 2001) and ΔCt = Ct target gene − Ct reference gene.
Statistical analyses
Statistical differences between the two experimental groups were determined using Student’s t test (two-tailed) or the Mann–Whitney test (for data expressed as percentage). For multiple comparisons (as in the analysis of static insulin secretion and RT-qPCR), we used one-way ANOVA followed by Bonferroni post-test. For statistical analysis of the frequency of cytoarchitecture change, the Chi-square test was used. All statistical analyses were performed using the GraphPad Prism Software version 5.00 (GraphPad Software, USA). Data are presented as means + SEM, and the significance level was set at p < 0.05.
Results
Mice became obese and diabetic after prolonged exposure to high-fat diet, although still displayed compensatory beta cell expansion
Figures 1 and 2 depict the data on metabolic parameters of the model presented. HF diet-fed mice displayed significantly higher weight gain (170 %) (p < 0.0001) in comparison with the control group (37 %) at the end of the diet administration period. Additionally, animals fed a HF diet for 8 months showed a significant increase in postprandial and fasting glycemia (Fig. 1b, c, respectively) as well as a significant increase in plasma insulin relative to controls (Fig. 1d). These metabolic changes were associated with significant insulin peripheral resistance (Fig. 1e; area under the curve of ITT).

C57BL/6 mice become obese and diabetic after prolonged high-fat diet (HFd). HFd for 8 months induced a significant increase (***p < 0.0001) in the body weight of mice in relation to control mice fed a regular diet a (control, n = 38; HF diet, n = 37). Postprandial and fasting blood glucose (panels b and c, control n = 15–22; HF diet n = 16–22) as well as plasma insulin levels (at fed state) (panel d, control n = 25; HF n = 25) were also significantly increased (*p < 0.020 in b; ***p < 0.0001 in c and **p < 0.006 in d) in HFd mice. The curve of the insulin tolerance test (ITT) showed larger (***p = 0.0001) AUC values in HFd mice e (control n = 8; HF n = 8). All data are expressed as means + SEM

Pancreatic islets from diabetic mice show impairment of glucose-stimulated insulin secretion associated with a decreased intracellular calcium oscillations. a Islets from chow-fed control (open bars) or HFd-fed mice (black bars) were exposed to either 2.8 or 16.7 mM glucose for 60 min. Islets from HFd mice (black bars) showed comparable values of basal insulin secretion in the presence of 2.8 mM glucose (G 2.8), but a significant decrease in insulin release when stimulated by 16.7 mM glucose (G 16.7) (**p < 0.01). The bars represent the means + SEM of 3 independent experiments (23–30 batches of 5 islets isolated from 6 mice per group). b Representative curves of cytoplasmic calcium oscillations induced by glucose in islets from control (gray line) or HFd-fed mice (black line). Before KRBB containing 11 mM glucose, the islets were perfused with KRBB containing 2.8 mM glucose (not shown). c Frequency of glucose-induced calcium oscillations during 11 mM glucose and after the first peak. d Augment in cytoplasmic calcium induced by 11 mM glucose observed as the first peak in the figure b. e Area under the curve of 11 mM glucose subtracted from basal values. Data are means + SEM of 4 different mice. It was registered 2–4 islets from each mouse. *p < 0.05 versus Control; two-tailed unpaired t test in c
Isolated islets from mice of the two experimental groups show no significant difference in insulin release after exposure to basal glucose concentration (2.8 mM). However, the isolated pancreatic islets of HF diet- treated animals showed a significant impairment of insulin secretory function when stimulated with 16.7 mM glucose (3.9 ± 0.4 ng/mL insulin/h) as compared to controls (7.1 ± 1.2 ng/mL insulin/h, p < 0.01). In addition, islets isolated from HF diet-fed mice presented less glucose-induced oscillations in cytoplasmic calcium after the first peak provoked by the increase in glucose concentration (1.20 ± 0.23) than islets from control mice (1.95 ± 0.08; p < 0.05). Also, we observed a tendency (p = 0.057) of a delay in cytoplasmic calcium increment (first peak) induced by 11 mM glucose in islets from HF diet-fed mice compared to islets from control mice (1.56 ± 0.13 vs. 1.19 ± 0.12 min, respectively). No differences in the amplitude of intracellular calcium or in the area under the curve were observed between HF diet-fed and control mice.
Histology of islets from control (Fig. 3a) and HF diet-fed (Fig. 3b) mice showed a similar morphology (ovate to rounded) with a parenchyma consisting of endocrine cells interspersed with capillaries. Immunohistochemistry of pancreatic islets for insulin (red) and glucagon (green) (Fig. 3c, d) followed by a semi-quantitative analysis showed no significant difference in islet cytoarchitecture between the groups (e, f). A significant increase in the relative volume of beta cells per pancreas (p < 0.036 Fig. 3i) and in the percentage of beta cells per islet in the treated group was observed (p < 0.003; Fig. 3j) as compared with the controls. This suggests an increase in beta cell mass as visualized in pancreas sections processed for insulin immunoperoxidase (Fig. 3g, control group vs. Fig. 3h, HF diet group). This apparent increase in beta cell mass after 8-month HF diet treatment is associated with significant increase in number [HFd 83.5 ± 7.9 (246) vs. Control 54.5 ± 4.3 cells/islet (203 islets)] and size [HFd 144.7 ± 4.1 μm2 (246) vs. Control 125.4 ± 2.7 μm2 (203 islets)] of beta cells in comparison with control animals (p < 0.003). There was also a significant decrease in the relative area of alpha cells in the islets of diabetic mice [11.06 ± 0.84 % (66) when compared with islets from the control group 15.82 ± 1.24 % (85)] (p < 0.003).

Prolonged exposure to HFd induces beta cell area expansion but no marked alteration to islet cytoarchitecture in mice. No marked changes in islet histology (a, b) and cytoarchitecture (differential distribution of insulin-positive beta cells (red) and glucagon-positive alpha cells (green) (c–f) were seen in mice after HFd in comparison with controls (255 islets analyzed of 4 animals/group). However, a significant increase in beta cell relative volume per pancreas (i) (*p < 0.036) and in the percentage of beta cells per islet (j) (**p < 0.003) was observed, indicating an expansion of beta cell mass, as seen in pancreas sections processed by immunoperoxidase for insulin (arrows) after HFd (h) relative to controls (g) (6 pancreas sections/animal; n = 4–6 animals/group). Scale bars a–b and c–d 50 µm; g–h 500 µm
Altogether, the data indicate that prolonged HF diet exposure induces metabolic disturbances indicative of T2DM. While diabetics, the mice still display partial compensatory beta cell expansion (hyperplasia/hypertrophy) associated with impairment of intracellular calcium signaling and insulin secretion.
Cell distribution of intercellular junction-associated proteins and F-actin in pancreatic islets of diabetic animals
Immunofluorescence for AJ-associated (E-, N-, VE-cadherins) (Figs. 4, 5, 6) and adaptor proteins (ZO-1, α- and β-catenins) (Fig. 7) showed strong intercellular labeling of islet cells. Among the junctional proteins analyzed, only N-cadherin was specific to the endocrine pancreas, being detected in both beta (insulin-secreting) and alpha (glucagon-secreting) cells (Fig. 5e, f), while the others proteins were observed in both endocrine and exocrine parenchyma. VE-cadherin (Fig. 6a–d) and ZO-1 (Fig. 7a–d) were detected in the endothelium, being the VE-cadherin exclusively to the vascular compartment, as expected (Ahrens et al. 2003), and in the case of ZO-1, its association with vessels was confirmed by double immunolabeling with CD31 (Fig. 7a, inset), a recognized endothelial marker (Shim et al. 2015).

Prolonged HFd decreases junctional E-cadherin, which is not accompanied by changes in protein level in beta cells of diabetic mice. Confocal images a–d of E-cadherin (green, a and c) co-labeled with insulin (red, b and d) in pancreatic islets of control (panels a and b) and HFd mice (panels c and d). Note that 8 months of exposure to HFd diminishes E-cadherin labeling (c) at the intercellular contact region of beta cells (c, inset arrow) relative to controls (a, inset arrow) that was confirmed quantitatively (graph in e, ***p < 0.0001). All images are representative of islets of 6 animals per group from 3 independent experiments. Insets are zoomed images (1 × Zoom). Scale bars 50 µm. Lysates of isolated islets were immunoblotted for E-cadherin (panel f). After stripping, the same membrane was reblotted for anti-β-actin, for loading control (panel g). Islets from HFd mice showed no significant changes in E-cadherin levels in comparison with control islets (h). The bars represent the means + SEM of 5 membranes from 5 independent experiments (n = 6 mice per group/experiment)

Prolonged HFd induces N-cadherin redistribution, which is not accompanied by significant change in protein level in beta cells of diabetic mice. Images a and b show confocal views of N-cadherin in pancreatic islets of control (a) and HFd mice (b). Note that 8-month HFd induces redistribution of N-cadherin from the intercellular region to the cytoplasm of beta cells (arrows), as revealed by a significant decreased immunofluorescence at the cell–cell contact region (c) associated with an increased area of cells displaying diffuse cytoplasm labeling (d) in HFd islets as compared to control islets (***p < 0.0001). In e and f, co-localization of N-cadherin (green) and insulin (red) and glucagon (blue). All images are representative of islets of 5 animals per group from 3 independent experiments. Scale bars 50 µm. Lysates of isolated islets were immunoblotted for N-cadherin (panel e). β-actin served as a loading control (panel f). Islets from HFd mice showed no significant changes in N-cadherin levels relative to controls (g). Bars represent the means + SEM of 6 membranes from 6 independent experiments (n = 4 mice per group/experiment)

Increased level of intra-islet blood vessel-associated VE-cadherin was seen after prolonged exposure to HFd. Images a–d show VE-cadherin (green, a and c) and insulin immunostaining (red, b and d) in pancreatic islets of control (panels a and b) and HFd mice (panels c and d) by confocal microscopy. VE-cadherin was exclusively found in endothelial cells. HFd induces enhanced islet labeling of VE-cadherin (c) in comparison with control group (a). Quantitative assessment confirmed this (e, **p < 0.004) suggesting increased islet vascularization in diabetic mice. All images are representative of islets of 4–5 animals per group from 3 independent experiments. Scale bars 50 µm. Lysates from isolated islets were immunoblotted for VE-cadherin (panel f). β-actin served as a loading control (panel g). Islets from HFd mice show a significant increase in VE-cadherin protein levels relative to controls (h). The bars represent the means + SEM of 8 membranes from 5 independent experiments (n = 6 mice per group/experiment)

Prolonged HFd decreases α-catenin junctional content without altering ZO-1 or β-catenin labeling in beta cells of diabetic mice. Images a–n show ZO-1 (green, a and c), α-catenin (green, f and h), β-catenin (green, k and m) and the corresponding insulin co-immunostaining (in red) in pancreatic islets of control (panels b, g, l) and HFd mice (panels d, i, n) by confocal microscopy. Note that HFd islets have reduced labeling for α-catenin at the intercellular contact region of beta cells (h, i, inset, arrow) in comparison with control group (f, g, inset, arrow) that was confirmed quantitatively (graph in j, ***p < 0.001). However, no significant difference in ZO-1 (b vs. d and graph in e) and β-catenin islet labeling (l vs. n and graph in o) was observed. Inset in a shows colocalization of ZO-1 (green) and CD31 (magenta), an endothelial marker. All images are representative of islets of 4–6 animals per group from 5 independent experiments. Insets are zoomed images (1× Zoom). Scale bars 50 µm
Figure 4 shows images of islets that were double immunolabeled for E-cadherin (green, 4a, c) and insulin (red, 4b, d). The panels a and b show the same islet from control group, whereas the c and d figures present the same representative islet for the diabetic group. Note that islets from diabetic mice showed a decrease in E-cadherin labeling at the intercellular region (Fig. 4c) that was statistically significant in comparison with control islets as revealed by semi-quantitative analysis (p < 0.0001; Fig. 4e), indicating possible internalization of this CAM. Nevertheless, as shown in Fig. 4f–h, immunodetection of E-cadherin in pancreatic islets lysates showed no significant difference in the total protein content of this adhesion molecule between groups. The apparent discrepancy between the immunohistochemistry and the Western blot data could be explained by nonrecognition or decreased antibody binding after immunofluorescence reaction as result of modification of the E-cadherin molecule structure during internalization (for instance by phosphorylation; Collares-Buzato et al. 1998; Lawler et al. 2009; Singh et al. 2009), and/or “washing out” of intracellular soluble E-cadherin fraction due to relatively poor preservation after acetone fixation (Hoetelmans et al. 2001).
Islets from diabetic mice showed a cellular redistribution of N-cadherin from the intercellular site toward the cytoplasm (Fig. 5b). This was confirmed quantitatively, as a significant decrease in intercellular labeling of islet cells (Fig. 5c; p < 0.0001) as well as a significant increase in islet area displaying diffuse cytoplasmic labeling (Fig. 5d; p < 0.0001) were observed in HF-diet-fed mice. As mentioned before, both endocrine cells, beta- (e) and alpha (f) cells, expressed this CAM. No significant difference in total islet content of N-cadherin was observed in islet homogenates of diabetic and control mice (g–i).
As shown in Fig. 6, an increase in VE-cadherin staining in pancreatic islets of diabetic animals (Fig. 6c) was seen as compared to the controls (Fig. 6a). This was confirmed by quantitative measure of the labeling area of VE-cadherin (in pixels) normalized by islet area (Fig. 6e). Accordingly, a higher cell content for this CAM was detected in islet homogenates of diabetic mice relative to controls, by immunoblotting (Fig. 6f–h).
As depicted in Fig. 7, islets from diabetic mice showed a significant decrease in α-catenin immunolabeling at the intercellular contact region of beta cells as compared to islets of control mice (Fig. 7f, g). This was confirmed by quantitation of the junctional fluorescence degree within the islets from both experimental groups (Fig. 7j). However, ZO-1 (control, a–b; diabetic, c–d) and β-catenin (control, k–l; diabetic m–n) showed no statistically significant changes in islet distribution and total fluorescence degree between the two groups (Fig. 7e, o, respectively).
Islet homogenates from control and HF diet-fed mice were analyzed for ZO-1 (Fig. 8a–c), α-catenin (Fig. 8d–f) and β-catenin (Fig. 8g–i) protein expression by immunoblotting that showed no significant difference in the islet content of these proteins between the experimental groups.

No significant change in islet content of ZO-1, α- and β-catenins was seen in diabetic mice. Lysates of isolated islets were immunoblotted for ZO-1 (a), α-catenin (d) and β-catenin (g). β-actin was used as a loading control (b, e, h). Islets from HFd mice show no significant changes in ZO-1 (c), α-catenin (f) and β-catenin levels (i) in comparison with control islets. The bars represent the means + SEM of 7 membranes from 7 independent experiments (n = 8 mice per group/experiment)
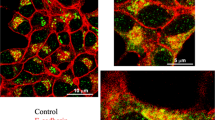
Since it has recently been suggested that cadherin-mediated adhesion, particularly through E- and N-cadherins, induces reorganization of cortical actin in beta cells (Parnaud et al. 2015), we went to see the F-actin distribution in pancreas sections of both experimental groups. As shown in Fig. 9, islet cells from diabetic mice displayed a significantly increased actin staining at the cell-to-cell contact region (Fig. 9b, inset) in comparison with those from control animals (Fig. 9a, inset), which was confirmed quantitatively (Fig. 9c).

Increased cell contact-associated F-actin was observed in islet cells of diabetic mice. Confocal images a and b show F-actin labeling with TRITC-conjugated phalloidin in pancreatic islets of control (a) and HFd mice (b). Within the islet, cytoskeletal F-actin was found in endothelial cells (arrowheads indicate vessels) as well as at the contact region between islet cells (insets, arrows). Note an increased actin labeling in islet cells of diabetic mice (b, inset) as compared to control islet cells (a, inset). Quantitative assessment confirmed this (c, ****p < 0.0001). All images are representative of islets of 7–8 animals per group from 3 independent experiments. Insets are zoomed images (1× Zoom). Scale bars 50 µm
RNAm expression of junctional proteins and insulin in pancreatic islets
Gene expression of junctional proteins, namely E-, N-, VE-cadherin, ZO-1, α-catenin, β-catenin, as well as insulin (isoforms 1 and 2) was assessed by qPCR in pools of pancreatic islets from control and HF diet-fed mice (Fig. 10). There was a statistically significant increase in gene expression of cdh5 (VE-cadherin, p < 0.05), tjp1 (ZO-1, p < 0.05), ins1 and ins2 (p < 0.001).

Effect of long-term HFd on the RNAm expression of junctional proteins and insulin in pancreatic islets from mice. mRNA expression was determined by qPCR in lysates of isolated islets from HFd diabetic and control mice. HFd islets showed a significant increase in cdh5 (VE-cadherin, *p < 0.05), tjp1 (ZO-1, *p < 0.05), ins1 and ins2 (**p < 0.001). Bars represent the means + SD of 6 mice per experiment from 3 to 4 independent experiments
Discussion
The main focus of this study was to investigate the impact of long-term exposure to HF diet on the distribution/expression of CAMs and associated proteins in pancreatic islet cells of mice. Modified diets, rich in fat, have been extensively used to induce obesity in rodents in order to study related metabolic alterations (Ahrén and Pacini 2002; Herrera, 2009; Oliveira et al. 2015; Peyot et al. 2010; Sandu et al. 2005; Surwit et al. 1988; Winzell and Ahrén 2004). Nevertheless, a flat comparison among these studies is usually difficult due to the differences found in the experiments with respect to nutritional composition and fat content of the diets (Sandu et al. 2005; Shirakawa et al. 2011) combined with differences in animal strains (Rossmeisl et al. 2003; Surwit et al. 1988), gender (Gómez-Pérez et al. 2008; Oliveira et al. 2015), and time frame of exposure to the diet used (Sone and Kagawa 2005; Oliveira et al. 2015). Taken this in consideration and the fact that there are relatively few works employing modified diets for prolonged periods (>6 months), we started this study by characterizing our animal model with respect to its metabolic state as well as the islet structure and beta cell function.
Severe impairment of the glucose homeostasis was observed in 8-month HF diet-fed animals that displayed, besides obesity, a significant insulin resistance associated with high fasting and postprandial hyperglycemia and moderate hyperinsulinemia. Morphological analysis of the endocrine pancreas of the HF diet-fed mice revealed no significant changes of islet appearance or cytoarchitecture, parameters that have been shown to be markedly altered in case of severe beta cell dysfunction and diabetes (Gomez-Dumm et al. 1990; Kai et al. 2013; Kilimnik et al. 2011; Kim et al. 2009; Sandu et al. 2005). However, a compensatory expansion of beta cell relative area per pancreas, at the expense of a decrease in alpha cell area, was observed in mice on prolonged HF diet. The beta cell expansion, which was due to beta cell hyperplasia and hypertrophy, is probably a response to a greater demand for insulin to maintain homeostasis and counteract insulin resistance of peripheral tissues in the HFd-treated mice (Kahn et al. 2006; Oliveira et al. 2015; Prentki and Nolan 2006).
Although we did not observe severe morphological changes in islets from HFd-fed mice, beta cells from these animals showed significant impairment of glucose-stimulated insulin secretion, when stimulated with 16.7 mM glucose. A similar result was reported by others, showing a lower insulin response to high glucose concentrations in islets of female C57 mice fed a HF diet for 6 months, in comparison with controls (Winzell et al. 2003). Intriguingly, in our animals, we found by qPCR that insulin 1 and insulin 2 mRNAs were markedly upregulated (fivefold the control level), that cannot be explained merely by the increase in proportion of beta cells within the islet observed, since this was relatively subtle (representing only an increment of 4.7 % above the control). Therefore, these data suggest a post-translational block or a failure in one or more steps of the insulin stimulus-secretion coupling process after prolonged HF diet.
One of the crucial steps for insulin secretion is an increase in intracellular calcium (Ashcroft and Rorsman 1989). In beta cells, calcium influx has an oscillatory pattern which is synchronized with the rhythm of insulin secretion and contributes to its normal pulsatile pattern (Gilon et al. 1993). Loss of regularity of basal plasma insulin oscillations is a well-known feature of type 2 diabetic patients (Bergsten 2000). In this study, we demonstrate for the first time that long-term exposure to HF diet significantly diminished the frequency of Ca2+ oscillations induced by glucose in isolated islets. Accordingly, it has been reported that islets from ob/ob mice display a desynchronization in calcium oscillations which was associated with perturbation of the pulsatility of insulin release (Ravier et al. 2002). Therefore, the alteration in intracellular calcium reported herein may one of the contributor factors to the reduction of insulin secretion observed in our HF diet-fed diabetic mice.
As compared with previous studies (using shorter-time exposure to HFd, such as 1–4 months)(Oliveira et al. 2015; Sone and Kagawa 2005), we described herein that 8-month treatment with a high-fat-containing diet induced more severe metabolic changes. This included a relatively high fast hyperglycemia (>130 mg/dL) associated with marked impairment of the stimulated insulin release, changes that were not reported after short exposure to HFd (Oliveira et al. 2015; Sone and Kagawa 2005). Nevertheless, in contrast with animals treated for 12 months with HFd, that displayed a significant decreased beta cell proliferation and mass (Sone and Kagawa 2005), our treated mice showed beta cell expansion and moderate hyperinsulinemia. Therefore, 8-month exposure to HFd resulted in an established stage of T2DM in mice but still associated with partial compensatory beta cell expansion.
After metabolic characterization of our animal model, we went to investigate the islet distribution and content of cell junctional proteins (i.e., E-, N-, VE-cadherins, α and β-catenins and ZO-1) and the cytoskeletal F-actin to establish a possible link between T2DM-associated beta cell dysfunction and alteration/impairment of the CAM-mediated cell–cell interactions within the endocrine pancreas. In control mice, we found that the CAMs and anchoring proteins studied (i.e., E-, N-, VE-cadherins and ZO-1, α- and β-catenins) were relatively well expressed at the intercellular site of islet cells. We observed that ZO-1 and cadherins/catenins are also expressed by cells of the exocrine parenchyma with the exception of N-cadherin that was exclusive to islet endocrine cells. Further, VE-cadherin, a well-known endothelial-associated cadherin (Ahrens et al. 2003), was detected only in blood vessels found in both exocrine and endocrine pancreas.
In diabetic HF-fed mice, we observed a significant decrease in intercellular content of E-, N-cadherin and α-catenin within pancreatic islets by immunohistochemistry. E-cadherin has been extensively studied in the endocrine pancreas. Several studies have highlighted the importance of this adhesion molecule for insulin secretion and beta cell mass by means of down- or overexpressing this CAM in vitro and in vivo conditions. Increased expression of E-cadherin at the intercellular region or E-cadherin/E-cadherin engagement are associated with augmented insulin secretion, total protein and/or mRNA content of pre-pro-insulin (Bosco et al. 2007; Calabrese et al. 2004; Carvell et al. 2007; Hauge-Evans et al. 1999; Parnaud et al. 2015; Yamagata et al. 2002). MIN6 pseudoislets, which express relatively more E-cadherin, show a higher insulin secretion than when these cells are organized as monolayers (Calabrese et al. 2004; Hauge-Evans et al. 1999). In addition, exposure of these cells to anti-E-cadherin neutralizing antibody inhibits glucose-stimulated insulin secretion from isolated islets (Calabrese et al. 2004; Rogers et al. 2007). Although currently under debate, the role of E-cadherin in the insulin secretory process may be indirect, influencing islet architecture, actin cytoskeleton remodeling or guaranteeing the structure of GJs, and thus intercellular communication (Carvell et al. 2007; Parnaud et al. 2015; Rogers et al. 2007). Furthermore, E-cadherin appears to mediate proliferation and survival of beta cells, where low expression is associated with high proliferation capacity, involving reduction in p27Kip1 levels, a cyclin-dependent kinase inhibitor (Carvell et al. 2007; Wakae-Takada et al. 2013), while high expression of this CAM results in beta cell protection against apoptosis induced by cytotoxic agents such as pro-inflammatory cytokines (Ngamjariyawat et al. 2013; Parnaud et al. 2011; Shirakawa et al. 2011). Yet, a possible activation of the canonical Wnt pathway may also be involved in cell proliferation mediated by E-cadherin (Wakae-Takada et al. 2013). Therefore, data from the present study show a decreased junctional E-cadherin content in conjunction with reduced junctional α-catenin in endocrine islet cells of diabetic mice may be related to the expansion (hyperplasia) of beta cells and reduced insulin secretory response (see below) found in these animals.
N-cadherin, whose function is not so well established in the endocrine pancreas, appears to perform a similar function to E-cadherin as regards insulin secretion and beta cell survival (Johansson et al. 2010; Parnaud et al. 2011, 2015). In human pancreas, N-cadherin is restricted to beta cells and appears to be important for maintenance and viability of this cell type (Parnaud et al. 2011). Nevertheless, according to our data on double labeling with glucagon, N-cadherin seems to be also expressed by non-beta cells (alpha cells) at least in mice. N-cadherin knockout mice displayed a relatively normal pancreatic morphogenesis and organogenesis but an impaired insulin secretion associated with a reduced number of beta cell secretory granules (Johansson et al. 2010). A direct role of N-cadherin, along with E-cadherin, in the insulin secretion process has been elegantly confirmed by Parnaud et al. (2015). By using chimeric proteins made of functional N- and E-cadherin ectodomains fused to Fc fragment of immunoglobulin, they demonstrated that cadherin-to-cadherin engagement significantly increased insulin secretion in single beta cells in vitro. This phenomenon involved a decrease in cortical actin barrier at cell–cell contacting area that in turn would facilitate the access of insulin granules to the plasma membrane and, therefore, insulin release (Parnaud et al. 2015). In order to investigate whether similar mechanism linking cadherin adhesion and actin remodeling would be observed in our in vivo model, we performed the phalloidin-labeling F-actin in pancreas sections of control and diabetic mice. Interestingly, we noticed an increase in F-actin labeling at beta cell contact region, which inversely correlated with the cadherin junctional content. Therefore, the decreased junctional E- and N-cadherin content observed in our diabetic mice is in agreement with their decreased secretory beta cell capacity, through possibly actin cytoskeleton remodeling.
Besides this decrease in junctional content, we found an increase in immunoreaction for N-cadherin at the cytoplasm in islet cells of diabetic mice. It is possible that the increase in cytoplasm content of this cadherin is result of a mere internalization of this protein from the intercellular compartment during this state, or alternatively the cytoplasmic N-cadherin may represent a pool of this molecule that is associated with cell structures or organelles (i.e., insulin granules; Johansson et al. 2010) whose function is yet to be determined.
The adhesive function of cadherins depends on their association with catenins (alpha, beta and gamma catenin and p120) and, in the absence of tight junction, also with ZO-1 (Aberle et al. 1996; Lewis et al. 1997; Miravet et al. 2003; Perez-Moreno et al. 2003; Yin and Green 2004). Previous studies have shown that α/β-catenins and ZO-1 do not function solely as structural components, but also mediate many cellular processes such as differentiation and proliferation in other tissues (Benmerah et al. 2003; Conacci-Sorrell et al. 2002; Murtaugh 2008; Spadaro et al. 2012) as well as in beta cells (Figeac et al. 2009). Our group demonstrated that the maturation of the insulin secretory machinery in vitro and in vivo occurs in parallel with increased expression of catenins and ZO-1 in pancreatic islets from newborn rats (Collares-Buzato et al. 2004; Santos-Silva et al. 2012). However, in the present work, we found no changes in the distribution and cellular content of β-catenin and ZO-1 in the islets of diabetic animals. Only α-catenin showed a decrease in immunostaining in the intercellular region in beta cells.
Additionally, we observed a significant increase in VE-cadherin immunoreactivity, islet protein content and RNAm expression in pancreatic islets of diabetic mice. VE-cadherin is expressed by endothelial cells and found exclusively at the adherens junction of endothelium of other organs (Ahrens et al. 2003; Breier et al. 1996; Corada et al. 1999; Dejana et al. 2001) and also in vessels of pancreas as reported here. ZO-1 also showed a similar staining pattern to VE-cadherin, suggesting an association of this protein with the vascular compartment in the pancreas (as confirmed by the co-localization with CD31), in addition to its presence at the intercellular contact region of pancreatic endocrine cells. Interestingly, we observed also an increase in mRNA expression of ZO-1 in the islets of HFd-fed mice. All together, these data strongly suggest an increase in vascularization of pancreatic islets in diabetic mice. Pancreatic islets are highly vascularized, which is directly related to its endocrine function. Several animal models of insulin resistance demonstrate vessel neogenesis (angiogenesis) and/or dilatation of existing vessels within islets (Agudo et al. 2012; Dai et al. 2013). Endothelial cells are involved with oxygen and nutrient supply to the endocrine cells but also appear to induce insulin expression, promote cell proliferation, and produce vasoactive, angiogenic and growth factors (reviewed by Zanone et al. 2008). Conversely, the beta cell-derived VEGF-A (vascular endothelial growth factor) induces capillary network formation and increases fenestration of the endothelium, resulting in increased vascular permeability (Zanone et al. 2008). Additionally, it has been shown that hyperglycemia leads to an increase in blood flow and islet capillary pressure (Zanone et al. 2008). This increase could, over time, contribute to the damage of the endothelium and thickening of the capillary wall, thereby decreasing perfusion of the islets. It is noteworthy that hyperglycemia induces the production of NO (nitric oxide) by the endothelium, which may result in cytotoxicity to the islets and thus impair insulin secretion (Henningsson et al. 2002; Zanone et al. 2008). Therefore, increased vascularization (as revealed by increased VE-cadherin islet expression) may be associated with the beta cell hyperplasia (considering the positive feedback of the endothelium upon beta cell proliferation) (Duvillie et al. 2002; Zanone et al. 2008) as well as with the impairment of insulin secretion (considering the release of deleterious endothelial factors under high glucose concentration) (Zanone et al. 2008) found in our diabetic mice.
In conclusion, our results showed that C57BL/6 mice after long-term exposure to HFd displayed metabolic manifestations of T2DM associated with an impaired glucose-stimulated insulin secretion by beta cells. The present work showed for the first time that the distribution/expression of N-, E- and VE-cadherins as well as α-catenin and F-actin in islet cells were significantly altered in obese and diabetic mice, which may be relevant to the T2DM pathogenesis. However, it remains to be determined whether these alterations in junctional protein distribution in the islets from HFd-fed mice are a cause or effect of beta cell changes seen during the pre-diabetic and diabetic condition. Jonas et al. (1999) demonstrated that chronic exposure to hyperglycemia triggers loss of beta cell differentiation in an animal model of diabetes. This suggests that alterations in the present study may be a consequence of cell dedifferentiation as this process is directly associated with junctional protein loss in other organs (Cannito et al. 2010; Luebke-Wheeler et al. 2009; Luther et al. 2005). However, experimental evidence indicates that junctional proteins may interfere directly with intercellular communication (Calabrese et al. 2004; Carvell et al. 2007; Rogers et al. 2007), beta cell proliferation/survival (Carvell et al. 2007; Parnaud et al. 2011; Wakae-Takada et al. 2013), and biosynthesis and release of insulin (Calabrese et al. 2004; Green et al. 2015; Hauge-Evans et al. 1999; Johansson et al. 2010; Parnaud et al. 2015). Therefore, a causal effect of junctional protein changes in diabetes-associated beta cell dysfunction is also plausible. One possible mechanism was suggested herein linking, for the first time, a relatively low E- and N-cadherin junctional content, a high submembranous actin content and impaired glucose-induced insulin release of beta cells in diabetic mice. Future studies employing transgenic and/or knockout mice for specific junctional proteins exposed to HFd may provide a valuable tool to investigate the exact role of beta cell–beta cell contact in the pathogenesis of T2DM.
References
Aberle H, Schwartz H, Kemler R (1996) Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem 61(4):514–523
Agudo J, Ayuso E, Jimenez V, Casellas A, Mallol C, Salavert A, Bosch F (2012) Vascular endothelial growth factor-mediated islet hypervascularization and inflammation contribute to progressive reduction of β-cell mass. Diabetes 61(11):2851–2861. doi:10.2337/db12-0134
Ahrén B, Pacini G (2002) Insufficient islet compensation to insulin resistance versus reduced glucose effectiveness in glucose-intolerant mice. Am J Physiol Endocrinol Metab 283(4):E738–E744
Ahrens T, Lambert M, Pertz O, Sasaki T, Schulthess T, Mège RM, Timpl R, Engel J (2003) Homoassociation of VE-cadherin follows a mechanism common to “classical” cadherins. J Mol Biol 325(4):733–742
Andralojc KM, Mercalli A, Nowak KW, Albarello L, Calcagno R, Luzi L, Bonifacio E, Doglioni C, Piemonti L (2009) Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia 52:486–493. doi:10.1007/s00125-008-1238-y
Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol 54:87–143
Benmerah A, Scott M, Poupon V, Marullo S (2003) Nuclear functions for plasma membrane associated proteins? Traffic 4:503–511
Bergsten P (2000) Pathophysiology of impaired pulsatile insulin release. Diabetes Metab Res Rev 16:179–191
Bosco D, Rouiller DG, Halban PA (2007) Differential expression of E-cadherin at the surface of rat beta-cells as a marker of functional heterogeneity. J Endocrinol 194(1):21–29
Breier G, Breviario F, Caveda L, Berthier R, Schnurch H, Gotsch U, Vestweber D, Risau W, Dejana E (1996) Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood 87:630–641
Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC (2005) Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 53(9):1087–1097
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52(1):102–110
Calabrese A, Caton D, Meda P (2004) Differentiating the effects of Cx36 and E-cadherin for proper insulin secretion of MIN6 cells. Exp Cell Res 294:379–391
Cannito S, Novo E, Bonzo LV, Busletta C, Colombatto S, Parola M (2010) Epithelial–mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal 12(12):1383–1430. doi:10.1089/ars.2009.2737
Carvalho CPF, Martins JC, Da Cunha DA, Boschero AC, Collares-Buzato CB (2006) Histomorphology and ultrastructure of pancreatic islet tissue during in vivo maturation of rat pancreas. Ann Anat 188:221–234
Carvalho CPF, Barbosa HC, Britan A, Santos-Silva JC, Boschero AC, Meda P, Collares-Buzato CB (2010) Beta cell coupling and connexin expression change during the functional maturation of rat pancreatic islets. Diabetologia 53(7):1428–1437. doi:10.1007/s00125-010-1726-8
Carvalho CP, Oliveira RB, Britan A, Santos-Silva JC, Boschero AC, Meda P, Collares-Buzato CB (2012) Impaired β-cell-β-cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am J Physiol Endocrinol Metab 303:144–145. doi:10.1152/ajpendo.00489.2011
Carvell MJ, Marsh PJ, Persaud SJ, Jones PM (2007) E-cadherin interactions regulate beta-cell proliferation in islet-like structures. Cell Physiol Biochem 20(5):617–626
Chang-Chen KJ, Mullur R, Bernal-Mizrachi E (2008) Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord 9(4):329–343. doi:10.1007/s11154-008-9101-5
Collares-Buzato CB, Jepson MA, Simmons NL, Hirst BH (1998) Increased tyrosine phosphorylation causes redistribution of adherens junction and tight junction proteins and perturbs paracellular barrier function in MDCK epithelia. Eur J Cell Biol 76:85–92
Collares-Buzato CB, Carvalho CPF, Furtado AG, Boschero AC (2004) Upregulation of the expression of tight and adherens junction-associated proteins during maturation of neonatal pancreatic islets in vitro. J Mol Histol 35:811–822
Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A (2002) The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest 109:987–991
Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM, Ward PA, Dejana E (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA 96:9815–9820
Coronel-Cruz C, Hernández-Tellez B, López-Vancell R, López-Vidal Y, Berumen J et al (2013) Connexin 30.2 is expressed in mouse pancreatic beta cells. Biochem Biophys Res Commun 438(4):772–777. doi:10.1016/j.bbrc.2013.06.100
Dai C, Brissov M, Reinert RB, Nyman L, Liu EH, Thompson C, Powers AC (2013) Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes 62(12):4144–4153. doi:10.2337/db12-1657
Dejana E, Spagnuolo R, Bazzoni G (2001) Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb Haemost 86:308–315
Drolet MC, Roussel E, Deshaies Y, Couet J, Arsenault M (2006) A high fat/high carbohydrate diet induces aortic valve disease in C57BL/6 J mice. J Am Coll Cardiol 47(4):850–855
Duvillie B, Currie C, Chrones T, Bucchini D, Jami J, Joshi RL, Hill DJ (2002) Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to beta-cell hyperplasia in mutant mice lacking insulin. Endocrinology 143:1530–1537
Ebnet K (2008) Organization of multiprotein complexes at cell-cell junction. Histochem Cell Biol 130:1–20. doi:10.1007/s00418-008-0418-7
Esni F, Täljedal IB, Perl AK, Cremer H, Christofori G, Semb H (1999) Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J Cell Biol 144(2):325–337
Figeac FBU, Faro M, Chelali N, Portha B, Movassat J (2009) Neonatal growth and regeneration of β-cells are regulated by the Wnt/β-catenin signaling in normal and diabetic rats. Am J Physiol Endocrinol Metab 298:245–256. doi:10.1152/ajpendo.00538.2009
Gilon P, Shepherd RM, Henquin JC (1993) Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidenced in single pancreatic islets. J Biol Chem 268:22265–22268
Gomez-Dumm CL, Semino MC, Gagliardino JJ (1990) Sequential morphological changes in pancreatic islets of spontaneously diabetic rats. Pancreas 5(5):533–539
Gómez-Pérez Y, Amengual-Cladera E, Català-Niell A, Thomàs-Moyà E, Gianotti M, Proenza AM, Lladó I (2008) Gender dimorphism in high fat diet-induced insulin resistance in skeletal muscle of aged rats. Cell Physiol Biochem 22:539–548
Green AD, Vasu S, McClenaghan NH, Flatt PR (2015) Pseudoislet formation enhances gene expression, insulin secretion and cytoprotective mechanisms of clonal human insulin-secreting 1.1B4 cells. Pflugers Arch 467(10):2219–2228. doi:10.1007/s00424-014-1681-1
Harris TJ, Tepass U (2010) Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol 11(7):502–514. doi:10.1038/nrm2927
Hauge-Evans AC, Squires PE, Persaud SJ, Jones PM (1999) Pancreatic beta-cell-to-beta-cell interactions are required for integrated responses to nutrient stimuli: enhanced Ca2+ and insulin secretory responses of MIN6 pseudoislets. Diabetes 48(7):1402–1408
Henningsson R, Salehi A, Lundquist I (2002) Role of nitric oxide synthase isoforms in glucose-stimulated insulin release. Am J Physiol Cell Physiol 283:296–304
Herrera PL (2009) Transgenic and other experimental models of pancreas and islet damage. Diabetes Obes Metab 11(4):81–86. doi:10.1111/j.1463-1326.2009.01104.x
Hoetelmans RW, Prins FA, Cornelese-ten Velde I, van der Meer J, van de Velde CJ, van Dierendonck JH (2001) Effects of acetone, methanol, or paraformaldehyde on cellular structure, visualized by reflection contrast microscopy and transmission and scanning electron microscopy. Appl Immunohistochem Mol Morphol 9:346–351
Jain R, Lammert E (2009) Cell–cell interactions in the endocrine pancreas. Diabetes Obes Metab 11(Suppl. 4):159–167. doi:10.1111/j.1463-1326.2009.01102.x
Johansson JK, Voss U, Kesavan G, Kostetskii I, Wierup N, Radice GL, Semb H (2010) N-cadherin is dispensable for pancreas development but required for beta-cell granule turnover. Genesis 48(6):374–381. doi:10.1002/dvg.20628
Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, Weir GC (1999) Chronic hyperglycemia triggers loss of pancreatic cell differentiation in an animal model of diabetes. J Biol Chem 274(20):14112–14121
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 14:840–846
Kai AKL, Lam AKM, Chen Y, Tai ACP, Zhang X, Lai AKW, Yeung PKK, Tam S, Wang J, Lam KS, Vanhoutte PM, Bos JL, Chung SSM, Xu A, Chung SK (2013) Exchange protein activated by cAMP 1 (Epac1)-deficient mice develop β-cell dysfunction and metabolic syndrome. FASEB J 27:4122–4135
Kilimnik G, Zhao B, Jo J, Periwal V, Witkowski P, Misawa R, Hara M (2011) Altered islet composition and disproportionate loss of large islets in patients with type 2 diabetes. PLoS One 6(11):e27445. doi:10.1371/journal.pone.0027445
Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M (2009) Islet architecture: a comparative study. Islets 1(2):129–136. doi:10.4161/isl.1.2.9480
Lawler K, O’Sullivan G, Long A, Kenny D (2009) Shear stress induces internalization of E-cadherin and invasiveness in metastatic oesophageal cancer cells by a Src-dependent pathway. Cancer Sci 100:1082–1087
Leite AR, Carvalho CP, Furtado AG, Barbosa HC, Boschero AC, Collares-Buzato CB (2005) Co-expression and regulation of connexins 36 and 43 in cultured neonatal rat pancreatic islets. Can J Physiol Pharmacol 83:142–151
Lewis JE, Wah JK 3rd, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ (1997) Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol 136(4):919–934
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T) method. Methods 25(4):402–408
Luebke-Wheeler JL, Nedredal G, Yee L, Amiot BP, Nyberg SL (2009) E-cadherin protects primary hepatocyte spheroids from cell death by a caspase-independent mechanism. Cell Transplant 18(12):1281–1287. doi:10.3727/096368909X474258
Luther MJ, Davies E, Muller D, Harrison M, Bone AJ, Persaud SJ, Jones PM (2005) Cell-to-cell contact influences proliferative marker expression and apoptosis in MIN6 cells grown in islet-like structures. Am J Physiol Endocrinol Metab 288(3):E502–E509
Meda P (2013) Protein-mediated interactions of pancreatic islet cells. Scientifica 2013:621249. doi:10.1155/2013/621249
Mese G, Richard G, White TW (2007) Gap junctions: basic structure and functions. J Invest Dermatol 127(11):2516–2524
Miravet S, Piedra J, Castaño J, Raurell I, Francí C, Duñach M, García de Herreros A (2003) Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol 23(20):7391–7402
Murtaugh LC (2008) The what, where, when and how of Wnt/beta-catenin signaling in pancreas development. Organogenesis 4(2):81–86
Ngamjariyawat A, Turpaev K, Vasylovska S, Kozlova EN, Welsh N (2013) Co-culture of neural crest stem cells (NCSC) and insulin producing beta-TC6 cells results in cadherin junctions and protection against cytokine-induced beta-cell death. PLoS One 8(4):e61828. doi:10.1371/journal.pone.0061828
Nlend RN, Michon L, Bavamian S, Boucard N, Caille D, Cancela J, Charollais A, Charpantier E, Klee P, Peyrou M, Populaire C, Zulianello L, Meda P (2006) Connexin36 and pancreatic beta cell functions. Arch Physiol Biochem 112:74–81
Oliveira RB, Maschio DA, Carvalho CPF, Collares-Buzato CB (2015) Influence of gender and time diet exposure on endocrine pancreas remodeling in response to high fat diet-induced metabolic disturbances in mice. Ann Anat 200:88–97. doi:10.1016/j.aanat.2015.01.007
Orci L (1976) The microanatomy of the islets of Langerhans. Metabolism 25:1303–1313
Parnaud G, Gonelle-Gispert C, Morel P, Giovannoni L, Muller YD, Meier R, Borot S, Berney T, Bosco D (2011) Cadherin engagement protects human beta-cells from apoptosis. Endocrinology 152(12):4601–4609. doi:10.1210/en.2011-1286
Parnaud G, Lavallard V, Bedat B, Matthey-Doret D, Morel P, Berney T, Bosco D (2015) Cadherin engagement improves insulin secretion of single human β-cells. Diabetes 64:887–896. doi:10.2337/db14-0257
Perez-Moreno M, Jamora C, Fuchs E (2003) Sticky business: orchestrating cellular signals at adherens junctions. Cell 112(4):535–548
Peyot M-L, Pepin E, Lamontagne J, Latour MG, Zarrouki B, Lussier R, Pineda M, Jetton TL, Madiraju MSR, Joly E, Prentki M (2010) B-cell failure in diet-induced obese mice stratified according to body weight gain: secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes 59:2178–2187
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116:1802–1812
Ramakers C, Ruijter JM, Deprez RH, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339(1):62–66
Ravier MA, Sehlin J, Henquin JC (2002) Disorganization of cytoplasmic Ca(2+) oscillations and pulsatile insulin secretion in islets from ob/ob mice. Diabetologia 45(8):1154–1163
Rieck S, White P, Schug J, Fox AJ, Smirnova O, Gao N, Kaestner KH (2009) The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol 23(10):1702–1712. doi:10.1210/me.2009-0144
Rogers GJ, Hodgkin MN, Squires PE (2007) E-cadherin and cell adhesion: a role in architecture and function in the pancreatic islet. Cell Physiol Biochem 20(6):987–994
Rossmeisl M, Rim JS, Koza RA, Kozak LP (2003) Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes 52:1958–1966
Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H (2005) Insulin resistance and type 2 diabetes in high-fat–fed mice are linked to high glycotoxin intake. Diabetes 54:2314–2319
Santos-Silva JC, Carvalho CP, de Oliveira RB, Boschero AC, Collares-Buzato CB (2012) Cell-to-cell contact dependence and junctional protein content are correlated with in vivo maturation of pancreatic beta cells. Can J Physiol Pharmacol 90:837–850. doi:10.1139/y2012-064
Shafrir E, Ziv E, Mosthaf L (1999) Nutritionally induced insulin resistance and receptor defect leading to beta-cell failure in animal models. Ann N Y Acad Sci 892:223–246
Shim Y, Nam MH, Hyuk SW, Yoon SY, Song JM (2015) Concurrent hypermulticolor monitoring of CD31, CD34, CD45 and CD146 endothelial progenitor cell markers for acute myocardial infarction. Anal Chim Acta 853:501–507. doi:10.1016/j.aca.2014.10.036
Shirakawa J, Amo K, Ohminami H, Orime K, Togashi Y, Ito Y, Terauchi Y (2011) Protective effects of dipeptidyl peptidase-4 (DPP-4) inhibitor against increased β cell apoptosis induced by dietary sucrose and linoleic acid in mice with diabetes. J Biol Chem 286(29):25467–25476. doi:10.1074/jbc.M110.217216
Singh R, Lei P, Andreadis ST (2009) PKC-delta binds to E-cadherin and mediates EGF-induced cell scattering. Exp Cell Res 315:2899–2913. doi:10.1016/j.yexcr.2009.07.002
Sone H, Kagawa Y (2005) Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48:58–67
Spadaro D, Tapia R, Pulimeno P, Citi S (2012) The control of gene expression and cell proliferation by the epithelial apical junctional complex. Essays Biochem 53:83–93. doi:10.1042/bse0530083
Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN (1988) Diet-induced type II diabetes in C57BL/6 J mice. Diabetes 37(9):1163–1167
Tripathy D, Chavez AO (2010) Defects in insulin secretion and action in the pathogenesis of type 2 diabetes mellitus. Curr Diab Rep 10(3):184–191. doi:10.1007/s11892-010-0115-5
Wakae-Takada N, Xuan S, Watanabe K, Meda P, Leib RL (2013) Molecular basis for the regulation of islet beta cell mass in mice: the role of E-cadherin. Diabetologia 56(4):856–866. doi:10.1007/s00125-012-2824-6
Winzell MS, Ahrén B (2004) The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53(suppl 3):215–219
Winzell MS, Holm C, Ahrén B (2003) Downregulation of islet hormone-sensitive lipase during long-term high-fat feeding. Biochem Biophys Res Commun 304:273–278
Yamagata K, Nammo T, Moriwaki M, Ihara A, Iizuka K, Yang Q, Satoh T, Li M, Uenaka R, Okita K, Iwahashi H, Zhu Q, Cao Y, Imagawa A, Tochino Y, Hanafusa T, Miyagawa J, Matsuzawa Y (2002) Overexpression of dominant-negative mutant hepatocyte nuclear factor-1 alpha in pancreatic beta-cells causes abnormal islet architecture with decreased expression of E-cadherin, reduced beta-cell proliferation, and diabetes. Diabetes 51(1):114–123
Yin T, Green KJ (2004) Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol 15(6):665–677
Zanone MM, Favaro E, Camussi G (2008) From endothelial to beta cells: insights into pancreatic islet microendothelium. Curr Diabetes Rev 4(1):1–9
Acknowledgments
The authors thank Célia Garcia for technical assistance, Luiza Martinez Perdigueiro for helping with morphometric analysis, and Profs. Antonio Carlos Boschero and Everardo Carneiro for the use of their laboratory facilities for RIE and intracellular calcium measurements. We thank the staff of the Hemocentro (Medical School) and Life Sciences Core Facility (LaCTAD) from University of Campinas (UNICAMP) for helping with confocal microscopy imaging. The confocal scanning laser microscopes used were acquired under the Grants # 2000/05137-4 and 2009/54129-9. This work was funded by grants from FAPESP (Grant # 2010/50789-1), CNPq, FAEPEX/UNICAMP and CAPES/PROEX (Brazil). VTFLF was recipient of a PhD CAPES-DINTER fellowship (Brazil). CBC-B are supported by a Research Fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil, Grants # 307163/2012-1; 304991/2015-5). This work has been presented at the 2014 Annual ASCB/IFCB Meeting and partially published in abstract form (Mol Biol Cel 25, 448 (Abstract # P591).
Author information
Authors and Affiliations
Corresponding author
Additional information
Viviane Tannuri F. L. Falcão and Daniela A. Maschio have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Falcão, V.T.F.L., Maschio, D.A., de Fontes, C.C. et al. Reduced insulin secretion function is associated with pancreatic islet redistribution of cell adhesion molecules (CAMs) in diabetic mice after prolonged high-fat diet. Histochem Cell Biol 146, 13–31 (2016). https://doi.org/10.1007/s00418-016-1428-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-016-1428-5