
Abstract
Lipid deposits can injury the kidney of diabetic patients and models. Sterol regulatory element binding protein-1 (SREBP-1) is transcription factor regulating the synthesis of fatty acid and triglyceride. At present whether the expression of SREBP-1 makes some effects on the lipid accumulation in diabetic kidney is not still clear completely. The purpose of our in vivo and in vitro study is to investigate the relationship between the expression of SREBP-1 and lipid abnormal metabolism in the type 1 diabetic rats and explore to inhibit SREBP-1 gene expression by RNA interfere in human renal proximal tubular epithelial cells line (HKC cells). The animal experiment showed that triglyceride and SREBP-1 were up-regulated in proximal tubule of diabetic rats’ kidney, which may result in increase of transforming growth factor-β1 (TGF-β1) and accumulation of extracellular matrix (ECM). The further HKC cells experiment confirmed SREBP-1 increasing resulted into lipid droplet formation. The expression of fatty acid synthase (FAS) in HKC cells transfected with specific plasmid for SREBP-1 gene was significantly more than that of the cells transfected with the control plasmid pcDNA3.1 and that of the untransfected cells. Simultaneously, up-regulation of TGF-β1 and fibronectin, an ECM glycoprotein, was evident in HKC cells transfected by specific SREBP-1 plasmid. Furthermore, we found that high glucose was a positive factor on the expression of SREBP-1 at protein and mRNA levels in HKC cells. High glucose makes effects on SREBP-1 in time-dependent manner, and the greatest effect was at 48 h. In addition, two effective eukaryotic expression plasmid vectors of shRNA aimed at SREBP-1 were designed and constructed successfully. Compared with the negative control plasmid group, the levels of the expression of SREBP-1 were inhibited by 24.11 and 36.15%, respectively, at mRNA level, 20.80 and 37.59%, respectively, at precursor segment of protein level, and 38.12 and 52.24%, respectively, at mature segment of protein level at 48 h after transfection. In vivo and in vitro study suggested that high glucose caused increasing SREBP-1 mRNA and protein in renal proximal tubule epithelial cells of type 1 diabetic rats. Increasing SREBP-1 plays an important role in the pathogenesis of renal lipid accumulation by up-regulation of FAS and ECM accumulation by inducing TGF-β1 expression. The application of vector-mediated RNAi could markedly inhibit the expression of SREBP-1 in HKC cells, which is a promising tool for future research into the mechanisms of renal lipid accumulation in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy is an important chronic complication of diabetes mellitus. Many factors are involved in the pathogenesis of this disease, for example, hyperglycemia, growth factors (Prasad et al. 2007), inflammatory cytokines, oxidative stress (Beisswenger et al. 2005) and advanced glycation end products (Suzuki et al. 2006). At present abnormal lipid accumulation has been testified to injury the kidney of diabetic patients and animal models (Ruan et al. 2004).
Sterol regulatory element-binding proteins (SREBPs) are members of the basic helix-loop-helix leucine zipper family of transcription factors (Brown and Goldstein 1997, 1999). SREBPs are synthesized as 1,150-amino acid precursor proteins that remain bound to the endoplasmic reticulum. Upon sterol deprivation, the precursor protein undergoes a sequential two-step cleavage process to release the NH2-terminal portion (Sakai et al. 1996). This NH2-terminal, mature segment then enters the nucleus and activates the transcription of genes involved in cholesterol and fatty acid synthesis by binding to sterol regulatory elements or to palindromic sequences called E-boxes within their promoter regions (Kim et al. 1995).
Currently, there are three isoforms of SREBP that have been characterized. SREBP-1a and -1c are derived from a single gene through the use of alternate promoters that give rise to different first exons (Brown and Goldstein 1999), and SREBP-1a is the more common isoform found in cultured cells and is a stronger activator of transcription than SREBP-1c because of a longer transactivating domain (Shimano et al. 1997; Shimomura et al. 1997). SREBP-2 is derived from a different gene and is known to be actively involved in the transcription of cholesterogenic enzymes. It has been shown that all cultured cells analyzed to date exclusively express SREBP-2 and the -1a isoform of SREBP-1, whereas most organs, including the liver, express predominantly SREBP-2 and the -1c isoform of SREBP-1 (Shimomura et al. 1997). Genes involved in cholesterol synthesis and uptake, regulated by SREBPs, include the low density lipoprotein receptor, 3-hydroxy-3-methylglutaryl-CoA synthase, 3-hydroxy-3-methylglutaryl-CoA reductase, squalene synthase (Guan et al. 1997; Sato et al. 1996). Fatty acid synthetic genes such as acetyl-CoA carboxylase, fatty acid synthase (FAS) (Magana and Osborne 1996) stearoyl-CoA desaturase-1 and -2 (SCD) (Tabor et al. 1998) ATP citrate lyase (ACL) (Sato et al. 2000), S14 (Mater et al. 1999), and glycerol-3-phosphate acyltransferase (Ericsson et al. 1997) are also directly activated by SREBPs.
Some renal injuries have been reported to involve triglyceride accumulation and high expression of SREBP-1 (Jiang et al. 2005; Saito et al. 2005). Renal SREBP-1 expression is increased in diabetes and that SREBP-1 plays an important role in the increased lipid synthesis, TG accumulation, mesangial expansion, glomerulosclerosis, and proteinuria by increasing the expression of transforming growth factor β1 (TGF-β1) and vascular endothelial growth factor (Sun et al. 2002). The study on PEPCK-promoter transgenic mice showed that overexpressing nuclear SREBP-1c exhibited enhancement of proteinuria with mesangial proliferation and matrix accumulation, mimicking diabetic nephropathy. These suggested that activation of glomerular SREBP-1c could contribute to emergence and/or progression of diabetic nephropathy (Ishigaki et al. 2007). However, the effects of expression of SREBP-1 in renal tubular cells on lipid droplet formation and renal tubule interstitial fibrosis were no well known.
RNA interference (RNAi) is a specific and powerful tool that is used to silence gene expression in the study of gene function and in the development of therapeutic strategies (Novina et al. 2002). Sustained RNAi can be achieved by constructing recombinant plasmid that synthesizes short-hairpin RNA (shRNA). A shRNA becomes siRNA, the final and functional product, through enzyme digestion of Dicer (Sui et al. 2002).
In this study, we investigated the function of tubular SREBP-1 expression in regulation of tubular lipid metabolism, growth factors secretion and tubule interstitial fibrosis using STZ-induced diabetic rats and HKC cells. Furthermore, we constructed two specific shRNA plasmid aimed at SREBP-1 gene and transfected them into HKC cells respectively for the first time to explore the effects on SREBP-1 expression and provide an effective gene silencing method.
Materials and methods
Reagents and chemicals
Oil Red O and streptozotocin (STZ) were purchased from Sigma Chemical Co. In situ hybridization kit for detecting SREBP-1 mRNA was from Tianjin Haoyang Co. Triglyceride detecting kit was obtained from Dongou Biotechnology Co. Polymer Detection System for Immuno-Histological Staining was bought from Zhongshan Golden Bridge Biotechnology Co. Lipofectamine 2000 was purchased from Invitrogen Co. Restriction enzyme BamHI, HindIII and SalI were bought from TaKaRa Co. The following products were from Santa Cruz Biotechnology Inc.: rabbit anti-SREBP-1 antibody (K-10; sc-367), rabbit anti-TGF-β1 antibody (sc-146), mouse anti-β-actin antibody (sc-47778). Rabbit anti-FN antibody was purchased from Beijing Biosynthesis Biotechnology Co.
Expression vectors and recombinant plasmid construction
pcDNA3.1-SC1 plasmid containing the gene for mature segment of human SREBP-1 protein was a gift from the professor Timothy F. Osborne, California University, USA. The empty expression vector pcDNA3.1 and E. coil strain DH5 alpha were preserved in our laboratory. The plasmid vector of pGenesil-1 was purchased from Wuhan Jingsai Co. which contains human U6 promoter and green fluorescence protein (GFP) for RNAi research. Two targeted sequences aimed at SREBP-1 gene were designed and the single-stranded DNA oligonucleotides for shRNA designated SREBP1-1 and SREBP1-2 were chemically synthesized (Table 1) and annealed to form double-stranded DNA. After pGenesil-1 vector was digested with restriction enzyme of BamHI and HindIII at 37°C for 3 h, the double-stranded DNA was cloned downstream of U6 promoter in the expression vector by T4 ligase at 16°C overnight. The recombinant plasmids were amplified and chosen by transfected into E. coil strain DH5 alpha and extracted using the kit from Promega Co. If the plasmid was constructed successfully, one 400 bp segment will be produced by digestion with SalI because of an enzyme site of SalI designed into the single-stranded DNA oligonucleotides. Finally, two constructs that were designated pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2 respectively were sequence-verified before use. pGenesil-1-SREBP1-1 targets the sequence: 5′-GCCTGACCATCTGTGAGAA-3′ and pGenesil-1-SREBP1-2 targets the sequence: 5′-CCAGCAGCTCCATTGACAA-3′. Meanwhile, a vector containing the following non-specific siRNA was designed as negative control (HK), of which the sequence was 5′-GACTTCATAAGGCGCATGC-3′.
Animals
The 50 Wistar rats were bought from the experimental animal center of Hebei Medical University. The animals were housed on an alternating 12 h light and dark cycle with room temperature of 20 ± 3°C and fed with the standard diet. Twenty-five rats were chosen randomly as normal group, and the rest (25 rats) as model of diabetic mellitus. The STZ-induced rats were injected intraperitoneally with 60 mg/kg body weight STZ in 50 mM sodium citrate solution (pH 4.5) to make type 1 diabetic models, and rats in the control group were injected with 50 mM sodium citrate solution. Only animals with plasma glucose concentrations >16.7 mM 1 week after the induction of STZ were considered diabetic (Bloch et al. 2007). Throughout the experimental period, tail vein blood glucose levels were measured every day and one rat regained normal serum glucose. For this experiment, we studied 24 normal rats and 24 diabetic rats. Normal control rats and diabetic rats were raised for 1, 2, 4 and 8 weeks, respectively, until they were sacrificed.
At the end of the study, rats were deeply anesthetized with chloral hydrate and blood samples were obtained through abdominal aorta. Renal cortex was removed, cleaned, washed and immediately saved in buffered neutral formalin for histopathological sections, or saved in liquid nitrogen for frozen sections and extraction of protein.
Cell culture and transient transfection
Human renal proximal tubular epithelial cells line (HKC) was kindly provided by Professor Chen Xiang-mei, Division of Nephropathy, 301 Hospital, Beijing, China. HKC cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (Gibco PRL,USA) containing penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively) and 10% fetal bovine serum (FBS), with a normal d-glucose concentration of 5 mM. Prior to use, cells at 80% confluence were incubated in serum-free medium for 24 h. In order to examine the direct effects of SREBP-1 on lipid droplet formation, HKC cells were randomly divided into three groups: untransfection normal control group, pcDNA3.1 control group and pcDNA3.1-SC1 group. At 48 h after transfection, the HKC cells were collected and analyzed for lipid droplets, SREBP-1, FAS, TGF-β1, FN expression. To investigate the effects of glucose on protein and mRNA expression of SREBP-1, serum-deprived HKC cells were cultured in DMEM containing 5 mM glucose (normal glucose) or 30 mM glucose (high glucose) for 12, 24, 48 and 72 h. At the end of each time point lipid droplets, SREBP-1, TGF-β1 and FN expression were examined by Oil Red O staining, Western blot and RT-PCR. For RNAi research, HKC cells were randomly divided into four groups: normal control group, HK control group, pGenesil-1-SREBP1-1 group and pGenesil-1-SREBP1-2 group. Total RNA and protein of the cells after 48 h transfection were extracted. Lipid droplet, SREBP-1, FAS, TGF-β1 and FN expression was assessed.
Transient transfection of HKC cells was carried out using Lipofectamine 2000 according to the manufacturer’s instruction. Briefly, HKC cells were cultured in six-well plates and the medium was changed the following day until 80% confluence was achieved. The cells were transfected with 4.0 μg plasmid DNA by 10 μl lipofectamine 2000 in 2 ml serum-free DMEM medium. At 6 h after transfection the medium was replaced by normal DMEM medium with 10% FBS for normal control untransfection group, pcDNA3.1 group and pcDNA3.1-SC1 group. For RNAi research, cells were grown in high glucose DMEM medium (30 mM glucose) with 10% FBS for an additional 48 h. At 48 h after transfection, fluorescent microscopy was used to examine GFP expression and then the cells were collected for the extraction of protein and total RNA. All experiments were repeated three times.
Measurement of TG content in renal tissue
The renal cortex lipid was extracted according to the Folch method (Folch et al. 1957). The TG was detected with the kit from Zhejiang Dongou Company.
Oil Red O staining
Accumulation of neutral fat in kidney of diabetic rats and HKC cells was examined by Oil Red O (St Louis, MO, USA) staining. Tissue sections and cultured cells were fixed for 30 min in 4% paraformaldehyde (in PBS), and stained for 15 min in 1% Oil Red O (in 60% isopropanol). Then, the sections were washed with 70% alcohol for 5 s to remove background staining. Finally, the cells were rinsed in tap water and counterstained with Harris hematoxylin (10 s). The stained sections were imaged with an Olympus microscope and examined in a blinded manner by the renal pathologist.
Immunohistochemistry, immunocytochemistry and quantitative image analysis
PV9000 Polymer Detection System for Immuno-Histological Staining was purchased from Zhongshan Biological Inc., Beijing, China. The procedure was performed according to the instruction of staining system. The cortex tissue was resected from kidney and fixed immediately in 4 % formaldehyde. Dehydration, transparence, embedding and section were performed according to the standard procedure. Antigen recovery was processed with microwave. The sections were blocked by pretreatment with 3% H2O2 for 10 min at 37°C. Finally, the sections were incubated respectively with primary antibodies against SREBP-1 (1:100, Santa Cruz Biotechnology, CA, USA), TGF-β1 (1:100, Santa Cruz Biotechnology, CA, USA) and FN (1:100, Beijing Biosynthesis Biotechnology Co. Ltd., Beijing, China) overnight at 4°C. At the following day, the sections were washed with PBS for 5 min ×3 and incubated with reagent 1 (polymer helper) for 30 min at 37°C. After incubated with reagent 2 (polyperoxidase-anti-mouse/rabbit IgG) for 30 min at 37°C, the sections were stained with diaminobenizidine (DAB). Negative controls were obtained by replacing specific antibody with PBS.
Cells were cultured on the cover glasses in six-well plates for 12, 24, 48 and 72 h, immediately washed three times with cold PBS and fixed with 75% ethanol for 30 min. Washed three times with PBS, cover slips with cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. Then, cells were blocked by pretreatment with 3% H2O2 for 10 min at 37°C. Finally HKC cells were incubated respectively with rabbit anti-SREBP-1 polyclonal antibody (1:100, Santa Cruz Biotechnology, CA, USA) overnight at 4°C. At the following day, cells were washed with PBS for three times and incubated with polymer helper for 30 min at 37°C. Thereafter, HKC cells were washed three times with PBS and then treated for 30 min at 37°C with polyperoxidase-anti-mouse/rabbit IgG. After washed with PBS, cover slips were stained with DAB. Negative controls were obtained by replacing specific antibody with PBS.
Brownish yellow granules in cytoplasm and nuclei were interpreted as positive region. The sections were imaged with Olympus microscope and characterized quantitatively by digital image analysis using the Image Pro-Plus 5.0 image-analysis software (Media Cybernetics, Silver Spring, MD) by using the method introduced by Chen et al. (1999). A series of 10 random images on several sections was taken for each immunostained parameter to obtain a mean value for statistical comparison. Staining was defined via color intensity that was applied equally to all images, and then measurements were obtained. The intensity of the labeling was determined by the computer program and gave a gray value ranging from zero (black) to 256 (white). Immunocytochemical parameters assessed in the area detected include mean stained area, mean intensity of stain and mean integrated optical density (mean IOD). Mean integral calculus of the mean stained area times the intensity of stain in each pixel in the area indicates the total amount of staining material in that area. All sections were inspected independently by two pathologists.
In situ hybridization
Frozen sections were used for in situ hybridization to detect the mRNA of SREBP-1. The sections were fixed with 4% formaldehyde in 0.1% DEPC and filled with 0.3% Triton X-100 solution for 10 min at room temperature. Tissues were incubated for 3 min at 37°C in 1 mg/ml proteinase K followed by blocking with 3% H2O2 solution for about 20 min at 37°C. After equilibration in 2 × SSC, the sections were prehybridized for 1 h at 37°C. Hybridization was carried out for 4 h at 37°C, and then the slides were washed with 2 × SSC for 5 min × 3 at 37°C. The sections were stained with DAB and negative controls were performed with PBS. Brownish yellow granules in the cytoplasm were considered as positive region. The sections were imaged with Olympus microscope and semiquantificationally analyzed through Image Pro-Plus 5.0 software by the renal pathologist. Sequences of oligonucleotide probes were respectively:
-
1.
5′-CTCAGGAAGAAACGTGTCAAGAAGTGC-3′
-
2.
5′-CCACAAGAAGTAGATCACACAGGAGCA-3′
-
3.
5′-TGGCTCCGTCTGTCTTCATGGCTGTCA-3′
Western blot
HKC cells were seeded in 100-cm2 flasks. Twenty-four hours after plating, cells were incubated with substances for different time intervals. Confluent cell layers were washed with PBS and lysed for 30 min at 4°C with 1% Nonidet P-40, 0.1% Triton X-100, 30 mM sodium phosphate (pH 7.4) containing 1 mM sodium orthovanadate, 2.5 mM Tris–HCl (pH 7.5), 100 mM NaCl and 10 μg/mL of leupeptin, aprotinin. Subsequently, the homogenate was centrifuged at 13,000 g for 30 min at 4°C. Insoluble material was removed and the supernatant was collected. Protein was quantified with the Bio-Rad protein colorimetric assay. The renal cortex protein was extracted and quantified according to the previous procedure.
After addition of sample buffer to the cellular extract and boiling samples at 95°C for 5 min, protein was separated on 7.5% SDS-PAGE gel. Protein was transferred onto PVDF membrane (Millipore Corporation, Bedford, MA) and the membrane was blocked for 1 h at room temperature with 5% BSA (bovine serum albumin) in Tris-buffered saline containing 0.05% Tween 20 (TBST). Next, blots were washed and incubated overnight at 4°C in TBST containing 1% BSA with rabbit primary antibodies for SREBP-1 (1:500, Santa Cruz Biotechnology, CA, USA), TGF-β1 (1:500, Santa Cruz Biotechnology, CA, USA), FN (1:500, Beijing Biosynthesis Biotechnology Co. Ltd., China) and β-actin (1:100, Santa Cruz Biotechnology, CA, USA). Membranes were washed three times with TBST, incubated with goat anti-rabbit or mouse horseradish peroxidase-conjugated secondary antibody (1:5,000 dilution in TBST containing 1% BSA, Santa Cruz Biotechnology) for 120 min at room temperature and then washed three times with TBST. After chemiluminescence reaction (Pierce, Rockford, IL), bands were detected by exposing the blots to X-ray films for the appropriate time period. For quantitative analysis, bands were detected and evaluated densitometrically with LabWorks software (UVP Laboratory Products, Upland, CA, USA), normalized for β-actin density.
Semi-quantitative RT-PCR
Total RNA was extracted from HKC cells with TRIzol reagent (Invitrogen Co., CA, USA) according to the instructions of the manufacturer. RNA samples were quantified by the measurement of optic absorbance (A) at 260 nm in a spectrophotometer, with the A260/A280 ratio ranging from 1.8 to 2.0, which indicates a high purity of extracted RNA. RNA concentrations were calculated according to A260. Total RNA (2 μg) was reverse transcribed using random primers and M-MLV at 42°C for 1 h and then heated to 94°C for 5 min in a total reaction volume of 20 μl. Equal amounts of the product of the reverse transcription reaction were subjected to PCR amplification. We co-amplified the housekeeping gene GAPDH to allow for a semi-quantifiable comparison of the PCR products. The PCR amplification began with a 5-min denaturation at 95°C, followed by 40 cycles of denaturation at 95°C for 45 s, annealing at 55°C for 45 s and extension at 72°C for 60 s. The final extension was set for 10 min at 72°C. After amplification, 5 μl of each PCR reaction product was electrophoresed on a 1.5% (w/v) agarose gel containing ethidium bromide (0.5 μg/ml). The mRNA levels of SREBP-1 and FAS were normalized with GAPDH mRNA level. All PCR primers were synthesized by Aoke Biotech, China. The sequences and the amplified lengths are shown as follows for HKC cells study: SREBP-1 (sense: GCTGAGTGCCCTGAACC; antisense: CCATCCACGAAGAAACG 237 bp), FAS (sense: TATGCTTCTTCGTGCAGCAGT; antisense: GTGGATGATGCTGATGATGGA 392 bp) and GAPDH (sense: TCCCATCACCATCTTCCA; antisense: CATCACGCCACAGTTTCC 390 bp).
Masson staining
Renal tubulointerstitial fibrosis is mainly featured as the produce of extracellular matrix (ECM) and Masson staining was used to observe the accumulation of ECM.
Statistical analysis
All data were expressed as mean ± standard deviation (SD) and analyzed with SPSS 11.0 for Windows. The values are normally distributed and one-way analysis of variance (ANOVA) with Student–Newman–Keuls test was used to determine statistically significant differences within and between groups. Student’s t test was performed to assess the statistical significance between the two sets of data. P < 0.05 was considered statistically significant.
Results
Triglyceride accumulation was located in the renal proximal tubular epithelium in type 1 diabetic rats
Compared with normal control group rats, TG content of renal cortex was elevated markedly in the diabetic rats fed respectively for 1, 2, 4 and 8 weeks (Table 2). The location study of triglyceride was carried out with the method of Oil Red O staining and the results revealed that red lipid droplets appeared in the renal proximal tubule epithelium, not glomerulus in the kidney of diabetic rats fed for 4 and 8 weeks. However, no lipid droplet was found in the kidney of normal control rats, 1 and 2 weeks diabetic rats (Fig. 1).

Oil Red O staining of renal cortex in normal control rats and diabetic rats fed for 8 weeks. a Normal control group ×100. b Normal control group ×400. c Diabetic group ×100. d Diabetic group ×400
Overexpression of SREBP-1 may induce to the triglyceride content increase and ECM accumulation in renal proximal tubule epithelium of type 1 diabetic rats
The expression of SREBP-1 protein was investigated by immunohistochemistry and the results showed that SREBP-1 was located in the cytoplasm and increased in the renal proximal tubule epithelial cells of diabetic rats. SREBP-1 expression of diabetic rats was higher than that of normal rats at all time spots (1, 2, 4, 8 weeks) through semi-quantitative image analysis. The expression of SREBP-1 in diabetic rats fed for 2 weeks was the strongest and the final mean IOD of immunohistochemitry images was 80019.17 ± 5698.50 that is 2.43 times than that of normal control rats (Table 3). SREBP-1 protein has two kinds of forms: precursor segment and mature segment. To further investigate whether altered SREBP-1 involved in both of them, we explored SREBP-1 protein expression by Western blot. We found that the precursor and mature segments both showed high expression in diabetic rats’ kidney at all time spots. After fed for 2 weeks, the diabetic rats showed highest expression of precursor and mature segments and bands IOD ratio (SREBP-1/β-actin) were 0.673 ± 0.027 and 0.670 ± 0.028 respectively. In situ hybridization was used to detect SREBP-1 mRNA in both diabetic rats and normal control rats at four time spots. The mRNA of SREBP-1 was found to increase in the renal cortex of diabetic rats (Table 4) and was located in the cytoplasm of renal proximal tubular cells. On the contrary, renal tubular cells of control normal rats showed only mild stained (Fig. 2).

SREBP-1 expression increased in diabetic rats’ renal proximal tubule in mRNA and protein levels at four time points. a Immunohistochemical staining for SREBP-1 protein in renal tissue sections of normal control rats fed for 2 weeks ×400. b Immunohistochemical staining for SREBP-1 protein in renal tissue sections of diabetic rats fed for 2 weeks ×400. c Cell lysates were subjected to SDS-PAGE and Western blot analysis using SREBP-1 antibody. β-actin staining of blots after transfer revealed equivalent loading of total protein. d Precursor segment of SREBP-1 protein was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls. All values are means ± SD of three separate experiments. e Mature segment of SREBP-1 protein was quantified using LabWorks software. f The expression of SREBP-1 mRNA in kidney of normal control rats fed for 2 weeks was detected by in situ hybridization ×400. g The expression of SREBP-1 mRNA in kidney of diabetic rats fed for 2 weeks was detected by in situ hybridization ×400
Masson staining revealed the partial accumulation ECM in renal tubules in diabetic rats fed for 8 weeks. However, no significant ECM was found in kidney of normal control rats and diabetic rats fed for 1, 2 and 4 weeks (Fig. 3a, b). Fibronectin (FN) is a large modular glycoprotein that is a polymeric fibrillar network in the ECM (Albrecht et al. 1999). Immunohistochemistry was used to investigate the expression of FN in pathological tissue sections. The result testified that FN was located in cytoplasm of renal proximal tubular cells. Compared with corresponding normal control rats, expression of FN increased significantly in diabetic rats fed for 4 and 8 weeks. However, no difference was found between normal control rats and diabetic rats fed for 1 and 2 weeks (Fig. 3c, d) (Table 5). TGF-β1 plays an important role in fibrogenesis (Chai et al. 2003). We investigated, with immunohistochemical methods, the location and expression of TGF-β1. The finding of location study revealed that TGF-β1 immunostaining was in the cytoplasm of renal proximal tubular cells. Compared with normal control group, there was marked increase in renal cortex of diabetic rats at all four spots (Fig. 3e, f) (Table 6).

Masson staining and immunohistochemical staining for FN and TGF-β1 in normal control rats and diabetic rats fed for 8 weeks. a Masson staining of normal rats ×400. b Masson staining of diabetic rats ×400. c Immunohistochemical staining for FN in normal control rats ×400. d Immunohistochemical staining for FN in diabetic rats ×400. e Immunohistochemical staining for TGF-β1 in normal control rats ×400. f Immunohistochemical staining for TGF-β1 in diabetic rats ×400
pcDNA3.1-SC1 plasmid transfection confirmed the effects of SREBP-1 expression on lipid droplet formation and ECM produce in HKC cells
In order to confirm the effects of SREBP-1 gene on lipid metabolism in renal proximal tubule epithelial cells, we carried out in vitro research by transfection of specific plasmid for SREBP-1 gene into HKC cells for the first time. The expression of SREBP-1 mRNA and protein were analyzed by RT-PCR and Western blot. IOD ratio of SREBP-1 mRNA bands normalized by internal control GAPDH was 0.20 ± 0.09 for control group, 0.30 ± 0.07 for pcDNA3.1 group, 1.24 ± 0.10 for pcDNA3.1-SC1 group. Compared with normal control group, the SREBP-1 mRNA expression was increased by 6.158 times in pcDNA3.1-SC1 transfection group. The results of Western blot demonstrated that normalized by β-actin, IOD ratio of SREBP-1 protein was 0.93 ± 0.21 for normal control group, 1.16 ± 0.25 for pcDNA3.1 group, 3.09 ± 0.25 for pcDNA3.1-SC1 group. Compared with normal control group, the protein expression was up-regulated markedly by transfection of pcDNA3.1-SC1 plasmid (Fig. 4). Oil Red O staining revealed that overexpression of SREBP-1 in HKC cells by transfection of pcDNA3.1-SC1 plasmid resulted into lipid droplet formation that was not seen in normal control HKC cells. FAS was a key enzyme for fatty acid synthesis. To investigate whether it was involved in the regulation of SREBP-1 on triglyceride accumulation, we detected the FAS mRNA expression in HKC cells transfected with SREBP-1 specific plasmid by RT-PCR. The results showed that FAS mRNA expression in HKC cells transfected with pcDNA3.1-SC1 was significantly more than that in untransfected HKC cells (Fig. 5).

Transient transfection of HKC cells with pcDNA3.1-SC1 plasmid significantly increased SREBP-1 mRNA and protein. a Total cellular RNA from pcDNA3.1-SC1 or vector transfected HKC cells was subjected to RT-PCR analyses for SREBP-1. GAPDH probe was used as the loading control. b SREBP-1 RNA levels were quantified using LabWorks software. RNA levels were normalized to respective GAPDH controls. All values are means ± SD of three separate experiments. c Lysates of HKC cells transfected with SREBP-1 expression vector (pcDNA3.1-SC1) or control vector were subjected to SDS-PAGE and Western blot analysis using antibody against mature segment of SREBP-1 protein. β-actin staining of blots after transfer revealed equivalent loading of total protein. d SREBP-1 protein levels were quantified using LabWorks software. Protein levels were normalized to respective β-actin controls

Transient transfection of HKC cells with pcDNA3.1-SC1 plasmid significantly increased FAS mRNA and lipid droplets. a Oil Red O staining in normal untransfected HKC cells ×400. b Oil Red O staining in vector transfected HKC cells ×400. c Oil Red O staining in pcDNA3.1-SC1 plasmid transfected HKC cells ×400. d Oil Red O staining in pcDNA3.1-SC1 plasmid transfected HKC cells ×800. e Total cellular RNA from pcDNA3.1-SC1 or vector transfected HKC cells was subjected to RT-PCR analyses for FAS. GAPDH probe was used as the loading control. Results presented are representative of three separate experiments. f FAS RNA levels were quantified using LabWorks software. RNA levels were normalized to respective GAPDH controls
Simultaneously, TGF-β1 and FN protein was detected to explore the effects of SREBP-1 expression on ECM produce. The results of Western blot showed that after transfection with SREBP-1 plasmid for 48 h the HKC cells presented high expression of TGF-β1 and FN protein. These revealed that SREBP-1 increase led to the secretion of TGF-β1 and accumulation of ECM in renal tubular cells (Fig. 6).

Transient transfection of HKC cells with pcDNA3.1-SC1 plasmid significantly increased FN and TGF-β1 expression. a Cell lysates were subjected to SDS-PAGE and Western blot analysis using FN and TGF-β1 antibody. β-actin staining of blots after transfer revealed equivalent loading of total protein. b FN protein levels were quantified using LabWorks software. c TGF-β1 protein levels were quantified using LabWorks software
High glucose modulates SREBP-1 protein expression in HKC cells
From the results of rats experiment, we presumed that overexpression of SREBP-1 in renal proximal tubular cells originated from high glucose condition. In order to confirm this hypothesis, HKC cells were stimulated with high glucose and the expression of SREBP-1 gene was detected. HKC cells were cultured in media containing 5 mM glucose (normal glucose level) or 30 mM glucose (high glucose level) respectively for 12, 24, 48 and 72 h. We performed Oil-Red O staining, which revealed the accumulation of neutral fat in cytoplasm. We found that lipid droplets started to appear at 48 h under the stimulation of high glucose (Fig. 7a, b). The results of immunocytochemistry showed that the addition of high glucose led to significant elevation of SREBP-1 protein located in cytoplasm and nuclei. As can be seen in Fig. 7c, d and Table 7, comparison with normal glucose group, high glucose group showed much larger expression of SREBP-1 (P < 0.01) at the corresponding time point.

High glucose stimulated lipid droplet formation and up-regulation of SREBP-1 in HKC cells. a Oil Red O staining of HKC cells in 72 h normal glucose group ×400. b Oil Red O staining of HKC cells in 72 h high glucose group ×400. c Immunocytochemistry of SREBP-1 protein in 48 h normal glucose group ×400. d Immunocytochemistry of SREBP-1 protein in 48 h high glucose group ×400
To study the impact of glucose on the precursor and mature nuclear forms of SREBP-1, total protein extracts were prepared from HKC cells that had been cultured for 12, 24, 48, 72 h in DMEM containing 5 mM glucose or 30 mM glucose. Equal amounts of proteins were separated by 7.5% SDS/PAGE and subjected to Western blot analysis. As Fig. 8a, b and c show, immunoblotting studies revealed that precursor and mature forms of SREBP-1 protein were significantly higher in HKC cells of high glucose group than controls at all time points. The intensity of SREBP-1 precursor bands in high glucose group was 4.9 times more than that of normal glucose group. Furthermore, the mature form expression in high glucose group was 6.5 times more than that of normal control group at 48 h. We also performed experiments to detect the existence of a time-dependent manner of glucose-induced changes in the precursor and nuclear forms of SREBP-1. The precursor content of SREBP-1 increased gradually after high glucose addition. This increase reached the maximum after 48 h and the density ratio of SREBP-1 precursor/β-actin was 2.334. The changes in the mature form were parallel to the changes in precursor form of SREBP-1 in the HKC cells treated with high glucose. The density ratio of SREBP-1 mature form/β-actin was 1.101 at 48 h that was also maximal.

High glucose increased SREBP-1 expression in mRNA and protein levels in HKC cells. a Cell lysates were subjected to SDS-PAGE and Western blot analysis using SREBP-1 antibody. β-actin staining of blots after transfer revealed equivalent loading of total protein. b Precursor segment of SREBP-1 protein was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls. All values are means ± SD of three separate experiments. c Mature segment of SREBP-1 protein was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls. d Total cellular RNA from HKC cells under stimulation of high glucose or normal glucose was subjected to RT-PCR analyses for SREBP-1. GAPDH probe was used as the loading control. Results presented are representative of three separate experiments. e SREBP-1 RNA levels were quantified using LabWorks software. RNA levels were normalized to respective GAPDH controls
Increased expression of SREBP-1 mRNA was further confirmed by RT-PCR. We first assessed the effects of high glucose on SREBP-1 mRNA expression. Being consistent with the results observed at the protein level, semi-quantitative RT-PCR analysis also showed a significant increase in SREBP-1 mRNA expression under high glucose condition (30 mM) at all time points (Fig. 8d, e).
Under stimulation of high glucose for 48 h and 72 h, HKC cells showed more expression of TGF-β1 and FN than that of normal glucose group. However, HKC cells treated with high glucose for 12 and 24 h showed no difference in TGF-β1 and FN expression with normal glucose group. These finding revealed that high glucose may result in TGF-β1 secretion and ECM produce through up-regulation of SREBP-1(Fig. 9).

High glucose increased FN and TGF-β1 expression in HKC cells. a Lysates of HKC cells were subjected to SDS-PAGE and Western blot analysis using antibodies against FN and TGF-β1. β-actin staining of blots after transfer revealed equivalent loading of total protein. b FN protein was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls. c TGF-β1 protein was quantified using LabWorks software
Recombinant shRNA plasmid inhibited SREBP-1 gene expression in HKC cells
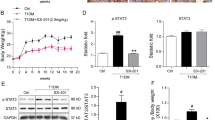
We performed SREBP-1 gene silencing research with the method of vector-mediated RNAi in HKC cells for the first time. Restriction endonuclease digestion of two recombinant shRNA plasmids by SalI got a 400 bp DNA fragment, respectively. Besides, the sequencing-identification also confirmed that the target sequence had inserted into the predicted site precisely and the recombining plasmids were successfully constructed (Fig. 10). At 48 h after transfection, GFP expression was observed though the comparative analysis of bright field image and corresponding fluorescent image and about 40% HKC cells were transfected successfully (Fig. 11). Meanwhile, the SREBP-1 mRNA and protein were inspected using the methods of RT-PCR and Western blot. Compared with HK group, the SREBP-1 mRNA was markedly inhibited by 24.11, 36.15%, respectively, in recombinant plasmid groups. The results of Western blot revealed that IOD ratio of precursor segment of SREBP-1 protein bands (SREBP-1/β-actin) was 0.83 ± 0.03 for normal control group, 0.80 ± 0.02 for HK group, 0.63 ± 0.02 for pGenesil-1-SREBP1-1 group and 0.50 ± 0.03 for pGenesil-1-SREBP1-2 group. IOD ratio of mature segment of SREBP-1 protein bands (SREBP-1/β-actin) was 1.18 ± 0.02 for normal control group, 1.20 ± 0.03 for HK group, 0.75 ± 0.02 for pGenesil-1-SREBP1-1 group and 0.58 ± 0.03 for pGenesil-1-SREBP1-2 group. IOD ratios in recombinant plasmid groups were significantly lower than that of normal control group and HK negative control group (Fig. 12). The results of Oil Red O staining revealed that significant lipid droplets were found in HKC cells of untransfection group and HK group under stimulation of high glucose for 48 h, on the contrary, the effects of high glucose on lipid droplet formation was inhibited by transfection of specific SREBP-1 shRNA plasmid for 48 h. No lipid droplet was found in HKC cells of pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2 groups. Similarly, compared with HK group, FAS mRNA expression respectively decreased by 10.68 and 33.18% in HKC cells of pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2 groups. Furthermore, TGF-β1 and FN protein were detected by the method of Western blot and it can be found that they both decreased in HKC cells of shRNA plasmid groups (Fig. 13).

Two recombinant shRNA plasmids were constructed successfully. a Restriction enzyme cleavage of recombinant plasmid by SalI. b Sequencing-identification of pGenesil-1-SREBP1-1 recombinant plasmid. c Sequencing-identification of pGenesil-1-SREBP1-2 recombinant plasmid

Transient transfection of HKC cells with recombinant plasmids (pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2) significantly caused GFP expression which means successful transfection. a Bright field image in HKC cells transfected with pGenesil-1-SREBP1-1 ×200. b Fluorescent image in HKC cells transfected with pGenesil-1-SREBP1-1 ×200. c Bright field image in HKC cells transfected with pGenesil-1-SREBP1-2 ×200. d Fluorescent image in HKC cells transfected with pGenesil-1-SREBP1-2 ×200

Transient transfection of HKC cells with recombinant shRNA plasmids (pGenesil-1-SREBP1-1, pGenesil-1-SREBP1-2) respectively significantly decreased SREBP-1 RNA and protein. a Total cellular RNA from recombinant shRNA plasmid or control vector transfected HKC cells was subjected to RT-PCR analyses for SREBP-1. GAPDH probe was used as the loading control. Results presented are representative of three separate experiments. b SREBP-1 RNA levels were quantified using LabWorks software. RNA levels were normalized to respective GAPDH controls. c Lysates of HKC cells transfected with recombinant shRNA plasmids (pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2) or control vector were subjected to SDS-PAGE and Western blot analysis using antibody against SREBP-1 protein. β-actin staining of blots after transfer revealed equivalent loading of total protein. d Precursor segment of SREBP-1 protein level was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls. e Mature segment of SREBP-1 protein level was quantified using LabWorks software. Protein levels were normalized to respective β-actin controls

Transient transfection of HKC cells with recombinant shRNA plasmids (pGenesil-1-SREBP1-1, pGenesil-1-SREBP1-2) decreased lipid droplet, FAS, FN and TGF-β1. a Oil Red O staining in untransfected HKC cells ×400. b Oil Red O staining in pGenesil-1-HK control plasmid transfected HKC cells ×400. c Oil Red O staining in pGenesil-1-SREBP1-1 plasmid transfected HKC cells ×400. d Oil Red O staining in pGenesil-1-SREBP1-2 plasmid transfected HKC cells ×400. e Total cellular RNA from HKC cells was subjected to RT-PCR analyses for FAS. GAPDH probe was used as the loading control. f FAS RNA were quantified using LabWorks software. RNA levels were normalized to respective GAPDH controls. g Cell lysates were subjected to SDS-PAGE and Western blot analysis using TGF-β1 and FN antibodies. β-actin staining of blots after transfer revealed equivalent loading of total protein. h TGF-β1 protein levels were quantified using LabWorks software. i FN protein levels were quantified using LabWorks software
Discussion
The presence of lipid deposits in the kidney of diabetic patients and experimental animals has been described by several investigators and some studies testified that lipid deposits play an important role in the pathogenesis of diabetic nephropathy (Guijarro et al. 1995; Lee et al. 1991). SREBP-1 plays a prominent role in regulating the synthesis of fatty acid. Some researcher testified that activation of SREBP-1 resulted in intramyocellular triglyceride (IMTG) increase in the Zucker diabetic fatty rats (ZDF) (Nadeau et al. 2007). To investigate whether SREBP-1 makes effects on triglyceride accumulation in the kidney of diabetic rats is the objective of our study.
We investigated the triglyceride content of kidney of diabetic rats and found that triglyceride markedly increased. In addition, location research of lipid accumulation was performed by Oil Red O staining and the results showed that lipid droplets presented in the renal proximal tubular epithelium. These are similar to the finding of Sun et al. (2002). Furthermore, we study SREBP-1 expression in kidney of diabetic mellitus. Overexpression of SREBP-1 was found to be mostly located in cytoplasm of kidney proximal tubule epithelial cells. The glomerular cells weren’t almost found stained. There indicated that increasing SREBP-1 of renal proximal tubular cells may be associated with abnormal triglyceride deposits. Some studies found that SREBP-1a transgenic mice (Sun et al. 2002), Akita and OVE26 diabetic mice (Proctor et al. 2006) presented lipid droplets in glomerulus; however, PEPCK-promoter transgenic mice (Ishigaki et al. 2007) and STZ-induced diabetic mice (Sun et al. 2002) showed overexpression of SREBP-1 in glomerulus without evident lipid droplets. The location difference indicated that animal race, gene change and high glucose may be involved in SREBP-1 expression and lipid droplet formation in diabetic kidney.
We subsequently performed studies by Western blot to observe expression of precursor and mature segment of SREBP-1 protein. Our research demonstrated that both precursor and mature segment were up-regulated. The result is similar to Szolkiewicz M’ study that supposed SREBP transcription factors play an important role in disturbed lipid metabolism in renal failure (Szolkiewicz et al. 2007). The finding of in situ hybridization that the renal proximal tubular cells in diabetic rats showed increasing SREBP-1 mRNA suggested overexpression of SREBP-1 protein may be caused by up-regulation of mRNA. Meanwhile, detection of in situ hybridization also confirmed renal proximal tubular epithelium location of SREBP-1 expression, not renal glomerulus. Therefore, the conclusion can be drawn from the serial location experiments that lipid accumulation of early diabetic renal tissue derived from excess lipid droplets caused by increasing of SREBP-1 gene in renal proximal tubular cells. Accumulation of ECM and fibrosis is the important character of diabetic nephropathy (Sowers et al. 2007). Our results showed the increase of TGF-β1 and FN in renal tubular cells of diabetic rats. Masson staining also confirmed partial accumulation of ECM. From these, we can conclude that SREBP-1 expression and lipid droplets may play an important role in renal tubulointerstitial ECM deposit in diabetic nephropathy.
Our animal experiment suggested that up-regulation of SREBP-1 may result in renal tissue triglyceride accumulation through lipid droplet formation of renal proximal tubular cells in diabetic rats’ kidney. To determine whether SREBP-1 expression was associated directly with triglyceride accumulation in renal proximal tubule epithelium, the specific plasmid for mature segment of SREBP-1 protein was transfected into HKC cells by Lipofectamine 2000. Overexpression of SREBP-1 was confirmed by Western blot and RT-PCR, then, there is a marked up-regulation of FAS mRNA that mediates fatty acid synthesis, resulting in the accumulation of lipid droplets. Sun found similar results in SREBP-1a transgenic mice that there was an 8-fold increase in SREBP-1 protein expression and the triglyceride content of renal cortex was 2–3-fold higher in Tg-SREBP-1a mice than in wild-type littermates (Sun et al. 2002). So our results indicated that SREBP-1 expression can result into lipid droplets in HKC cells through up-regulation of FAS mRNA. Consequently, the detecting of TGF-β1 and FN showed the same results as animal experiment. SREBP-1 specific plasmid can upregulate the secretion of TGF-β1 and FN in HKC cells through inducing cellular lipid droplets.
Many factors were involved in the SREBP-1 expression regulation such as polyunsaturated fatty acid (Sampath and Ntambi 2005), Intermittent hypoxia (Li et al. 2005), LXR (Pawar et al. 2003), insulin (Dif et al. 2006). Some researchers found high glucose take effects on SREBP-1 expression. INS-1 cells treated with high glucose resulted in SREBP-1c activation, lipid accumulation, impaired glucose-stimulated insulin secretion and apoptosis. These lipotoxic effects of high glucose were largely prevented by induction of a dominant-negative mutant of SREBP-1c, suggesting SREBP-1c is a major factor responsible for beta cell glucolipotoxicity (Wang et al. 2005). Sun also found SREBP-1 activation caused by high glucose in mesangial cells (Sun et al. 2002). To determine high glucose potentiation of SREBP-1-induced triglyceride accumulation in renal proximal tubule epithelium, we performed the experiment using HKC cells. Our in vitro experimental data demonstrated that high glucose significantly increased both precursor and nuclear forms of SREBP-1 protein in HKC cells. The increase in mature form of SREBP-1 accompanying increase in precursor form raised the possibility that the synthesis of the SREBP-1 precursor form was most likely due to an increase in SREBP-1 mRNA. To check this possibility, we measured the amounts of mRNA for SREBP-1 by RT-PCR of RNA isolated from HKC cells extracts. The data revealed that SREBP-1 mRNA significantly increased in HKC cells treated with high glucose. The results showed that high glucose first induced SREBP-1 mRNA up-regulation, which then led to SREBP-1 protein increase. The influence of glucose on SREBP-1 protein has been further proposed not to induce SREBP-1 protein expression after 6 h incubation (Foretz et al. 1999; Wang et al. 2005). We compared precursor and mature forms of SREBP-1 protein in HKC cells cultured in either low (5 mM) or high (30 mM) glucose-containing media for 12, 24, 48 and 72 h. These glucose concentrations are commonly used to estimate the effects of glucose in cell culture studies and also reflect glucose level of human plasma in physiological and diabetic conditions. The present study testified that high glucose can affect the expression of precursor and mature forms of SREBP-1 protein from 12 to 72 h in HKC cells, and the effect was greatest at the high glucose concentration at 48 h. The results suggested that the induction might be mediated through some metabolites of glucose and that it took some time to have the effect to reach its peak. The discrepancies in time-course effects of high glucose between our study and those of other researcher’s may be due to different sensitivities to glucose in different cells and tissues.
RNAi, also known as RNA interference, is a powerful tool for silencing gene expression through short interfering 21 23-mer double-strand RNA segments (siRNA) that guide mRNA degradation in a sequence-specific fashion (Soutschek et al. 2004). Compared with other RNAi methods, such as RNAi molecular synthesis in vitro, construction of expression frame, shRNA can directly construct RNAi DNA template expression vector in vitro, and it has better stability. shRNA in mammal cells can not only stably block the expression of target genes, also stably inherit to filial generation, and it has replaced traditional gene knock-out technique to establish transgenic cell lines and transgenic animals. Therefore, constructing shRNA expression vector has become the main method for RNAi technology at present. We successfully constructed two recombinant expression plasmids for SREBP-1, pGenesil-1-SREBP1-1 and pGenesil-1-SREBP1-2, performed the study of RNA interference on inhibiting overexpression of SREBP-1 by transfection of eukaryotic expression vector in HKC cells under high glucose condition. The results showed that at 48 h after transfection, the expression of SREBP-1 mRNA was inhibited by 24.11 and 36.15% respectively and provided a theoretical basis and technical reserves for future animal experiment. The consequence of SREBP-1 inhibition was explored and FAS expression and lipid droplets were decreased. Similarly, both TGF-β1 and FN was decreased by inhibition of SREBP-1 expression. It is meaning to avoid the injury of lipid deposit and through specific RNAi plasmid aimed at SREBP-1 gene.
In summary, our results indicate high glucose caused increasing SREBP-1 mRNA and protein of renal proximal tubular epithelium in type 1 diabetic rats. Increasing SREBP-1 plays an important role in the pathogenesis of renal triglyceride accumulation by up-regulation of FAS. Cellular lipid droplets can induce ECM produce by up-regulation of TGF-β1 that is crucial in pathogenesis of diabetic nephropathy. The application of vector-mediated RNAi could markedly inhibit the expression of SREBP-1 in HKC cells, decrease lipid droplet formation and accumulation of ECM which is a promising tool for future research into the mechanisms of renal lipid accumulation in vivo.
References
Albrecht M, Renneberg H, Wennemuth G, Möschler O, Janssen M, Aumüller G, Konrad L (1999) Fibronectin in human prostatic cells in vivo and in vitro: expression, distribution, and pathological significance. Histochem Cell Biol 112:51–61
Beisswenger PJ, Drummond KS, Nelson RG, Howell SK, Szwergold BS, Mauer M (2005) Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 54:3274–3281
Bloch K, Shichman E, Vorobeychik M, Bloch D, Vardi P (2007) Catalase expression in pancreatic alpha cells of diabetic and non-diabetic mice. Histochem Cell Biol 127:227–232
Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340
Brown MS, Goldstein JL (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA 96:11041–11048
Chai Q, Krag S, Chai S, Ledet T, Wogensen L (2003) Localisation and phenotypical characterisation of collagen-producing cells in TGF-beta 1-induced renal interstitial fibrosis. Histochem Cell Biol 119:267–280
Chen MK, Strande LF, Beierle EA, Kain MS, Geldziler BD, Doolin EJ (1999) Fas-mediated induction of hepatocyte apoptosis in a neuroblastoma and hepatocyte coculture model. J Surg Res 84:82–87
Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E (2006) Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J 400:179–188
Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA (1997) Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J Biol Chem 272:7298–7305
Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509
Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Lièpvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferré P, Foufelle F (1999) ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol 19:3760–3768
Guan G, Dai PH, Osborne TF, Kim JB, Shechter I (1997) Multiple sequence elements are involved in the transcriptional regulation of the human squalene synthase gene. J Biol Chem 272:10295–10302
Guijarro C, Kasiske BL, Kim Y, O’Donnell MP, Lee HS, Keane WF (1995) Early glomerular changes in rats with dietary-induced hypercholesterolemia. Am J Kidney Dis 26:152–161
Ishigaki N, Yamamoto T, Shimizu Y, Kobayashi K, Yatoh S, Sone H, Takahashi A, Suzuki H, Yamagata K, Yamada N, Shimano H (2007) Involvement of glomerular SREBP-1c in diabetic nephropathy. Biochem Biophys Res Commun 364:502–508
Jiang T, Liebman SE, Lucia MS, Li J, Levi M (2005) Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int 68:2608–2620
Kim JB, Spotts GD, Halvorsen YD, Shih HM, Ellenberger T, Towle HC, Spiegelman BM (1995) Dual DNA binding specificity of ADD1/SREBP1 controlled by a single amino acid in the basic helix-loop-helix domain. Mol Cell Biol 15:2582–2588
Lee HS, Lee JS, Koh HI, Ko KW (1991) Intraglomerular lipid deposition in routine biopsies. Clin Nephrol 36:67–75
Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, Marino RL, Rodriguez A, Hubbard WC, O’Donnell CP, Polotsky VY (2005) Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res 97:698–706
Magana MM, Osborne TF (1996) Two tandem binding sites for sterol regulatory element binding proteins are required for sterol regulation of fatty-acid synthase promoter. J Biol Chem 271:32689–32694
Mater MK, Thelen AP, Pan DA, Jump DB (1999) Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J Biol Chem 274:32725–32732
Nadeau KJ, Ehlers LB, Aguirre LE, Reusch JE, Draznin B (2007) Discordance between intramuscular triglyceride and insulin sensitivity in skeletal muscle of Zucker diabetic rats after treatment with fenofibrate and rosiglitazone. Diabetes Obes Metab 9:714–723
Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, Collman RG, Lieberman J, Shankar P, Sharp PA (2002) siRNA-directed inhibition of HIV-1 infection. Nat Med 8:681–686
Pawar A, Botolin D, Mangelsdorf DJ, Jump DB (2003) The role of liver X receptor-alpha in the fatty acid regulation of hepatic gene expression. J Biol Chem 278:40736–40743
Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK (2007) Association of TGFbeta1, TNFalpha, CCR2 and CCR5 gene polymorphisms in type-2 diabetes and renal insufficiency among Asian Indians. BMC Med Genet 8:1–6
Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M (2006) Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 55:2502–2509
Ruan XZ, Moorhead JF, Fernando R, Wheeler DC, Powis SH, Varghese Z (2004) Regulation of lipoprotein trafficking in the kidney: role of inflammatory mediators and transcription factors. Biochem Soc Trans 32:88–91
Saito K, Ishizaka N, Hara M, Matsuzaki G, Sata M, Mori I, Ohno M, Nagai R (2005) Lipid accumulation and transforming growth factor-beta upregulation in the kidneys of rats administered angiotensin II. Hypertension 46:1180–1185
Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL (1996) Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85:1037–1046
Sampath H, Ntambi JM (2005) Polyunsaturated fatty acid regulation of genes of lipid metabolism. Annu Rev Nutr 25:317–340
Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, Maeda M (1996) Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J Biol Chem 271:26461–26464
Sato R, Okamoto A, Inoue J, Miyamoto W, Sakai Y, Emoto N, Shimano H, Maeda M (2000) Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J Biol Chem 275:12497–12502
Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL (1997) Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 99:846–854
Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS (1997) Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99:838–845
Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Röhl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP (2004) Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432:173–178
Sowers KM, Habibi J, Hayden MR (2007) Diabetic nephropathy and tubulointerstitial fibrosis in cardiometabolic syndrome and type 2 diabetes mellitus. J Cardiometab Syndr 2:143–148
Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC, Shi Y (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 99:5515–5520
Sun L, Halaihel N, Zhang W, Rogers T, Levi M (2002) Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem 277:18919–18927
Suzuki D, Toyoda M, Yamamoto N, Miyauchi M, Katoh M, Kimura M, Maruyama M, Honma M, Umezono T, Yagame M (2006) Relationship between the expression of advanced glycation end-products (AGE) and the receptor for AGE (RAGE) mRNA in diabetic nephropathy. Intern Med 45:435–441
Szolkiewicz M, Chmielewski M, Nogalska A, Stelmanska E, Swierczynski J, Rutkowski B (2007) The potential role of sterol regulatory element binding protein transcription factors in renal injury. J Ren Nutr 17:62–65
Tabor DE, Kim JB, Spiegelman BM, Edwards PA (1998) Transcriptional activation of the stearoyl-CoA desaturase 2 gene by sterol regulatory element-binding protein/adipocyte determination and differentiation factor 1. J Biol Chem 273:22052–22058
Wang H, Kouri G, Wollheim CB (2005) ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J Cell Sci 118:3905–3915
Acknowledgments
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work. This research was supported by a grant from Department of Science and Technology of Hebei Province of China (No.07276170).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jun, H., Song, Z., Chen, W. et al. In vivo and in vitro effects of SREBP-1 on diabetic renal tubular lipid accumulation and RNAi-mediated gene silencing study. Histochem Cell Biol 131, 327–345 (2009). https://doi.org/10.1007/s00418-008-0528-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0528-2