
Abstract
Copper is essential for a variety of important biological processes as a cofactor and regulator of many enzymes. Incorporation of copper into the secreted and plasma membrane-targeted cuproenzymes takes place in Golgi, a compartment central for normal copper homeostasis. The Golgi complex harbors copper-transporting ATPases, ATP7A and ATP7B that transfer copper from the cytosol into Golgi lumen for incorporation into copper-dependent enzymes. The Golgi complex also sends these ATPases to appropriate post-Golgi destinations to ensure correct Cu fluxes in the body and to avoid potentially toxic copper accumulation. Mutations in ATP7A or ATP7B or in the proteins that regulate their trafficking affect their exit from Golgi or subsequent retrieval to this organelle. This, in turn, disrupts the homeostatic Cu balance, resulting in copper deficiency (Menkes disease) or copper overload (Wilson disease). Research over the last decade has yielded significant insights into the enzymatic properties and cell biology of the copper ATPases. However, the mechanisms through which the Golgi regulates trafficking of ATP7A/7B and, therefore, maintains Cu homeostasis remain unclear. This review summarizes current data on the role of the Golgi in Cu metabolism and outlines questions and challenges that should be addressed to understand ATP7A and ATP7B trafficking mechanisms in health and disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Golgi complex operates as a central compartment in the biosynthetic pathway: it receives newly synthesized proteins from the endoplasmic reticulum (ER), provides machinery for their post-translational modifications (e.g., glycosylation, proteolytic cleavage) and sorts modified proteins toward appropriate post-Golgi destinations (Polishchuk and Mironov 2004; Wilson et al. 2011). Over the years, novel functions of the Golgi have gradually emerged. This organelle appears to serve as a hub for initiation of several signaling pathways as well as an important compartment involved in cell fate decision and lipid metabolism (Wilson et al. 2011). Studies over the last decade also revealed that the Golgi is a central station for cellular copper (Cu) homeostasis (La Fontaine and Mercer 2007; Lutsenko et al. 2007).
Cu is an essential but toxic metal. Its ability to undergo reversible oxidation from Cu(I) to Cu(II) under physiologic conditions is utilized by enzymes for critical biochemical processes that include cellular respiration, free radical detoxification, pigmentation, neuropeptide processing, cross-linking of collagen and elastin, and iron transport (Lutsenko 2010; Nevitt et al. 2012). However, oxidation of Cu(I) produces reactive oxygen species, and unchelated Cu can be toxic to cells. Therefore, cells evolved a complex network of regulatory mechanisms that operate to satisfy the metabolic demand for Cu and to control Cu levels both at the cellular and at the systemic levels. High-affinity transporter CTR1 imports Cu from extracellular space into the cytosol, where the metal is captured by cytosolic copper chaperones and shuttled toward different intracellular destinations (Fig. 1). The copper chaperone ATOX1 carries copper to the trans-Golgi network (TGN), where it is transferred to ATP7A/7B, which in turn load Cu into newly synthesized cuproenzymes that move through the secretory pathway (La Fontaine and Mercer 2007; Lutsenko et al. 2007).

Schematic depiction of copper distribution in a mammalian cell. Copper, taken up via CTR1 (red), is transferred to chaperones ATOX1, CCS and COX17, which ferry it (black dash arrows) to ATP7A/B (blue) in the Golgi, to Cu–Zn superoxide dismutase (magenta) in the cytosol and to cytochrome c oxidase (green) in the mitochondria, respectively. In the Golgi, ATP7A/B load Cu on newly synthetized cuproenzymes (orange ball), which traffic along the biosynthetic pathway (orange arrow). Significant increase in intracellular Cu induces export of ATP7A/B (blue arrow) toward post-Golgi compartments and plasma membrane where ATP7A/B drive the efflux of excessive Cu from the cell. Solid black arrows indicate Cu translocation across the membrane
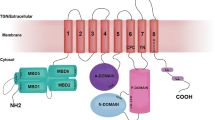
ATP7A and 7B are complex multispan membrane proteins that belong to a P1B-type ATPase family (Fig. 2a). The N-terminal portion of both proteins contains six metal-binding domains followed by eight transmembrane domains that form a copper translocation pathway to move Cu from the cytosol at the expense of ATP hydrolysis. Autophosphatase, nucleotide binding and phosphorylation domains in 2nd and 3rd cytosolic loops coordinate the catalytic activity of ATP7A/7B. Apart from their biosynthetic function in the TGN, both Cu ATPases exhibit a unique property to traffic out of the Golgi in response to the increasing cytosolic Cu to post-Golgi structures and then to the plasma membrane. This regulated trafficking serves multiple functions. In intestinal cells, it allows copper export from the enterocytes into the bloodstream for further distribution to other tissues (La Fontaine and Mercer 2007; Lutsenko et al. 2007). In hepatocytes, movement of ATP7B to post-Golgi Cu excretion sites serves to remove excessive Cu, which is toxic for the cell due to its high redox potential (La Fontaine and Mercer 2007; Lutsenko et al. 2007). In melanocytes, trafficking to melanosome is necessary to maintain the activity of tyrosinase in this specialized compartment (Setty et al. 2008). The inability of Cu ATPases to traffic in response to changing Cu levels and/or to pump Cu across the membrane results in severe aberrations of Cu metabolism, which are especially apparent in Menkes and Wilson diseases caused by mutations in ATP7A and ATP7B genes, respectively (de Bie et al. 2007; La Fontaine and Mercer 2007; Lutsenko et al. 2007).
Despite the importance of ATP7A/B trafficking in the maintenance of Cu homeostasis, the mechanisms of Cu ATPase targeting to the Golgi and their transport from this organelle remain poorly understood. In this review, we focus on outstanding mechanistic questions related to the interplay between Cu ATPases localization and function. Answering these questions, in our view, has the potential to open new avenues in ATP7A/B trafficking studies.
Localization and function of Cu ATPases in the Golgi
The main function of ATP7A/B in the Golgi is to supply Cu to newly synthesized cuproenzymes that move through the secretory pathway. ATP7A/B receive Cu from the chaperone ATOX1 through direct interactions (Lutsenko et al. 2007; Lutsenko 2010; Nevitt et al. 2012). Then, the Cu pumps transport the metal across the membrane into the Golgi lumen, where Cu is loaded on a number of enzymes with important functions in the central nervous system (dopamine β-hydroxylase, peptidylglycine α-amidating monooxygenase), connective tissue and blood vessel development (lysyl oxidase, superoxide dismutase 3), pigmentation (tyrosinase) as well as in iron and Cu transport (ceruloplasmin, hephaestin). The lack of biosynthetic ATP7A/B function in the Golgi leads to a number of severe symptoms that are manifested in Menkes or Wilson diseases (Table 1) (de Bie et al. 2007; Tumer and Moller 2010).
Within the Golgi stack, Cu ATPases reside mainly in the TGN compartment, as revealed by co-localization studies with different markers (Cobbold et al. 2002; La Fontaine et al. 1998, 2001) and immuno-EM labeling (La Fontaine et al. 1998, 2001, Hasan 2012 #391). Why ATP7A/B prefer the TGN to the earlier Golgi compartments is not entirely clear. It is possible that the lower pH environment in the TGN lumen facilitates the release of Cu from ATP7A/B to the cuproenzymes (Safaei et al. 2008). On the other hand, the TGN operates as the Golgi exit site where the cargo proteins undergo sorting and packaging into the post-Golgi transport carriers (De Matteis and Luini 2008; Luini et al. 2008; Polishchuk and Mironov 2004). Thus, in the case of intracellular Cu increase, TGN localization allows ATP7A/B to leave the Golgi rapidly and without the need to cross the entire Golgi stack in the cis-to-trans direction.
Several studies have been done to identify signals within the protein structure that retain Cu pumps in the Golgi. A 38 amino acid sequence containing the transmembrane domain 3 appears to be sufficient to support ATP7A localization in the Golgi complex (Francis et al. 1998). It is clear, though, that several other determinants contribute to TGN compartmentalization of ATP7A/B at basal Cu conditions. The di-leucine and tri-leucine endocytic motifs in the carboxyl-tails of ATP7A and ATP7B, respectively, are required for their efficient retrieval to the Golgi from the peripheral Cu excretion sites (Cater et al. 2006; Francis et al. 1999; Petris et al. 1998).
Given that copper stimulates Cu ATPase activity and also triggers the redistribution of ATP7A/7B from the TGN to vesicles, it is interesting to consider the role of copper transport in TGN targeting and retention. The catalytically inactive ATP7B-D1027A mutant (with a replacement of invariant aspartate in the conserved phosphorylation DKTG motif; see Fig. 2) remains within the TGN even at elevated Cu levels, similar to the E1064A Wilson disease-causing mutation that prevents exit from Golgi (Dmitriev et al. 2011). In contrast, studies of the disease-associated ATP7A isoforms identified mutations in the phosphatase A-domain that disrupted retention of the pump in the Golgi and induced its redistribution to the PM (Petris et al. 2002). These observations led to the suggestion that phosphorylation/dephosphorylation of aspartate during the catalytic cycle is directly coupled to trafficking events. Given a very different time scale for the enzyme turnover [hundreds per second (Lewis et al. 2012)] and the TGN exit (which is usually complete within hours), the direct causative link seems unlikely. However, the conformations that copper ATPases adopt in the absence of copper (and hence low transport activity) and in a copper-saturated state could be markedly different and could be easily distinguished by cellular retention and trafficking machineries.

Structure and localization of Cu-translocating ATPases. a Three-dimensional representation of the Menkes (ATP7A) and Wilson (ATP7B) Cu ATPases. Both proteins are predicted to have eight transmembrane (TM) domains, with most of the protein on the cytoplasmic side. They contain ATP-binding (red), phosphatase (green), and phosphorylation (blue domain), which regulate catalytic activity. N-terminal region comprises six copper-binding motifs (black balls) that interact with copper chaperonins and presumably deliver copper to the channel. In addition, CPC motif in the 6th transmembrane domain plays a key role in Cu translocation along the channel, while LL signal in the C-tail is required for ATP7A/B endocytosis. b Cu-dependent compartmentalization of ATP7B. At low Cu conditions, endogenous ATP7B in HepG2 cells exhibits significant overlap with the TGN marker Golgin-97. Upon Cu increase, ATP7B moves from the TGN to the peripheral vesicles and, to some extent, to the cell surface, where Cu excretion takes place
The idea that protein conformations of Cu ATPases contribute significantly to ATP7A/7B targeting and trafficking received further support in recent studies analyzing the role of kinase-mediated (non-catalytic) phosphorylation in the Golgi targeting of ATP7A/B. Serine cluster at positions 340–341 favors Golgi localization of ATP7B as substitution at these positions to alanine (or any other residue) induces ATP7B redistribution to peripheral vesicles (Hasan et al. 2012). In contrast, the mutations of S1469 within the C-terminal part of ATP7A and S478/481/1121/1453 in ATP7B have been reported to keep the protein in the Golgi (Pilankatta et al. 2011; Veldhuis et al. 2009). The important role of the precise inter-domain contacts is also evident from studies on the N-terminal region of ATP7B, where mutation of a single residue Y44A not only diminishes the retention of ATP7B in the TGN, but also causes mis-sorting of ATP7B to a basolateral membrane (the normal destination of ATP7B in polarized cells is apical membrane) (Braiterman et al. 2009).
The above studies have raised questions about the specific role of a kinase-mediated phosphorylation in ATP7A/7B compartmentalization. Earlier reports have shown that both Cu ATPases have a basal level of phosphorylation and became hyperphosphorylated in response to copper elevation (Vanderwerf et al. 2001). This phosphorylation is distinct from the catalytic phosphorylation of aspartate and involves serine residues. Variation of phosphorylation status may change conformation/inter-domain interactions in Cu ATPases and therefore enable or prevent their interaction with the components of the post-Golgi trafficking machinery. Unfortunately, the kinases that execute phosphorylation at the traffic-relevant sites of either ATP7a or ATP7B are yet to be identified. The TGN-associated PKD1 represents an attractive candidate, as its activity is required for ATP7B export from the Golgi (Pilankatta et al. 2011). On the other hand, PKD inhibitors do not impact trafficking of ATP7A (Cobbold et al. 2002).
Finally, to reach the Golgi destination, newly synthesized ATP7A/B proteins must be properly folded. Investigation of Wilson disease-causing ATP7B mutants (including the most clinically frequent H1069Q and R778L) revealed their strong retention in the ER (Forbes and Cox 2000; Payne et al. 1998). Despite the residual Cu-translocating activity, retention and degradation of these mutants in the ER do not allow them to load Cu on the enzymes and to remove excess metal from the cell. Thus, identification of the molecules that improve the ER-to-Golgi export of these ATP7B mutants represents an important task as its application could benefit a large group of Wilson disease patients.
Trafficking of Cu ATPase from the Golgi
Apparently, the ability to exit from the Golgi toward Cu excretion sites (Fig. 2b) was developed by ATP7A and ATP7B during the evolution of high eukaryotes. Yeast regulates their intracellular Cu levels mainly through the metal responsive expression of genes operating in Cu influx, efflux and storage rather than through relocation of the proteins between different compartments (Nevitt et al. 2012). Indeed, the yeast homolog of ATP7A/B, Ccc2, never moves away from the Golgi where it performs, almost exclusively, biosynthetic functions (Yuan et al. 1997). Compared to mammalian orthologues, Ccc2 is a smaller protein: it contains only 2 N-terminal metal-binding domains instead of 6 and lacks targeting signals for the apical or basolateral delivery in polarized cells. This observation suggests that domains, missing in yeast, may determine the ability of ATP7A/B to traffic in response to Cu.
An important challenge faced by mammals in maintaining whole-body Cu homeostasis is associated with the development of Cu sensing and Cu transport mechanisms beyond the level of individual cells. At the organismal level, these mechanisms have to accommodate not only multiple cell types in various organs with different metabolic demands for Cu, but also the special and temporal separation of major Cu consumption sites. The post-Golgi trafficking of Cu ATPases to distinct apical/basolateral membranes in polarized cells and the differential expression of Cu ATPases in tissues and organs allow higher vertebrates to deal with systemic changes in Cu levels (Lutsenko 2010; Nevitt et al. 2012).
ATP7A is expressed in most tissues (except adult hepatocytes), but its most important function is in the small intestine and the choroid plexus. In enterocytes, ATP7A receives adsorbed dietary Cu and moves from the Golgi to the basolateral surface of the cells to release Cu toward portal circulation (Fig. 3). Mutations, which result in a loss of the ATP7A protein, its transport activity or a failure to traffic out of the Golgi, do not allow Cu to move beyond the intestinal barrier. As a result, severe Cu deficiency is observed in most tissues of Menkes disease patients, with a notable exception of intestine and kidneys (de Bie et al. 2007; Lutsenko et al. 2007; Lutsenko 2010). In contrast to ATP7A, ATP7B is highly expressed in the liver and present at lower levels in many other organs. In the liver, ATP7B receives Cu from the portal circulation and utilizes it in the Golgi for metallation of ceruloplasmin, which uses copper for regulation of iron balance. Cu elevation beyond a certain threshold activates ATP7B export from the Golgi to the “vesicular compartment” and biliary surface in the apical part of hepatocytes (Fig. 3). There, ATP7B supports the efflux of excess Cu into the biliary flow for further elimination of the metal from the body. In Wilson disease, the lack of ATP7B activity and/or delivery of the pump to the apical domain of hepatocytes induces a marked accumulation of Cu in hepatocytes, development of morphologic and metabolic abnormalities, culminating in liver failure (de Bie et al. 2007; Lutsenko et al. 2007, 2010).

Schematic representation of copper homeostasis in the body. Cu is absorbed through the apical CTR1 channel by the enterocytes in the small intestine and effluxed across the basolateral surface of these cells by ATP7A into the portal circulation. The latter process requires ATP7A trafficking from the Golgi to the basolateral surface of the cells (blue arrow). Lack of ATP7B function in Menkes disease patients (green bar) results in accumulation of Cu in the enterocytes and overall copper deficiency in the body. Most of the newly absorbed copper is normally taken up by the hepatocytes in the liver, where Cu is loaded in the TGN by ATP7B on newly synthesized ceruloplasmin, the principal Cu carrier in the blood. In cases of Cu overload, ATP7B traffics to Cu excretion sites (orange arrow), i.e., apical (canalicular) membrane and associated vesicular structures. Mutations in the ATP7B gene that affect activity and trafficking of the corresponding protein block Cu delivery to ceruloplasmin and its efflux into the bile (red bars). As a result, Cu accumulates in the liver and causes its toxicosis in Wilson disease patients
Despite the fundamental importance of ATP7A/B post-Golgi trafficking for the regulation of Cu balance in the body, many mechanistic questions are yet to be answered in full detail. These include the molecular basis of sorting within Golgi, the characteristics of post-Golgi routes for ATP7A or ATP7B, and the machinery involved in the anterograde, retrograde trafficking and recycling.
Several transport pathways, which are directed to the cell surface and/or endo-lysosomal system, originate from the TGN. The complexity of post-Golgi trafficking increases in polarized cells, as the cell surface-destined proteins have to be delivered to distinct domains (apical or basolateral) of cell membranes (De Matteis and Luini 2008; Muth and Caplan 2003; Rodriguez-Boulan et al. 2005). Earlier studies examined whether ATP7A reaches the PM via the post-Golgi pathway utilized by the constitutively secreted proteins. Insensitivity of ATP7A exit from the Golgi to inhibitors of constitutive secretion, such as a dominant negative PKD mutant, led to the suggestion that ATP7A is sorted from the TGN into the specific route regulated by Cu (Cobbold et al. 2002). On the other hand, ATP7A trafficking was suppressed by Cdc42 and PKA inhibitors, which were equally effective against constitutive cargo proteins (Cobbold et al. 2002). Thus, to firmly identify the ATP7A export route from the Golgi, the approach using molecular inhibitors should be complemented with other methods (see below).
While some aspects of ATP7A post-Golgi trafficking are understood, the mechanisms of ATP7B export from the Golgi remain poorly studied. With the exception of the role of PKD in ATP7B trafficking from the Golgi (Pilankatta et al. 2011), there are no studies identifying molecular players that support ATP7B export from the Golgi. Thus, the existing body of evidence does not allow us to assign ATP7B to any well-studied pathway or identify its itinerary as a newly identified post-Golgi route.
In this context, it will be important to understand whether ATP7A or ATP7B can be packed into the post-Golgi carriers containing cargoes moving through the constitutive pathway (such as VSVG, CD8, GPI-anchored proteins, Na/K-ATPase) (De Matteis and Luini 2008; Polishchuk and Mironov 2004). The biogenesis and morphology of such carriers has been extensively characterized (Polishchuk et al. 2000, 2003) as well as the methodologies that allow efficient analysis of their composition (Polishchuk et al. 2004). If ATP7A or ATP7B are found within the post-Golgi structures carrying known constitutive cargo proteins, then the role of Cu in inducing the TGN exit would be to allow the Cu ATPases to adopt the conformation necessary for incorporation into well-known constitutive TGN-to-PM route(s). The lack of co-localization with the conventional markers of the post-Golgi pathways, on the other hand, would indicate that ATP7A or ATP7B are transported along a specific route regulated by Cu. If the latter scenario turns out to be the case, it will be important to understand (1) how Cu mechanistically triggers the formation of Cu ATPase containing carriers from the TGN membranes and (2) whether any other cargo protein(s) utilize this ATP7A/B-specific post-Golgi pathway.
It is worth noting that, although both ATP7A and ATP7B are targeted to the TGN, a closer examination of their localization reveals significant segregation from conventional TGN markers, such as TGN38, TGN46 or Golgin 97 (Guo et al. 2005; Holloway et al. 2007; Nyasae et al. 2007). Therefore, the TGN regions containing Cu ATPases could constitute specific TGN subcompartments from where ATP7A or ATP7B exit toward distinct post-Golgi destinations (Guo et al. 2005; Holloway et al. 2007; Nyasae et al. 2007). A recent study revealed Arf1 to be involved in both the maintenance and biogenesis of ATP7A containing TGN membranes (Holloway et al. 2007). This finding may have an interesting implication, as Arf1 recruits clathrin adaptor proteins (mainly AP1, AP3, AP4 and GGAs) to the TGN, where these adaptors drive both sorting and trafficking events (Robinson and Bonifacino 2001). ATP7A and ATP7B possess, respectively, di-leucine and tri-leucine C-terminal motifs that can be recognized by adaptor proteins. However, recent studies have reported that the suppression of AP1, clathrin or GGA, while causing intracellular ATP7A redistribution, does not block ATP7A or ATP7B exit from the Golgi (Hirst et al. 2012; Holloway et al. 2013; Martinelli et al. 2013). The role of AP3 and AP4 in the TGN exit of ATP7A/7B as well as the biogenesis of the TGN subcompartment, where Cu ATPases reside, remains to be determined.
The other important issue that has to be addressed is whether ATP7A and ATP7B occupy the same TGN subcompartment or are sorted within the TGN into independent domains. Although ATP7A and ATP7B localization was studied in cell types where both proteins were expressed (La Fontaine and Mercer 2007), a detailed comparison of their localizations in the Golgi has not been made. Generation of high-resolution maps of ATP7A and ATP7B distribution over the TGN membranes (using advanced light microscopy and immuno-EM) would allow new insights into the mechanisms of Cu ATPase trafficking and sorting at the TGN level.
Polarized cells utilize ATP7A or ATP7B function at the basolateral or apical surface, respectively. The need to deliver copper transporters to these distinct domains constitutes additional complexity in the trafficking mechanisms. Site-directed mutagenesis revealed the requirement for a dileucine (1487LL1488) motif and the PDZ target (1497DTAL1500) domain in the C-terminus of ATP7A for localization at the basolateral membrane (Greenough et al. 2004). In contrast, the apical targeting of ATP7B has been shown to rely on a novel N-terminal (37FAFDNVGYE45) sequence that is absent from the corresponding region of ATP7A (Braiterman et al. 2009). However, the mechanistic details of how these signals are utilized by the trafficking machinery and where the sorting of ATP7A and ATP7B is executed remain unclear.
In general, the TGN operates as a main sorting station along the secretory pathway from where cargo proteins are delivered to their target compartments and surface domains (De Matteis and Luini 2008; Polishchuk and Mironov 2004; Rodriguez-Boulan et al. 2005). Several apical and several basolateral routes emerge from the TGN, and their number may vary significantly in different cell types or even during polarization of the same cell (Muth and Caplan 2003; Rodriguez-Boulan et al. 2005). In addition to the TGN, several endocytic compartments are thought to perform sorting functions along the secretory pathway (Mellman and Nelson 2008; Rodriguez-Boulan et al. 2005). Specific apical or basolateral cargo proteins, which follow either direct TGN-to-PM or “through-endosome” exocytic routes, were identified as well as selective molecular tools that allow the interception of trafficking along the individual routes (Mellman and Nelson 2008; Rodriguez-Boulan et al. 2005). The analysis of ATP7A or ATP7B colocalization with such cargo markers in the post-Golgi carriers and the treatments with specific molecular inhibitors should allow us to determine (1) which pathway is utilized by Cu ATPase to get from the TGN to the correct surface domain and (2) whether this pathway involves an endocytic intermediate.
Among the sorting endocytic stations, the so-called AP1-B recycling compartment plays an extensive role in basolateral targeting of several membrane proteins such as VSVG, transferrin receptor and LDL receptor (Folsch 2008; Gonzalez and Rodriguez-Boulan 2009) and could be involved in the basolateral sorting of ATP7A. Clathrin adaptor complex AP-1B (containing epithelial-specific mu1b subunit) drives sorting and transports events in this compartment and, therefore, defines its identity (Folsch 2008; Gonzalez and Rodriguez-Boulan 2009). Interestingly, high expression levels of mu1b subunit of AP-1B were found in a number of tissues (kidney, intestine, placenta, mammary gland) (Ohno et al. 1999) where both ATP7A and ATP7B were detected (La Fontaine and Mercer 2007; Lutsenko et al. 2007). In these tissues, Cu pumps undergo delivery to the opposite membrane domains where they execute specific functions (La Fontaine and Mercer 2007). However, whether AP-1B is required for correct delivery of either ATP7A or ATP7B remains unclear. ATP7B trafficking and sorting is unlikely to involve AP-1B because hepatocytes do not express this adaptor (Ohno et al. 1999), yet target ATP7B to the apical (canalicular) surface (Guo et al. 2005; Roelofsen et al. 2000). Whether ATP7A trafficking requires AP-1B activity also remains to be tested. Some membrane proteins with similar multispan topology (like Na/K-ATPase) bypass the AP-1B compartment on their route from the Golgi to the basolateral surface (Farr et al. 2009). On the other hand, expression of inactive Rab22 arrests ATP7A within the post-Golgi recycling station (Holloway et al. 2013). Rab22 is known to cooperate in the regulation of recycling with Arf6 (Weigert et al. 2004), which in turn recruits AP-1B to the membranes (Shteyn et al. 2011). Therefore, the engagement of post-Golgi recycling AP-1B station in ATP7A basolateral delivery cannot be ruled out but has to be verified using ablation of mu1b in polarized kidney or intestinal cells.
In response to copper elevation, Cu ATPase ATP7B moves from the TGN to large cytosolic vesicles (Cater et al. 2006; Roelofsen et al. 2000). This observation suggests that the endocytic intermediate is almost certainly involved in the post-Golgi trafficking of ATP7B toward apical surface of hepatic cells. Although the ATP7B-containing vesicles remind endosomes, the earlier immuno-EM microscopy did not detect a significant overlap of these ATP7B-containing structures with the conventional endo-lysosomal markers (La Fontaine et al. 2001). It has been proposed (although not yet shown) that ATP7B pumps Cu inside these vesicles and that vesicles fuse with the canalicular surface of hepatocytes to expel Cu from their lumen directly into the bile (Cater et al. 2006). Whether ATP7B is delivered to the apical canalicular membrane of hepatocytes during vesicle exocytosis remains an issue of ongoing debate (Cater et al. 2006; Roelofsen et al. 2000). Several studies fail to detect ATP7B at the canalicular membrane of polarized hepatic cells (Cater et al. 2006), while others provided compelling evidence that ATP7B reaches the apical surface of hepatocytes (Guo et al. 2005; Roelofsen et al. 2000). Determining whether ATP7B is present at the apical membrane (either transiently during vesicle fusion or for a longer time to mediate copper export) is essential in order to understand copper homeostasis in the liver.
From the technical point of view, it is difficult to investigate a coupling between ATP7B trafficking and Cu excretion and to determine whether these events are coordinated. The lack of comprehensive data on the molecular composition and, therefore, identity of the ATP7B-containing vesicles does not allow to judge, which molecular players may be involved in the delivery of ATP7B from the Golgi to vesicles and then from vesicles to the plasma membrane. The isolation of ATP7B vesicles combined with the characterization of their proteome would circumvent this obstacle as it would provide testable targets to investigate the ATP7B trafficking to and from the vesicles.
Several features of ATP7B vesicles are similar to secretory granules or specific lysosome-related organelles that release their content in response to stimuli (Raposo et al. 2007). Like the above organelles, ATP7B vesicles are employed in storage (as they accumulate Cu) and undergo exocytosis upon specific stimulus (increase in Cu concentration) (Cater et al. 2006). Given these behavioral similarities, it would be interesting to understand (1) whether ATP7B vesicles share some components of the molecular machinery with the exocytic storage organelles and (2) what is the specific signaling mechanism that links changes in intracellular copper with vesicle exocytosis. Interestingly, ATP7A also undergoes redistribution from the TGN to the specific post-Golgi vesicular structures upon exposure of enterocytes to Cu (Nyasae et al. 2007). Therefore, the existence of a Cu-sensitive post-Golgi storage station may represent a common feature in trafficking of both ATP7A and ATP7B. This vesicular pool may provide additional sorting of ATP7A/7B toward the plasma membrane (when copper is elevated), back to the TGN (when copper becomes depleted) and/or forward Cu ATPases for lysosomal degradation at the end of their life span. Differential phosphorylation by kinases (reported for both ATP7A and ATP7B) may play a key role in such sorting along with specialized adaptor proteins such as COMMD1. The existence of a specialized vesicular compartment may be especially useful in neurons where a robust response at the synaptic cleft may require rapid vesicular fusion rather than slow trafficking of ATP7A/7B from the TGN. Participation of the ATP7A-containing vesicles in rapid fusion (independent of trafficking from the TGN) is seen in response to activation of the NMDA receptor (Schlief et al. 2005).
Despite numerous gaps in our understanding of the mechanisms responsible for ATP7A and ATP7B trafficking, the regulatory role of Cu in these processes has been firmly established by many studies (Hung et al. 1997; Petris et al. 1996). It is thought that Cu stabilizes ATP7A and ATP7B in a conformation favorable for interaction with the components of membrane trafficking machinery; this allows for the export of Cu ATPases from the Golgi and their delivery toward post-Golgi destinations. Several Cu-dependent binding partners of ATP7A and 7B were predicted using yeast two-hybrid screen (La Fontaine and Mercer 2007); none of them belongs to conventional traffic machineries and so far their role in mammalian cells has not been explored in detail. One potentially interesting candidate is p62 subunit of dynactin–dynein microtubule motor complex. p62 interacts with ATP7B in the presence of high Cu (Lim et al. 2006) and, therefore, being in complex with dynein motor, can potentially pull ATP7B-enriched membranes along the MTs away from the bulk of the TGN. Whether and how other components of membrane budding/fission machinery can be triggered by changes in Cu levels remains unclear. One unexplored possibility is that Cu-induced structural changes in ATP7A/7B open binding sites for lipids, such as cholesterol or sphingomyelin, may influence Cu ATPase sorting within the TGN subdomains or initiate assembly of trafficking machinery. The ability of COMMD1 protein (a protein with a known role in hepatic copper balance) to specifically detect phosphotidyl inositols (PIPs) in vitro has been experimentally demonstrated (Burkhead et al. 2009), but the role of PIPs in ATP7A trafficking from Golgi and along the secretory pathway remains unexplored.
Recent bioinformatics analyses suggest that about 1 % of the entire eukaryotic proteome is composed of putative Cu-binding proteins (Andreini et al. 2008), suggesting that the list of Cu-dependent regulators of ATP7A and B trafficking is likely to be expanded. To this end, generation of ATP7A and ATP7B interactomes in low and high Cu would provide a valuable tool for the identification of new molecules involved in Cu ATPase trafficking.
Retrieval of ATP7A and ATP7B to the Golgi
After elimination of excess Cu at the cell surface, ATP7A and ATP7B return to the Golgi where they switch their activities toward metallation of the newly synthetized proteins. It has been convincingly demonstrated that the C-terminal di-leucine or tri-leucine signatures in ATP7A and ATP7B, respectively, are essential for the retrieval of proteins back to the Golgi (Braiterman et al. 2011; Francis et al. 1999; Petris et al. 1998). The ability of the leucine-based motifs to interact with clathrin adaptors (Robinson and Bonifacino 2001) led to the hypothesis that ATP7A and ATP7B undergo internalization through the clathrin-dependent pathway (Francis et al. 1999; Petris et al. 1998). However, the initial attempt to test this hypothesis experimentally led to the opposite conclusion (Cobbold et al. 2003), and only recently clathrin downregulation with RNAi indicated that ATP7A endocytosed from the cell surface via the clathrin-mediated pathway (Holloway et al. 2013). The importance of the di-leucine motif for recognition by endocytic machinery was recently confirmed in cells from patients with a new disorder of Cu metabolism, ATP7A-related distal motor neuropathy (Yi et al. 2012). The disease-causing mutation in ATP7A, P1386S, is located in the vicinity of the di-leucine motif and leads to a shift in steady-state ATP7A localization from the TGN to the cell surface.
Reduced retrieval of ATP7A and ATP7B to the Golgi was also observed when AP-1 function was suppressed (Hirst et al. 2012; Holloway et al. 2013). In a recent, very elegant study, Hirst and colleagues demonstrated that the inactivation of AP-1 components results in a depletion of both ATP7A and ATP7B from the clathrin-coated vesicles, which are likely directed from the endocytic compartment(s) to the Golgi (Hirst et al. 2012). The role of AP-1 in the endocytic trafficking of Cu ATPases to the Golgi was further confirmed by the study of mechanisms involved in the pathogenesis of MEDNIK syndrome, a congenital disorder with alterations in Cu homeostasis (Martinelli et al. 2013). This disease is caused by mutations in the AP1S1 gene encoding σ1A, the small subunit of AP-1. Fibroblasts from MEDNIK patients exhibit ATP7A mostly at the cell surface, even in the presence of Cu chelator, therefore indicating that the mutation severely compromised the retrieval of the Cu pump to the Golgi (Martinelli et al. 2013). The reduced amount of ATP7A in the Golgi could impair metallation of several Cu-dependent enzymes (see Table 1) and produce neurologic, metabolic, pigmentation and skin symptoms observed in MEDNIK patients and in a model system, such as Ap1s1 zebra fish (Martinelli et al. 2013; Montpetit et al. 2008).
Surprisingly, mutation of di- or tri-leucine motifs, as well as the suppression of clathrin/AP-1 functions, does not impact the export of ATP7A/B from the Golgi (Hirst et al. 2012; Holloway et al. 2013). This indicates that Cu ATPases take a clathrin-independent exit route(s) from the TGN. On the other hand, the above studies provide new insights into the nature of the pathway that carries Cu ATPases back to Golgi. Apart from ATP7A and ATP7B, AP-1 suppression eliminates from clathrin-coated vesicles several well-studied proteins, such as M6PR, Sortilin-1 and furin (Hirst et al. 2012), which recycle from the endosomes to the Golgi (Bonifacino and Rojas 2006). Therefore, it is likely that Cu ATPases get delivered to the Golgi from sorting endosomes through the pathway utilized by M6PR, Sortilin-1 and furin.
Overall, Cu ATPases exhibit interesting and distinct trafficking properties. Their export from the Golgi to the sites of Cu excretion seems to require a specific exocytic route, tightly regulated by Cu, whereas their retrieval back to the Golgi seems to proceed through a more common endocytic pathway.
Concluding remarks
The well-known role of ATP7A and ATP7B in the maintenance of Cu homeostasis has been recently expanded to their involvement in other processes, such as modulation of the Alzheimer disease phenotype, pathogen defense and anti-cancer drug resistance (Gupta and Lutsenko 2009; Wang et al. 2011). The list of these new functions continues to grow. Therefore, the Golgi complex, which operates as a hub for Cu ATPases, will remain an important focus of cell biology research on Cu metabolism. In this review, we pointed to the main challenges and questions related to Cu ATPase trafficking and hope that the need to answer these many intriguing questions will gain attention from both the copper and Golgi communities.
We believe that the advent of modern system biology approaches could help achieve real breakthroughs in the understanding of ATP7A and ATP7B trafficking pathways and mechanisms. Next-generation sequencing is likely to reveal new ATP7A and ATP7B mutants/variants as well as new genes associated with cell responses to Cu toxicity and deficiency (Fuchs et al. 2012); studies of new regulators will determine their impact on the localization and trafficking of Cu ATPases and expand the network of proteins involved in the regulation of copper metabolism. Similarly, proteomics/mass spectrometry approaches may help in the discovery of new ATP7A- and ATP7B-binding partners that regulate trafficking of Cu ATPases to and from the Golgi, as well as establish the role of posttranslational modifications in Cu ATPase targeting and sorting. Combining this approach with immunoisolation of ATP7A- and ATP7B-containing membranes would help characterize the post-Golgi compartments along the exocytic route(s) of both proteins. Finally, high-content microscopy screening of the siRNA or chemical libraries is expected to further expand or confirm a list of new molecular players that regulate ATP7A and/or ATP7B localization and activity. Taken together, such studies will provide new insights into the role of the Golgi in Cu homeostasis and will uncover new molecular targets for the development of next-generation therapeutic approaches to treat disorders associated with Cu imbalance.
References
Andreini C, Banci L, Bertini I, Rosato A (2008) Occurrence of copper proteins through the three domains of life: a bioinformatic approach. J Proteome Res 7:209–216
Bonifacino JS, Rojas R (2006) Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol 7:568–579
Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A (2009) Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am J Physiol Gastrointest Liver Physiol 296:G433–G444
Braiterman L, Nyasae L, Leves F, Hubbard AL (2011) Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking. Am J Physiol Gastrointest Liver Physiol 301:G69–G81
Burkhead JL, Morgan CT, Shinde U, Haddock G, Lutsenko S (2009) COMMD1 forms oligomeric complexes targeted to the endocytic membranes via specific interactions with phosphatidylinositol 4,5-bisphosphate. J Biol Chem 284:696–707
Cater MA, La Fontaine S, Shield K, Deal Y, Mercer JF (2006) ATP7B mediates vesicular sequestration of copper: insight into biliary copper excretion. Gastroenterology 130:493–506
Cobbold C, Ponnambalam S, Francis MJ, Monaco AP (2002) Novel membrane traffic steps regulate the exocytosis of the Menkes disease ATPase. Hum Mol Genet 11:2855–2866
Cobbold C, Coventry J, Ponnambalam S, Monaco AP (2003) The Menkes disease ATPase (ATP7A) is internalized via a Rac1-regulated, clathrin- and caveolae-independent pathway. Hum Mol Genet 12:1523–1533
de Bie P, Muller P, Wijmenga C, Klomp LW (2007) Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet 44:673–688
De Matteis MA, Luini A (2008) Exiting the Golgi complex. Nat Rev Mol Cell Biol 9:273–284
Dmitriev OY, Bhattacharjee A, Nokhrin S, Uhlemann EM, Lutsenko S (2011) Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and H1069Q mutants of copper-transporting ATPase ATP7B. J Biol Chem 286:16355–16362
Farr GA, Hull M, Mellman I, Caplan MJ (2009) Membrane proteins follow multiple pathways to the basolateral cell surface in polarized epithelial cells. J Cell Biol 186:269–282
Folsch H (2008) Regulation of membrane trafficking in polarized epithelial cells. Curr Opin Cell Biol 20:208–213
Forbes JR, Cox DW (2000) Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet 9:1927–1935
Francis MJ, Jones EE, Levy ER, Ponnambalam S, Chelly J, Monaco AP (1998) A Golgi localization signal identified in the Menkes recombinant protein. Hum Mol Genet 7:1245–1252
Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, Monaco AP (1999) Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci 112(Pt 11):1721–1732
Fuchs SA, Harakalova M, van Haaften G, van Hasselt PM, Cuppen E, Houwen RH (2012) Application of exome sequencing in the search for genetic causes of rare disorders of copper metabolism. Metallomics: Integr Biometal Sci 4:606–613
Gonzalez A, Rodriguez-Boulan E (2009) Clathrin and AP1B: key roles in basolateral trafficking through trans-endosomal routes. FEBS Lett 583:3784–3795
Greenough M, Pase L, Voskoboinik I, Petris MJ, O’Brien AW, Camakaris J (2004) Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells. Am J Physiol Cell Physiol 287:C1463–C1471
Guo Y, Nyasae L, Braiterman LT, Hubbard AL (2005) NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am J Physiol Gastrointest Liver Physiol 289:G904–G916
Gupta A, Lutsenko S (2009) Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem 1:1125–1142
Hasan NM, Gupta A, Polishchuk E, Yu CH, Polishchuk R, Dmitriev OY, Lutsenko S (2012) Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. J Biol Chem 287:36041–36050
Hirst J, Borner GH, Antrobus R, Peden AA, Hodson NA, Sahlender DA, Robinson MS (2012) Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr Biol Cb 22:1711–1716
Holloway ZG, Grabski R, Szul T, Styers ML, Coventry JA, Monaco AP, Sztul E (2007) Activation of ADP-ribosylation factor regulates biogenesis of the ATP7A-containing trans-Golgi network compartment and its Cu-induced trafficking. Am J Physiol Cell Physiol 293:C1753–C1767
Holloway ZG, Velayos-Baeza A, Howell GJ, Levecque C, Ponnambalam S, Sztul E, Monaco AP (2013) Trafficking of the Menkes copper transporter ATP7A is regulated by clathrin, AP-2, AP-1 and Rab22-dependent steps. Mol Biol Cell 24:1735–1748
Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem 272:21461–21466
La Fontaine S, Mercer JF (2007) Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys 463:149–167
La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Parton RG, Camakaris J, Mercer JF (1998) Functional analysis and intracellular localization of the human Menkes protein (MNK) stably expressed from a cDNA construct in Chinese hamster ovary cells (CHO-K1). Hum Mol Genet 7:1293–1300
La Fontaine S, Theophilos MB, Firth SD, Gould R, Parton RG, Mercer JF (2001) Effect of the toxic milk mutation (tx) on the function and intracellular localization of Wnd, the murine homologue of the Wilson copper ATPase. Hum Mol Genet 10:361–370
Lewis D, Pilankatta R, Inesi G, Bartolommei G, Moncelli MR, Tadini-Buoninsegni F (2012) Distinctive features of catalytic and transport mechanisms in mammalian sarco-endoplasmic reticulum Ca2+ATPase (SERCA) and Cu+(ATP7A/B) ATPases. J Biol Chem 287:32717–32727
Lim CM, Cater MA, Mercer JF, La Fontaine S (2006) Copper-dependent interaction of dynactin subunit p62 with the N terminus of ATP7B but not ATP7A. J Biol Chem 281:14006–14014
Luini A, Mironov AA, Polishchuk EV, Polishchuk RS (2008) Morphogenesis of post-Golgi transport carriers. Histochem Cell Biol 129:153–161
Lutsenko S (2010) Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol 14:211–217
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY (2007) Function and regulation of human copper-transporting ATPases. Physiol Rev 87:1011–1046
Martinelli D, Travaglini L, Drouin CA, Ceballos-Picot I, Rizza T, Bertini E, Carrozzo R, Petrini S, de Lonlay P, El Hachem M et al (2013) MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain: J neurol 136:872–881
Mellman I, Nelson WJ (2008) Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol 9:833–845
Montpetit A, Cote S, Brustein E, Drouin CA, Lapointe L, Boudreau M, Meloche C, Drouin R, Hudson TJ, Drapeau P, Cossette P (2008) Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. PLoS Genet 4:e1000296
Muth TR, Caplan MJ (2003) Transport protein trafficking in polarized cells. Annu Rev Cell Dev Biol 19:333–366
Nevitt T, Ohrvik H, Thiele DJ (2012) Charting the travels of copper in eukaryotes from yeast to mammals. Biochim Biophys Acta 1823:1580–1593
Nyasae L, Bustos R, Braiterman L, Eipper B, Hubbard A (2007) Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: copper-dependent redistribution between two intracellular sites. Am J Physiol Gastrointest Liver Physiol 292:G1181–G1194
Ohno H, Tomemori T, Nakatsu F, Okazaki Y, Aguilar RC, Foelsch H, Mellman I, Saito T, Shirasawa T, Bonifacino JS (1999) Mu1B, a novel adaptor medium chain expressed in polarized epithelial cells. FEBS Lett 449:215–220
Payne AS, Kelly EJ, Gitlin JD (1998) Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA 95:10854–10859
Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15:6084–6095
Petris MJ, Camakaris J, Greenough M, LaFontaine S, Mercer JF (1998) A C-terminal di-leucine is required for localization of the Menkes protein in the trans-Golgi network. Hum Mol Genet 7:2063–2071
Petris MJ, Voskoboinik I, Cater M, Smith K, Kim BE, Llanos RM, Strausak D, Camakaris J, Mercer JF (2002) Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. J Biol Chem 277:46736–46742
Pilankatta R, Lewis D, Inesi G (2011) Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J Biol Chem 286:7389–7396
Polishchuk RS, Mironov AA (2004) Structural aspects of Golgi function. Cell Mol Life Sci 61:146–158
Polishchuk RS, Polishchuk EV, Marra P, Alberti S, Buccione R, Luini A, Mironov AA (2000) Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J Cell Biol 148:45–58
Polishchuk EV, Di Pentima A, Luini A, Polishchuk RS (2003) Mechanism of constitutive export from the Golgi: bulk flow via the formation, protrusion, and en bloc cleavage of large trans-Golgi network tubular domains. Mol Biol Cell 14:4470–4485
Polishchuk R, Di Pentima A, Lippincott-Schwartz J (2004) Delivery of raft-associated, GPI-anchored proteins to the apical surface of polarized MDCK cells by a transcytotic pathway. Nat Cell Biol 6:297–307
Raposo G, Marks MS, Cutler DF (2007) Lysosome-related organelles: driving post-Golgi compartments into specialisation. Curr Opin Cell Biol 19:394–401
Robinson MS, Bonifacino JS (2001) Adaptor-related proteins. Curr Opin Cell Biol 13:444–453
Rodriguez-Boulan E, Kreitzer G, Musch A (2005) Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol 6:233–247
Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ (2000) Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119:782–793
Safaei R, Otani S, Larson BJ, Rasmussen ML, Howell SB (2008) Transport of cisplatin by the copper efflux transporter ATP7B. Mol Pharmacol 73:461–468
Schlief ML, Craig AM, Gitlin JD (2005) NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci: Off J Soc Neurosci 25:239–246
Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS (2008) Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 454:1142–1146
Shteyn E, Pigati L, Folsch H (2011) Arf6 regulates AP-1B-dependent sorting in polarized epithelial cells. J Cell Biol 194:873–887
Tumer Z, Moller LB (2010) Menkes disease. Eur J Hum Genet: EJHG 18:511–518
Vanderwerf SM, Cooper MJ, Stetsenko IV, Lutsenko S (2001) Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. J Biol Chem 276:36289–36294
Veldhuis NA, Valova VA, Gaeth AP, Palstra N, Hannan KM, Michell BJ, Kelly LE, Jennings I, Kemp BE, Pearson RB et al (2009) Phosphorylation regulates copper-responsive trafficking of the Menkes copper transporting P-type ATPase. Int J Biochem Cell Biol 41:2403–2412
Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ (2011) Advances in the understanding of mammalian copper transporters. Adv Nutr 2:129–137
Weigert R, Yeung AC, Li J, Donaldson JG (2004) Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell 15:3758–3770
Wilson C, Venditti R, Rega LR, Colanzi A, D’Angelo G, De Matteis MA (2011) The Golgi apparatus: an organelle with multiple complex functions. Biochem J 433:1–9
Yi L, Donsante A, Kennerson ML, Mercer JF, Garbern JY, Kaler SG (2012) Altered intracellular localization and valosin-containing protein (p97 VCP) interaction underlie ATP7A-related distal motor neuropathy. Hum Mol Genet 21:1794–1807
Yuan DS, Dancis A, Klausner RD (1997) Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem 272:25787–25793
Acknowledgments
We would like to acknowledge support from Telethon (grant # TGM11CB4) and AIRC (grant # IG 10233) to RP and from NIH to SL (grant # R01 DK 071865-07). We would also like to thank Ellen Abrams for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Polishchuk, R., Lutsenko, S. Golgi in copper homeostasis: a view from the membrane trafficking field. Histochem Cell Biol 140, 285–295 (2013). https://doi.org/10.1007/s00418-013-1123-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-013-1123-8