
Abstract
M2 macrophages showed large endocytotic structures, very different from classical macropinosomes that we named megapinosomes. As observed in the scanning electron microscope, megapinosome formation started with a large (diameter of several micrometers) invagination of the plasma membrane. When the invagination was almost completed, the remaining opening was closed by an actinomorphous centripetal arrangement of many (about 50–100) microvilli-like structures. In transmission electron microscopy using high-pressure freezing, we observed that the megapinosome was filled with a trabecular meshwork that originated from the highly structured plasma membrane. The trabecular meshwork was topologically part of the cytosol and separated from the extracellular fluid by a lipid bilayer. According to ultrastructural features, we could define different phases of megapinosome formation and decay. Megapinosomes became more frequent when M2 macrophages were inoculated with human cytomegalovirus. We did not find megapinosome formation in M1 macrophages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The basic function of endocytosis is the uptake of extracellular particles and/or fluid by invagination and fusion of the plasma membrane to form an internalized transport vesicle (Alberts et al. 2014). Pinocytosis refers specifically to the uptake of extracellular fluid, and it is currently divided into several subtypes (Conner and Schmid 2003; Mercer and Helenius 2009, 2012 and others). In the majority of cells, the unspecific uptake of fluid, solutes, and various ligands attached to the plasma membrane is primarily dependent on the process of macropinocytosis (Mercer and Helenius 2012). The vesicles formed by macropinocytosis are larger (up to 10 µm according to Mercer and Helenius 2012; and to Rasmussen and Vilhardt 2015) than the vesicles resulting from the other types of pinocytosis (50–150 nm according to Alberts et al. 2014). The structural characteristic feature of macropinocytosis is that membrane ruffles or lamellipodiae are produced which collapse back onto the cell. The tips of the tubular structures, thereby, fuse with the cell membrane, and a liquid-filled vesicle, the macropinosome, is formed (Lim and Gleeson 2011; Mercer and Helenius 2012; Alberts et al. 2014 and others). According to Mercer and Helenius (2012), referring to Doherty and McMahon (2009), however, “the classification of endocytic mechanism is still far from being complete.”
In this work, we observed and described large endocytic structures on macrophages that showed clear structural differences from the already known pinocytic structures and especially from macropinosomes. We, therefore, called them megapinosomes and the process leading to their formation megapinocytosis.
Two morphologically and functionally distinct types of macrophages were produced and analyzed in this study: the pro-inflammatory M1 macrophages and the anti-inflammatory M2 macrophages. As reported previously by Bayer et al. (2013), the two types of macrophages exhibited different susceptibilities to infection with the herpesvirus HCMV (human cytomegalovirus). M2 macrophages have a much higher susceptibility than M1 macrophages.
Materials and methods
Cell cultures
Human M1 and M2 macrophages were obtained as described by Bayer et al. (2013). Briefly monocytes were cultivated with either recombinant human granulocyte–macrophage colony-stimulating factor (rhGM-CSF) for polarization to M1 macrophages or rhM-CSF for M2 macrophages. Cells were incubated for 7 days in an incubator at 5 % CO2 and 37 °C. At day 3, half of the medium was changed and the growth factors replenished. Prior to infection, M1 and M2 macrophages were counted, resuspended in RPMI 10 % FCS without growth factors, and seeded in 96 wells. Then, sapphire disks (3 mm in diameter; Engineering Office M. Wohlwend GmbH, Sennwald, Switzerland) coated with approximately 20-nm layer of carbon (as described by Villinger et al. 2014) were added to allow the cells to adhere on them. For two experiments, cell cultures were inoculated with cell-free viral stocks of human cytomegalovirus (HCMV; Sinzger et al. 2008; endotheliotropic strain TB40E, kindly provided by C. Sinzger, University of Ulm, Germany, multiplicity of infection 50). For inoculation, the cell cultures were cooled to 4 °C, the virus was added, and the samples were kept cold (4 °C) for 1 h. Then cell cultures were washed three times with medium (kept at 4 °C) and then transferred to 37 °C and kept at this temperature for 5, 15, 30, or 90 min prior to preparation for electron microscopy as described below.
Scanning electron microscopy (SEM)
The sapphire disks with the adherent macrophages were prepared as described in Walther et al. (2010) with some modifications. The samples were fixed with 4 % paraformaldehyde and 2 % glutaraldehyde for 1 h. Samples were dehydrated in a graded series of propanol solutions and afterward critical point dried using CO2. Afterward, samples were coated with about 10 nm of carbon by electron beam evaporation. Secondary electron images were recorded with an Hitachi S-5200 field emission SEM (Hitachi, Tokyo, Japan) at an acceleration voltage of 4 kV. For stereo pairs (supplementary material), the sample was tilted by plus/minus 3.5°. This stereo pairs have also been used for the height diagrams (Figs. 2b, e, g, 3c), by measuring the parallaxis of surface features. According to Boyde (1970), the height difference z between two surface features was then calculated with the formula
where p is the difference in the parallaxis in between two surface features and 7° is the total tilting angle.
Transmission electron microscopy (TEM)
The sapphire disks with the cells on it were clamped in between two aluminum planchettes so that the cells were protected in a 100-µm-deep cavity (Buser and Walther 2008). Samples were high pressure frozen with a Wohlwend HPF Compact 01 high-pressure freezer (Engineering Office M. Wohlwend GmbH). Freeze substitution was performed as described by Walther and Ziegler (2002). The substitution medium consisted of acetone with 0.2 % osmium tetroxide, 0.1 % uranyl acetate, and 5 % of water for good contrast of the membranes. After substitution for 18 h from −90 °C to room temperature, samples were washed with acetone and gradually embedded in Epon. Ultrathin sections (75–80 nm) were cut parallel to the sapphire disk that served as a support for the adherent cells with a Leica Ultracut UCT ultramicrotome using a diamond knife (Diatome, Biel, Switzerland). To confirm that the data were comparable, sections in a distance of about 500 to 800 nm from the sapphire disk were used for Figs. 1, 4, and 5 and for the quantitative analysis. Sections were analyzed with a Jeol 1400 TEM (Jeol, Tokyo, Japan), and the images were digitally recorded with a Veleta camera (Olympus, Münster, Germany).

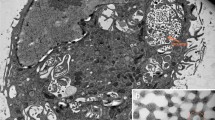
a A Megapinosome filled with the trabecular meshwork that irradiates from the cytosol (stars). b The trabecular meshwork (topologically equivalent to the cytosol) consists of knots and bridges. The bridges are concave. c Another megapinosome with another orientation of the trabecular meshwork. d A portion of a megapinosome with mostly longitudinal orientation of the filamentous structures
Each cell was first imaged as an overview (×6 k or ×8 k). Details were imaged at ×10, 12, 15, or 40 k.
For the serial section reconstruction shown in Fig. 6, serial sections with a thickness of 100 nm were collected and mounted on Formvar-coated single-slot copper grids as described in Villinger et al. (2014). Visualization of the megapinosome complex was done with the software Amira (FEI, Eindhoven, the Netherlands).
The presented results are based on quantitative and morphological analysis of 4603 electron microscopic images of 1981 macrophages. A total of 978 thin sections of M1 macrophages and 1003 thin sections of M2 macrophages were analyzed. We found megapinosome complexes on 409 (387 HCMV inoculated/22 MOCK) M2 macrophages. Some cells contained more than one megapinosome complex. For Table 1, only images of macrophages with only one megapinosome complex were used (in cell sections with more than one megapinosome complex, the elements of the complexes could not be clearly assigned to one complex; therefore, they were not considered for the statistical evaluation). In addition, for Table 1 only images of macrophages have been used, where no parts of the cell’s section were hidden by, e.g., the grid bar.
Quantitative particle counting (Fig. 7) Viral particles (virions, non-infectious enveloped particles, dense bodies, and naked capsids) could be clearly identified on the base of their characteristic ultrastructure. Extracellular and intracellular (inside vesicles or free in the cytoplasm) viral particles were differentially quantified using a recording sheet. We analyzed thin sections of 978 M1 macrophages and 1003 M2 macrophages. Morphological structures of megapinosomes in M2 macrophages: Morphological structures of megapinosomes or megapinosome complexes were defined and detected by means of questionnaire. Statistical analysis of the results was performed using Pearson’s Chi-square test, except for 90 min where the exact Fisher test was applied, since the expected frequency was less than 5. Differences with a P value of 0.05 were considered significant.
Immunofluorescence analysis of HCMV immediate-early proteins
Monoclonal antibodies (MAb) reactive against the immediate-early viral protein IE-1-2 (Argene-Biosoft, Varilhes, France) were chosen to detect viral proteins characteristic of the first phases of the HCMV replication cycle. M1 and M2 macrophages were seeded in µ-Slide 8 wells (Ibidi, Martinsried, Germany), and mock or TB40E infected (multiplicity of infection of 50). 24 h post-infection the cells were fixed with 4 % formaldehyde, permeabilized with 0.2 % Triton X-100, and probed with MAbs against IE-1-2, followed by incubation with Alexa 488-conjugated goat anti-mouse Ig (ICN Biomedical, Eschwege, Germany). Staining was detected using a Zeiss Axioskop 2 fluorescence microscope (Zeiss, Oberkochen, Germany).
Results
We found large endocytotic structures (diameter of several micrometers) in M2 macrophages that contained a trabecular meshwork of cytosolic elements and named them megapinosomes. Megapinosomes were frequently accompanied by additional vesicles that contribute to build a superstructure that we named megapinosome complex. When macrophages were inoculated with human cytomegalovirus (HCMV), these structures became more frequent in M2 macrophages but were not observed in M1 macrophages.
Megapinosome formation and decay
The megapinosome was filled with a trabecular meshwork. As shown in Fig. 1a, the trabecular meshwork was in continuity with the cytosol and was, therefore, topologically equivalent with the cytosol. The meshwork was confined toward the extracellular space and/or the lumen by a membrane bilayer. The meshwork consisted of knots and convex bridges with a diameter between 40 and 80 nm forming a coherent structure (Fig. 1b) with a regular distance between two knots of about 80–200 nm. Since the filamentous structures shown in Fig. 1c and d have the same diameter as the structures shown in Fig. 1b (40–80 nm), we concluded that they represent longitudinal sections of megapinosomes.
The structures found in the megapinosomes strongly resembled macrophages’ cell surface structures. Therefore, we investigated M2 macrophage surfaces with the SEM in order to search for early phases of megapinosome formation (Fig. 2a). The boxed area was enriched in microvilli-like structures and formed a depression, as visible in the height profile (Fig. 2b) of the macrophage surface that was calculated from the stereo pair shown in the supplementary material. The concave surfaces were covered with membrane ruffles and microvilli-like structures (Fig. 2c) that had the same diameter as the trabecular meshwork structures of megapinosomes shown in Fig. 1. Often microvilli-like structures rose at the edges of the ruffles (arrows in Fig. 2c), which is not typical for microvilli; therefore, we choose the expression “microvilli-like structures.” We assume that this depression represents an early phase of megapinosome formation. We also found macrophages with deeper depressions, or cavities (Fig. 2d; height profile in Fig. 2e), that again became clearly visible in the stereo pair and in the supplementary movie. For comparison, Fig. 2f shows a M1 macrophage without a depression on the cell surface (height profile Fig. 2g).

a SEM image of an early phase megapinosome formation in a M2 macrophage. Arrows indicate the position of the height diagram (b). The macrophage has a central depression. c Higher magnification of the boxed area at the center of the forming megapinosome. The central area is rich in microvilli-like structures. At the border, surface ruffles are dominating. Arrows point to microvilli-like structures originating from surface ruffles. d Represents a M2 macrophage at a later stage of megapinosome formation. At this stage, the central area is completely full of microvilli-like structures surrounded by ruffles and the depression is deeper (e). f, g Show a M1 macrophage lacking both area of surface depression and dense microvilli-like structures
Figure 3a and b shows a M2 macrophage with a putative later phase megapinocytosis. The depression of the cell surface forms a cavity that becomes surrounded by a centripetal actinomorphous arrangement of microvilli-like structures. The microvilli-like structures present at the peripheral rim as well as in the central deep portion of the cavity originate from membrane ruffles (Fig. 3b). Figure 3d represents a megapinosome open toward the extracellular space. We assume that this structure represents the same phase of megapinosome formation as shown in Fig. 3a, b. All together, these data suggest that the trabecular meshwork structures originate from surface microvilli-like structures and surface ruffles. Figure 3e, f represents the latest phase of megapinosome formation and closure. The centripetal arrangement of microvilli-like structures on the cell surface is now closed.

a M2 macrophage with a putative late phase of megapinosome formation. b Higher magnification of the large central depression that still contains microvilli-like structures. The height profile (c) indicates the depth of the depression (it becomes much clearer in the stereo pair and movie 1 in the supplementary files). A similar stage is shown in (d) on a TEM thin section showing an infolding with microvilli-like structures, still open to the extracellular space. e, f Represent the last stage of megapinosome formation, when the aperture is closed by an actinomorphous ring of microvilli-like structures, originating from membrane ruffles
A thin section of a polar megapinosome complex is shown in Fig. 4. The megapinosome complex consists of three zones. In the perinuclear portion, the vacuole (dark turquoise) is filled with the complex reticulated membrane structure constituting the trabecular meshwork. The dark gray content of this trabecular meshwork is topologically equivalent to the cytosol. In the central portion of the vacuole colored in middle turquoise, the meshwork is present but less dense. Finally, in the light turquoise area the meshwork is even less dense and had almost disappeared.

A Megapinosome complex (artificially colored). The polar structure showed in dark turquoise represents the dense trabecular meshwork in the vacuole. The meshwork is less dense in the middle turquoise area and had almost disappeared in the bright turquoise area
We hypothesize that megapinosomes have a complex life cycle and we propose to group the TEM images into a temporally ordered series of five phases. Phase 1 is characterized by a deep depression of the cell surface and an accumulation of microvilli-like structures. The following phases can be interpreted as a gradual fragmentation of this endocytotically formed megapinosome into smaller compartments. In phase 2 (Fig. 5a), the trabecular meshwork area is larger than the rest of the megapinosome complex. In phase 3 (Fig. 5b) on the contrary, the trabecular meshwork area is smaller than the rest of the megapinosome complex. In phase 4 (Fig. 5c), the trabecular meshwork is only rudimental or has completely disappeared. The megapinosome complex appears to have a polar organization from phase 2 to phase 4 (Fig. 5a–c). In phase 5, neither a trabecular meshwork nor a polarity of the megapinosome complex can be detected (Fig. 5d). In this phase, the megapinosome complex has completely decayed into vesicles, possibly endosomes.

Shows phases of the gradual fragmentation of the megapinosome. In an early phase, the trabecular meshwork area is larger than the rest of the complex (a). Later on (b), it becomes smaller than the rest of the complex. Then (c) the trabecular meshwork becomes rudimental or has disappeared. At last (d) no trabecular meshwork and, therefore, no polarity of the megapinosome complex can be detected. Virions are marked with an arrow. Some virions are boxed and enlarged in the insets
Figure 6 provides a 3D reconstruction of a serial section analysis of a cell with a megapinosome and a megapinosome complex. This cell shows a stage of megapinocytosis, where megapinosome and megapinosome complex are completely surrounded by cytoplasm in all sections; this is especially well visible in Movie 2 of the supplementary material. The reconstruction proves that megapinosomes exist without any connection to the extracellular space. Therefore, megapinocytosis is a real endocytotic process.

3D reconstruction of a serial section analysis of a cell with a megapinosome (red) a megapinosome complex (red and orange) and a nucleus (beige). This cell shows a stage of megapinocytosis, where megapinosome and megapinosome complex are completely surrounded by cytoplasm in all sections; this is especially well visible in Movie 2 of the supplementary material. The reconstruction proves that megapinosomes exist without any connection to the extracellular space. Therefore, megapinocytosis is a real endocytotic process
Table 1 shows a statistic evaluation of 143 images obtained from M2 macrophages at the different phases of megapinosome life cycle. Two megapinosome complexes showed phase 2, 22 megapinosome complex showed phase 3, 53 megapinosome complexes showed phase 4, and 66 megapinosome complexes showed phase 5. The average length diameter of the (more or less elliptic) megapinosomes’ section was 5.76 µm (SD = 2.7 µm). The spread was from 1.13 µm to 13.04 µm, also depending on the position of the section. In most cases, the megapinosome complexes exhibited a polar arrangement as shown in Fig. 4. A megapinosome was on one pole, from which small vesicles seemed to bud, that defined the other pole.
Megapinosomes and HCMV internalization
In order to obtain some insights about the potential biological role of these structures, we investigated megapinosome formation in M1 and M2 macrophages after inoculation with HCMV (Fig. 7).

Uptake of HCMV particles in M1 and M2 macrophages from 5 to 90 min after inoculation, as observed on electron microscopic images. Significantly more particles were taken up by M2 macrophages at all time points after inoculation. The majority of particles in M2 macrophages could be clearly assigned to megapinosome complexes. Considerably, less viral particles could be found in macropinosomes. N number of viral particles
A total of 1003 TEM images of M2 and 978 TEM images of M1 macrophages’ thin sections have been analyzed in this study. Out of 132 M2 cells from cultures not inoculated with HCMV, 22 cells (16.7 %) showed megapinosomes. Upon HCMV inoculation, the formation of megapinosomes was enhanced and 387 cells (44.4 %) out of 871 cells showed megapinosomes. In some cells, more than one megapinosome complex (in different stages of development) were observed. M1 macrophages with (736 cells) and without (242 cells) HCMV inoculation did not show megapinosome structures. We found 1561 viral particles taken up by 871 M2 macrophages in thin sections. 970 (62.1 %) of these 1561 viral particles could be clearly assigned to megapinosome complexes. Twenty-six of these were found in the cytoplasmic area of the megapinosome complex, which only consisted of a capsid, the envelope had most likely fused with the megapinosome membrane. In M1 macrophages, 736 cell thin sections have been analyzed and only 36 internalized viral particles and no capsids in the cytoplasm have been found.
The internalized viral particles could initiate the viral replication cycle and express the viral immediate-early proteins IE1-2. At 24 h post-infection, immunofluorescence analysis revealed that 44.5 % of M2 macrophages were infected versus only 1.6 % of M1 macrophages. (images not shown).
Discussion
We observed and described large endocytic structures present in M2 but not M1 macrophages. These structures showed clear differences to the already known pinocytotic structures and appeared distinct from already characterized macropinosomes. We, therefore, named them megapinosomes.
We found the following differences between megapinosomes and macropinosomes.
-
1.
We found megapinosomes filled with a dense trabecular meshwork. Macropinosomes are, however, usually described as fluid-filled endocytotic vacuoles (Swanson 2008) that do not show extensive internal, membrane-surrounded structures in electron microscopic images (Overmeyer et al. 2008).
-
2.
According to our observations on M2 macrophages, megapinosome formation starts with a large concave bending of the plasma membrane and an accumulation of microvilli-like structures. According to Mercer and Helenius (2012), however, macropinosome formation starts with enlargements of ruffles.
-
3.
According to our observation, the megapinosome becomes closed by an actinomorphous centripetal arrangement of many (about 50–100) microvilli-like structures. These microvilli-like structures seem to originate from the tips of membrane ruffles. According to Mercer and Helenius (2012), a macropinosome is closed by the collapse of a membrane ruffle on the cell surface.
-
4.
According to our observations, the average diameter of a megapinosome in an arbitrary thin section is about 4.4 µm. The largest diameter observed was 13 µm. According to the literature, the diameter of a macropinosome is “bigger than 0.2 µm” (Lewis 1931); “up to 5 µm” (Lim and Gleeson 2011); “0.5 to 10 µm” (Mercer and Helenius 2009). The diameter of macropinosome thin sections shown in electron microscopic images is usually less than 1 µm (Haspot et al. 2012; Maréchal et al. 2001) and, therefore, much smaller than the diameter of the megapinosome sections that we observed.
-
5.
A representative macrophage in Fig. 4 exhibits the structure that we named megapinosome together with other large vesicular structures previously identified as macropinosomes (Overmeyer et al. 2008; Haspot et al. 2012). This is another strong indication that megapinocytosis represents a kind of pinocytosis that can be clearly differentiated from macropinocytosis. It also indicates that the structural differences between megapinosomes and macropinosomes are not resulting from different electron microscopic preparation methods.
We, therefore, conclude that megapinocytosis is a pinocytotic uptake mechanism that needs to be clearly discerned from macropinocytosis.
Interestingly, megapinosome formation was enhanced when the M2 macrophages were inoculated with HCMV. In these cells, most viral particles were found in megapinosomes (970 out of 1561 counted intracellular particles). Megapinosome formation was only observed in M2 macrophages and not in M1 macrophages, and as also already shown by Bayer et al. (2013), viral uptake as well as infectivity with HCMV was considerably higher in M2 compared to M1 macrophages. In this context, it is of interest that Hoppstädter et al. (2015) also observed an increased uptake of silica nanoparticles by M2 compared to M1 macrophages. These data combined with our data suggest a function of megapinocytosis in viral infection as well as in the general uptake of small particles.
Origin of the trabecular network
Our hypothesis is that the trabecular network originates from cell surface structures (microvilli-like structures) that are then internalized during megapinocytosis. When we compare the TEM images of the trabecular network with SEM images of the cell surface, we find that the microvilli-like structures have a similar diameter as the knots in the trabecular network. However, a full understanding of the trabecular network structure requires a three-dimensional imaging method such as FIB-SEM tomography of high-pressure frozen and freeze-substituted samples (Villinger et al. 2012) which would be a promising approach for further studies.
References
Alberts B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K, Walter P (2014) Molecular biology of the cell, 6th edn. Garland Publishing, New York
Bayer C, Varani S, Wang L, Walther P, Zhou SX, Straschewski S, Bachem M, Söderberg-Naucler C, Mertens T, Frascaroli G (2013) Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J Virol 87:67–79
Boyde A (1970) Practical problems and methods in the three dimensional analysis of scanning electron microscope images. In: Johari O (ed) Scanning electron microscopy. IITRI, Chicago, pp 105–112
Buser C, Walther P (2008) Freeze-substitution: the addition of water to polar solvents enhances the retention of structure and acts at temperatures around −60°C. J Microsc 230:268–277
Conner DS, Schmid SL (2003) Regulated portals of entry into the cells. Nature 422:37–44
Doherty GJ, McMahon HT (2009) Mechanisms of endocytosis. Annu Rev Biochem 78:857–902
Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof YD, Pilet P, Bressolette-Bodin C, Halary F (2012) Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a ph-independent and cholesterol-dependent manner. PLoS ONE 7:e34795
Hoppstädter J, Seif M, Dembek A, Cavelius C, Huwer H, Kraegeloh A, Kiemer AK (2015) M2 polarization enhances silica nanoparticle uptake by macrophages. Front Pharmacol 23(6):55. doi:10.3389/fphar.2015.00055 (eCollection 2015)
Lewis WH (1931) Pinocytosis. Johns Hopkins Hosp Bull 49:17–27
Lim JP, Gleeson PA (2011) Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol 89:836–843
Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O (2001) Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol 75:11166–11177
Mercer J, Helenius A (2009) Virus entry by macropinocytosis. Nat Cell Biol 11:510–520
Mercer J, Helenius A (2012) Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15:490–499
Overmeyer JH, Kaul A, Johnson EE, Maltese A (2008) Active ras triggers death in glioblastome cells through hyperstimulation of macropinocytosis. Mol Cancer Res 6:965–977
Rasmussen I, Vilhardt F (2015) Macropinocytosis is the entry mechanism of amphotrophic murine leukemia virus. J Virol 89:1851–1866
Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B (2008) Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368
Swanson JA (2008) Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 9:639–649
Villinger C, Gregorius H, Kranz Ch, Höhn K, Münzberg C, von Wichert G, Mizaikoff B, Wanner G, Walther P (2012) FIB/SEM-tomography with TEM-like resolution for 3D imaging of high pressure frozen cells. Histochem Cell Biol 138:549–556
Villinger C, Schauflinger M, Gregorius H, Kranz C, Höhn K, Nafeey S, Walther P (2014) Three-dimensional imaging of adherent cells using FIB/SEM and STEM. In: Kuo J (ed) Methods in molecular biology. Springer, New York. doi:10.1007/978-1-62703-776-1_27
Walther P, Ziegler A (2002) Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J Microsc 208:3–10
Walther P, Wang L, Ließem S, Frascaroli G (2010) Viral infection of cells in culture—approaches for electron microscopy. Methods Cell Biol 96:603–618
Acknowledgments
We thank Renate Kunz, Anke Lüske, Eberhard Schmid, and Reinhard Weih for technical assistance, and Dr. Rudolph Reimer, HPI Hamburg, and Gregor Neusser, Ulm University, for their help with the analysis of the stereo pairs and the height profiles.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.

Figure S1
Anaglyph stereo pairs of Figs. 1 and 2 demonstrating the depressions in M2 macrophages. Figs S1A represents an early phase, B is a higher magnification of the boxed area, and C represents a later stage with an even deeper depression. Fig. S1D shows for comparison an M1 macrophage without depression. Fig S1E (F higher magnification) shows an M2 macrophage with a deep cavity most likely representing a late stage of megapinosome formation. (JPEG 1874 kb)
Animation of stereo images of a M1 macrophage without a surface depression, a M2 macrophage with an early phase megapinosome formation, a M2 macrophage with a later stage megapinosome. A M2 macrophage with a putative late stage megapinosome formation. 25 images were recorded with a tilt increment of 2°. Finally the megapinosome was closed by an actinomorphous ring of microvilli-like structures. Supplementary material 2 (M4 V 46585 kb)
Movie 2
3D reconstruction of a serial section analysis of a cell with a megapinosome (red) a megapinosome complex (red and orange) and a nucleus (beige). This cell shows a stage of megapinocytosis, where megapinosome and megapinosome complex are completely surrounded by cytoplasm in all sections. The reconstruction proves that megapinosomes exist without any connection to the extracellular space. Therefore, megapinocytosis is a real endocytotic process. (M4 V 34172 kb)
Movie 3
A polar large megapinosome complex. Also in this cell the megapinosome as well as the megapinosome complex are fully internalized into the cell without an opening to the cytoplasm. (M4 V 37636 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Bauer, A., Subramanian, N., Villinger, C. et al. Megapinocytosis: a novel endocytic pathway. Histochem Cell Biol 145, 617–627 (2016). https://doi.org/10.1007/s00418-015-1395-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-015-1395-2