
Abstract
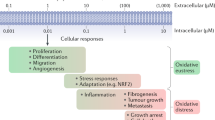
The presence and supposed roles of reactive oxygen species (ROS) were reported in literature in a myriad of instances. However, the breadth and depth of their involvement in cellular physiology and pathology, as well as their relationship to the redox environment can only be guessed from specialized reports. Whatever their circumstances of formation or consequences, ROS seem to be conspicuous components of intracellular milieu. We sought to verify this assertion, by collecting the available evidence derived from the most recent publications in the biomedical field. Unlike other reviews with similar objectives, we centered our analysis on the subcellular compartments, namely on organelles, grouped according to their major functions. Thus, plasma membrane is a major source of ROS through NAD(P)H oxidases located on either side. Enzymes of the same class displaying low activity, as well as their components, are also present free in cytoplasm, regulating the actin cytoskeleton and cell motility. Mitochondria can be a major source of ROS, mainly in processes leading to apoptosis. The protein synthetic pathway (endoplasmic reticulum and Golgi apparatus), including the nucleus, as well as protein turnover, are all exquisitely sensitive to ROS-related redox conditions. The same applies to the degradation pathways represented by lysosomes and peroxisomes. Therefore, ROS cannot be perceived anymore as a mere harmful consequence of external factors, or byproducts of altered cellular metabolism. This may explain why the indiscriminate use of anti-oxidants did not produce the expected “beneficial” results in many medical applications attempted so far, underlying the need for a deeper apprehension of the biological roles of ROS, particularly in the context of the higher cellular order of organelles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Overview
Formation of reactive oxygen species (ROS), in particular of superoxide (O2·−), in many—if not all—cellular systems is now beyond doubt, but their raison d’être remains unclear. Proposed explanations for their occurrence range from accidental byproducts of aerobic metabolism, to highly regulated and sophisticated signaling mechanisms. These rationalizations depend on the specific type of reaction in which ROS are envisaged. However, no unitary view emerged so far to provide a satisfactory explanatory framework for the role of ROS in biology.
A remarkable attempt, in this respect, was done by the late Manfred Saran who, in a posthumous hypothesis paper (Saran 2003), suggested that superoxide may be a universal cofactor of membrane functioning. Saran suggested that the ubiquitous and necessarily delocalized, weakly reactive, agent able to continuously alter the chemical and, not so surprisingly, the biomechanical stability of phospholipids, would be the NAD(P)H oxidase-derived O2·−. This would also explain the continuous production of low levels of lipid peroxides, as part of the tighter controlled lipid-based signaling machinery of membranes (Roberts and Morrow 2002). This position could be considered at odds with those that view ROS as the “evil” mediators of oxidative stress (the list is long and therefore we will cite only one recent example: Sadek et al. 2003), and complements another hypothesis that links the stress and inflammatory responses and ageing to life-long oxidative damage to components of the cell (Lane 2003). As usual, the truth is probably at the crossroads of these hypotheses.
Throughout the last decade, the role of ROS in controlling vital cellular processes became apparent, as necessary components in the normal functioning of cells (Aslan and Ozben 2003; Chiarugi 2003; Nathan 2003). Alternatively, they might be byproducts of metabolic processes. Regardless of their origin, when in too high an amount, ROS may interfere with the structure and function of biomolecules. Despite their apparent simplicity, ROS are thus many faceted chemical entities, from the point of view of their role within cells.
In this review we focus on the role of ROS and redox balance in the functions of cellular organelles. In looking at each organelle, we ask whether this requires a specific redox microenvironment, whether it is ROS-related, as well as why and how this is achieved. We then review the cellular consequences of the malfunction of organelles redox system, where we could find one. Finally, we analyze how increased “oxidative stress” inflicted upon cells affects the organelles.
There are numerous natural sources of oxidative stress, such as exposure to environmental oxidants or toxins (heavy metals, for instance), ionizing and UV irradiation, heat shock, and inflammation (Ermak and Davies 2002). Abnormally high levels of ROS may indeed lead to deregulation of redox-sensitive metabolic and signaling pathways. The targets are often proteins with redox-sensitive cysteine residues, which are oxidized to sulfenic acid moieties and mixed disulfides, thereby altering the signaling function of the protein (Droge 2003). We thus acknowledge that each organelle has a specific need for the reducing to oxidant balance, for its enzymes to work properly and to fulfill their role. Since numerous reviews have analyzed individual topics in higher detail, these will be referred to throughout this paper.
As a self-imposed limitation for space reasons, some fields in which ROS are known or supposed to be involved, such as the formation of lipid signaling molecules, the cellular effects of antioxidants, and the contributions to highly specialized mechanisms (for example, functioning of chromaffin granules), were intentionally omitted.
ROS biochemistry in a nutshell
The term “free radical” describes a chemical species that has one or more unpaired electrons, a definition that embraces the atom of hydrogen (one unpaired electron), most transitional metal ions, and the oxygen molecule (O2) (Halliwell 1991). O2, in its ground state, is in itself a radical, having two unpaired electrons. O2 readily reacts to form partially reduced species, which are generally short-lived and highly reactive, hence the idiom “reactive oxygen species” (not to be confused with “free radicals”). Some relevant ROS include:
-
1.
The superoxide anion (O2·−), formed through one-electron reduction of O2:
-
2.
Hydrogen peroxide (H2O2), which has no unpaired electrons, and thus is not a radical (Halliwell 1991). It is formed by several metabolic reactions, for instance the dismutation reaction of O2·− catalyzed by superoxide dismutases, and which has as intermediate the hydroperoxyl radical:
-
3.
The hydroxyl radical (·OH) that can be formed from either the superoxide anion (Haber-Weiss reaction) or from H2O2:
where Men+ is a metal ion. Probably the most frequent reaction in vivo is the Fe2+-catalyzed decomposition of H2O2, i.e., the Fenton reaction. ·OH is, perhaps, the most toxic form of oxygen. It is highly reactive, so it will react indiscriminately at, or close to, the site of its formation with most molecules it encounters. This is a reason for which this radical is unlikely to function as a signaling molecule, while both O2·− and H2O2, which are less reactive and therefore longer-lived, are more appropriate for intra- and even intercellular signaling (Saran 2003; on the specificity of reactive oxygen and nitrogen species as signaling intermediates, see Nathan 2003).
The study of ROS is often associated with the notion of “redox state” of cells. It has recently been pointed out that this is a poorly defined term (Schafer and Buettner 2001). These authors recommend the use of the phrase “redox environment” of a linked set of redox couples as found in a biological fluid, organelle, cell, or tissue. The redox environment would be defined as the summation of the products of reduction potential and reducing capacity of the linked redox couples present (Schafer and Buettner 2001). Such redox couples are NAD+/NADH, NADP+/NADPH, oxidized glutathione/reduced glutathione (GSSG/2GSH), oxidized thioredoxin/reduced thioredoxin, etc. The connection with ROS is the fact that the abovementioned oxygen species, in particular H2O2, interact with protein thiols or reduced glutathione (GSH), and thus contribute to the defining of cellular redox environment (Saran 2003). Glutathione is considered the major thiol-disulfide redox buffer of cells (Gilbert 1990), and the ratio GSSG/GSH is used to estimate the redox state of a given system (Schafer and Buettner 2001). Notably, the presence of membrane-surrounded organelles leads to compartmentation of the redox environment. We will further pursue the most remarkable features of cellular organelles in terms of their redox environment, as determined by ROS production and consumption, and how these two processes influence the equilibrium of the redox couples most relevant for each organelle.
Cellular redox homeostasis and organelles
The gate-keeper: plasma membrane
The flow of electrons and/or protons is an essential component of living state, and no component of the cell could function without it. Thus, it is no surprise that the plasma membrane (PM) is endowed with complex redox and pH regulating abilities. The protons cross the PM through numerous specialized molecules. Likewise, electrons are conducted within the PM. They contribute to the formation of transmembrane potential, a key element of biological control and signaling, whose discussion is beyond the scope of this review. Instead, we will call attention to the presence of molecular components of multiple redox chains, which are present on the external surface, attached to the internal face of the PM, or crossing it. In general, the surface of mammalian cells is exposed to an oxidizing environment, while the cytosol is reducing. Therefore, the environmental redox state, which in tissues could be fed with ROS from dysfunctional cells if not promptly removed (Skulachev 1996), has the potential to damage proteins, lipids, and carbohydrates, unless tightly regulated mechanisms are operational, and efficient anti-oxidant systems are in place.
Production of O2·− by the membrane-bound enzyme NADPH oxidase of phagocytes is a long-known phenomenon, generally assumed to help in killing bacterial intruders (Babior 1999). More than two decades ago, the same author hypothesized that “other cells–perhaps all cells–may contain an activable membrane oxidase whose purpose is entirely unrelated to host defense” (Babior 1982). Indeed, isoforms of NADPH oxidase were found in membranes of nearly every cell. In his hypothesis-based review (Saran 2003), Saran suggested that O2·− and its conjugate acid, HO2·, may have evolved under primordial conditions as regulators of membrane mechanics: by modifying cell membranes, they help other agents gain access to the hydrophobic region of phospholipid bilayers and hence contribute to lipid-dependent signaling cascades. Thus, O2·−/HO2· are proposed as indispensable adjuvants for the generation of cellular signals, for membrane transport, channel gating, and hence, in a global sense, for cell viability and growth (Saran 2003).
While the veracity of this proposition remains to be established, plasmalemmal NADH oxidoreductase- and NADPH oxidase-derived ROS are definitely involved in controlling PM functions, such as proton pumping and ion channels (Baker and Lawen 2000). The study of PM redox enzymes started by incidental use of artificial substrates, without knowledge of their natural counterpart. This is why to the present day some of these enzymes are described as acting on compounds absent in the biological environment, such as ferricyanide. Remarkable examples are the cell surface NADH oxidases: these are PM enzymes which, using NADH as the intracellular electron donor, are capable of reducing ferricyanide (Baker and Lawen 2000). Interestingly, the yeast PM NADH:ferricyanide reductase was found responsible for the transport of ferrous iron across the membrane (Dancis et al. 1990). This and several other yeast membrane proteins that have a role in iron uptake also bear homology to the mammalian gp91-phox protein, the PM component of phagocyte NAD(P)H oxidase (Georgatsou and Alexandraki 1999). It is assumed that mammalian PM NADH:ferricyanide reductase has many other roles: protection of oxidative stress, proliferation, and apoptosis (Baker and Lawen 2000). In fact, NAD(P)H-oxidase is more abundant on rapidly proliferating cells than on resting cells (Berridge and Tan 2000).
Trans-plasma membrane oxidoreductases that shift electrons from cytosolic NADH to external electron acceptors, such as oxygen, are widely involved in cellular redox control. In addition, external NAD(P)H oxidases have been demonstrated on intact cells and as eluted proteins, but the relationship between trans-plasma membrane NADH oxidoreductases and cell surface NAD(P)H oxidases is not known (Berridge and Tan 2000). The best-characterized NAD(P)H oxidase is the phagocytic enzyme that mediates the oxidative burst of activated neutrophils (Babior 1999). This is a multicomponent membrane complex, with flavocytochrome b558 in its catalytic core. It consists of a large glycoprotein gp91phox (phox for phagocyte oxidase; known as “beta subunit”) and a small protein p22phox (“alpha subunit”). The other components of the oxidase are water-soluble proteins of cytosolic origin, namely p67phox, p47phox, p40phox, and Rac1 (Rac2 is the counterpart in neutrophils). Upon stimulation, these components assemble with the membrane-bound flavocytochrome b558, become activated, and generate O2·− (Vignais 2002). Non-phagocytic homologues of gp91phox were found in endothelial cells (Al Mehdi et al. 1998; Bayraktutan et al. 2000; Hohler et al. 2000; Li and Shah 2002; Meyer et al. 1999; Parinandi et al. 2003), smooth muscle cells (Suh et al. 1999), and in other cells and tissues (Cheng et al. 2001; Geiszt et al. 2000; Kikuchi et al. 2000), including colon cancer cells (Geiszt et al. 2003a) and melanoma (Brar et al. 2001). These were designated by the common name Nox (for review, see Bokoch and Knaus 2003; Vignais 2002). Also, non-phagocytic homologues of p67phox and p47phox were described (Banfi et al. 2003; Geiszt et al. 2003b; Takeya et al. 2003). As opposed to the oxidative burst oxidase, these enzymes are constitutively active and seem to exist in preassembled complexes (Li and Shah 2002), but the amount of O2·− produced is much less than in phagocytes (Souza et al. 2002). In addition, while the phagocytic oxidase reduces molecular oxygen to O2·− using NADPH as the electron donor, the non-phagocytic enzyme may use either NADH or NADPH. The mechanism of O2·− production is better known from the study of the phagocytic oxidase: electrons pass from intracellular NADPH through a redox chain within the enzyme, to reduce extracellular O2 to O2·−. Electron flux is electrogenic, and rapidly depolarizes the membrane potential. Excessive depolarization can turn off electron transport by self-inhibition, but this is prevented by proton flux that balances the electron flux (DeCoursey 2003). The O2·− produced by these enzymes in non-phagocytic cells has potential roles in signaling related to growth and angiogenesis, immune function, hypoxic response, and oxidative modification of extracellular matrix proteins (for review, see Lambeth 2002).
In addition to transmembrane NAD(P)H oxidases, a family of cell surface NADH oxidases was described, called ECTO-NOX (Morre and Morre 2003). These enzymes exhibit both oxidative and protein disulfide isomerase (PDI)-like activities. The physiological substrates for the oxidative activity appear to be hydroquinones of the PM, such as reduced coenzyme Q10. ECTO-NOX proteins are growth-related and seem to regulate cellular volume (Morre and Morre 2003).
The presence of ROS-producing enzymes associated with PM means that a mechanism should be in place to protect against possible oxidative damage to proteins and lipidic components. PM, similar to mitochondria and Golgi apparatus, contains the lipophilic molecule coenzyme Q (CoQ, ubiquinone; Villalba and Navas 2000). CoQ is an intermediate electron carrier by shuttling them from intracellular reduced pyridine nucleotides to external impermeable oxidants (Crane et al. 1993). In addition, CoQ can be considered a central component of the PM antioxidant system (Villalba and Navas 2000). Oxidative stress may induce apoptosis through the release of ceramide from PM, which, in turn, activates the cascade of caspases, the enzymes responsible for the execution phase of apoptosis. CoQ effectively inhibits stress-induced apoptosis by reducing ceramide release (Navas et al. 2002).
Another effect of ROS interaction with PM components is the oxidation of PM proteins and lipids. Many of the proteins affected (mostly inactivated) by oxidation are involved in transmembrane transport of different ions and molecules, for example Na+/Ca2+ exchanger from synaptic membranes (Huschenbett et al. 1998), Ca2+-ATPase in alveolar macrophages (Hoyal et al. 1996), non-selective cation channels in hepatoma cells (Feranchak et al. 2003), and Na+ channels in isolated cardiac myocytes (Bhatnagar et al. 1990). Lipid peroxidation results in the release of 4-hydroxy-2,3-trans-nonenal (4HN; Benedetti et al. 1980), that has numerous damaging effects on membrane proteins, for instance, impairment of glutamate transport in cortical astrocytes (Blanc et al. 1998) or inhibition of several neutrophil PM ion transport ATPases (Siems et al. 2003). Selective oxidation of phosphatidylserine during apoptosis has an effect that is one of the hallmarks of apoptosis, namely the translocation of this phospholipid on the outer membrane leaflet. The mechanism involves inhibition of the enzyme responsible for phosphatidylserine maintenance on the inner membrane leaflet, the aminophospholipid transferase, by peroxidized phosphatidylserine (Tyurina et al. 2000).
In summary, a large number of oxidoreductases that use NADH or NADPH as electron donors are associated with the PM. The final electron acceptor is O2, whose one electron reduction leads to the formation of O2·−. The role of this ROS in phagocytes is to help kill invading bacteria, but in non-phagocytic cells this is much less understood; nevertheless it is associated with essential processes such as proliferation and apoptosis. Maintenance of an effective PM barrier implies to prevent potential damaging effects of ROS, a process in which CoQ plays a central role. When the protective capacity of the PM is exceeded, oxidant damage may result either directly, by inhibition of transmembrane transporters and other membrane proteins, or indirectly, mediated by lipid peroxidation products, such as 4HN.
The synthetic pathway: endoplasmic reticulum and Golgi apparatus
Endoplasmic reticulum (ER) is a continuous intracellular compartment, which performs multiple functions, supported by specialized domains. The rough ER is studded with ribosomes on the cytosolic surface and coordinates synthesis and folding of proteins, thereafter exported toward the Golgi apparatus by means of vesicle budding from the transitional elements (Palade 1975). Phospholipid and steroid synthesis takes place within the smooth ER, a compartment that may have other functions as well in different cells: detoxification (in the liver) or calcium sequestration (in muscle cells, where it is called sarcoplasmic reticulum) (for reviews, see, for example, Aridor and Balch 1999; Voeltz et al. 2002). A most remarkable characteristic of ER is the redox chemistry that takes place within its lumen. Proteins synthesized in ER are destined to either intracellular organelles, including ER itself, or to export. Intramolecular disulfide bonds stabilize the conformation of most of these proteins, a process that requires oxidation of free sulfhydryl moieties.
It was believed that intraluminal ER milieu ensures the feasibility of protein oxidative folding by maintaining a ratio of oxidized (GSSG) to reduced (GSH) glutathione higher than within the cytosol, in the ranges of approximately 1:1 to 3:1 versus approximately 30:1 to 100:1, respectively (Braakman et al. 1992; Hwang et al. 1992). The enzyme that catalyses formation, reduction, and isomerization of disulfide bonds in newly synthesized proteins is protein disulphide isomerase (PDI), a member of the thioredoxin family of proteins. The process of native disulfide formation involves both the oxidation of dithiols to disulfides as well as the rearrangement of non-native to native disulfide bonds. PDI is involved in both these steps (for a review of the mechanisms, see Kersteen and Raines 2003). Until recently, the source of oxidizing equivalents necessary for these reactions remained elusive, until it was found that in S. cerevisiae the native ER protein Ero1 transfers disulphide bonds to PDI (Frand and Kaiser 1998; Pollard et al. 1998; for review, see Frand et al. 2000). Human counterparts of the enzyme, Ero1-Lα and Ero1-Lβ, have similar roles (Mezghrani et al. 2001). Rat, mouse, and human homologues of Ero1-Lα but not of the isoform Ero1-Lβ are stimulated by hypoxia in rats in vivo and in rat, mouse, and human cell cultures. The temporal pattern of both gene and protein expression is very similar to that of genes triggered by the hypoxia-inducible transcription factor (HIF-1), thus placing Ero1-Lα among the family of classic oxygen-regulated genes (Gess et al. 2003).
Ero1 functions as an oxidase for PDI: oxidizing equivalents flow from Ero1p to substrate proteins via PDI, through direct dithiol–disulfide exchange between PDI and Ero1p (Frand and Kaiser 1999). This kinetic shuttling of oxidizing equivalents allows ER to support rapid disulfide formation while maintaining the ability to reduce and rearrange incorrect disulfide bonds (Tu et al. 2000). Thus, oxygen upregulation of Ero1-Lα expression is likely to maintain the transfer rate of oxidizing equivalents to PDI in situations of altered cellular redox state, induced by changes in cellular oxygen (Gess et al. 2003). In addition, Ero1 has a tightly associated flavin adenine dinucleotide (FAD) moiety and is highly responsive to small changes in physiological levels of free FAD (Tu and Weissman 2002; Tu et al. 2000). Another distinct flavoprotein, Erv2p, that can catalyze O2-dependent formation of disulphide bonds, was recently discovered in yeast (Sevier et al. 2001).
Ero1p-catalyzed disulfide formation is independent of glutathione, both in vivo and in vitro (Cuozzo and Kaiser 1999; Tu et al. 2000). These two studies demonstrated that, contrary to what it was believed, glutathione is not the source of oxidizing equivalents for oxidative protein folding within ER. Instead, it functions as a reductant of both PDI and of folding proteins, during the stepwise formation and re-formation of disulfide bonds, until the correct conformation is achieved. This conclusion is also supported by the finding of a bidirectional, saturable transport system of GSH in rat liver microsomal vesicles, while microsomal membranes were virtually non-permeable toward GSSG (Banhegyi et al. 1999). This result ruled out a previous supposition that the high GSSG:GSH ratio was obtained through preferential import of GSSG from the cytosol. In the membranes of skeletal muscle sarcoplasmic reticulum this transport was associated with the ryanodine receptor, while a different, unrelated system was postulated for ER-derived microsomes from other cell types (Csala et al. 2001, 2003). In addition, it was recently demonstrated by labeling with the thiol-specific monobromobimane, and subsequent separation by reversed-phase high-performance liquid chromatography, that more than half of the microsomal glutathione is present in mixed disulfides with proteins, and only the remainder is distributed between the reduced and oxidized forms of glutathione in the known ratio of 3:1 (Bass et al. 2004).
Thus, it became clear that the process of oxidative protein folding does not use GSSH as the source of oxidizing equivalents. This role is played, in fact, by molecular oxygen, which is used by Ero1p as its preferred terminal electron acceptor. Accordingly, Ero1p directly couples disulfide formation to the consumption of molecular oxygen, while its activity is modulated by free luminal FAD levels as well, potentially linking disulfide formation to cellular nutritional or metabolic status (Tu and Weissman 2002). However, this process may have as a consequence the formation of ROS (Tu and Weissman 2004). These latter authors made an interesting calculation, the result of which was that Ero1p-mediated oxidation could account for up to 25% of cellular ROS produced during protein synthesis (Tu and Weissman 2004).
Another important conclusion of these recent findings is that the pro-oxidant environment within ER is not there just to support, but also as a consequence of the oxidative formation of disulfide bonds necessary for correct protein folding. Moreover, the involvement in this process of both oxidation and reduction of protein sulfhydryls underscores the importance of a tight regulation of the ER redox milieu, since changes in either direction drastically interfere with ER functions. We will further examine some of the evidence that shows the effects of changes in the cellular redox status upon ER, and how this altering of ER functions affects, in return, cellular physiology.
A condition that has not been as widely studied as oxidative stress is that of reductive stress, defined as “an abnormally increased electron pressure or ‘reducing power’” and characterized by increased NADH:NAD+ ratio (Ghyczy and Boros 2001; Lipinski 2002). While misfolded proteins and unassembled chains of multimeric proteins are normally degraded by ER proteases, even the more stable, correctly folded proteins are degraded in reducing conditions in CHO cells (Young et al. 1993). Addition of dithiothreitol (DTT) or GSH to permeabilized cells promoted degradation of newly synthesized proteins, but not that of PDI or another major ER protein, GRP94, thus suggesting a highly controlled and specific process (Young et al. 1993). An unexpected player in the protection against reductive stress was found to be thioredoxin (Trotter and Grant 2002). Best known as antioxidants (Carmel-Harel and Storz 2000), yeast thioredoxins TRX1 and TRX2 were both required not only for resistance to the oxidant diamide, but also to the reductive agent DTT (Trotter and Grant 2002). Moreover, yeast lacking either or both of the two thioredoxins, or thioredoxin reductase, contained elevated levels not only of GSSG (expected), but also of GSH (unexpected). In turn, this increased GSH level led to inhibition of oxidative protein folding and accumulation of misfolded proteins, an effect known to induce a complex cellular reaction called the unfolded protein response (UPR; Gething and Sambrook 1992; Trotter and Grant 2002).
The UPR is an efficient “quality control” mechanism that has evolved within ER: only correctly folded molecules are allowed to reach their final destination. Accumulation of unfolded proteins triggers the UPR, which consists of: (a) inhibition of the α subunit of translation initiation factor 2 (eIF2α), thus shutting down translational supply of proteins to ER (Harding et al. 2003), and (b) induction of transcription of ER folding enzymes (Kohno et al. 1993; Shamu et al. 1994; for reviews of the mechanisms, see, for example, Brown et al. 2000; Mori 2003; Rutkowski and Kaufman 2004). Importantly, this complex sequence of events is induced by oxidative stress as well (Harding et al. 2003), and it was found that application of H2O2 induces specific oxidation of several essential ER proteins: PDI, Grp78, Grp94, ER60, and calnexin (van der Vlies et al. 2003a). Additionally, ER protein oxidation was observed in ageing mouse liver (Rabek et al. 2003), and was considered a mark of ageing (van der Vlies et al. 2003b). Another pathological situation where UPR was implicated is Alzheimer’s disease (Kudo et al. 2002).
Although some of the proteins synthesized in the rough ER are destined to remain associated with ER for its own functions, most of them travel further to the Golgi apparatus, and from there to various cellular compartments or to extracellular space (for excellent reviews and current concepts on the transfer of proteins from ER to Golgi, and Golgi structure and function, see Murshid and Presley 2004; Polishchuk and Mironov 2004; Ward and Brandizzi 2004). Interestingly, and contrary to what it is known about how ER maintains its own redox homeostasis and how it is affected by imbalances in the reducing:oxidizing equivalents interplay, we found very few studies addressing these questions to the Golgi apparatus. A good reason could be that processes within the Golgi compartment are simply independent of the redox state on the secretory pathway (Losch and Koch-Brandt 1995). Trafficking to, through, and from Golgi requires in fact a precise pH gradient that decreases from ER to Golgi to mature secretory granules, due to successive increases in the density of active H+ vacuolar ATPase (Losch and Koch-Brandt 1995; Machen et al. 2003). To our knowledge there is no indication as yet of if and how this ATPase may be affected by changes in the cellular redox status.
In turn, there are numerous reports describing the impact of high oxidative stress on the Golgi network in a specific cell and pathologic situation: neurons from patients or animal models of amyotrophic lateral sclerosis. This disease is characterized by a highly fragmented Golgi apparatus, and by oxidative stress due to mutations in Cu,Zn-superoxide dismutase (SOD; Fujita et al. 1999, 2000, 2002; Stieber et al. 1998, 2000). At the same time, inflicting oxidative stress on cells by using menadione or H2O2 reduces cell surface expression of the transferrin and low-density lipoproteins receptors, due to their blocking in the Golgi compartment (Malorni et al. 1993, 1998). In contrast, under similar conditions, namely use of H2O2 to induce oxidative stress in astrocytoma cells, the number and velocity of vesicles transporting apoE on the secretory pathway are increased (Dekroon and Armati 2002). This suggests a high specificity in the way different proteins transiting the Golgi apparatus are processed in pro-oxidant conditions, according to how their change in expression would benefit the cell. It is a challenge to understand how this discrimination in the transport of proteins from Golgi to the cell surface is achieved in the conditions of oxidative stress.
In conclusion, much knowledge was recently acquired about the role and maintenance of the pro-oxidant status within the ER lumen. GSH is preferentially transported through the ER membrane, and is used up in the redox cycle that proteins undergo until correct folding is achieved. PDI catalyses the oxidation of sulfhydryls and formation of disulfide bonds, using O2 as terminal electron acceptor and Ero1p as an intermediary. All these processes are regulated by oxygen availability and intracellular redox equilibrium. Either oxidative or reductive stress may affect protein synthesis and induce UPR, albeit by different mechanisms. This appears to be an adaptive response that helps cells to cope with an increased load of misfolded proteins. However, in ageing and in some pathological situations, such as Alzheimer’s disease, the ability of ER to handle the redox imbalance is overwhelmed. In contrast, Golgi apparatus may not depend, in its functioning, on a specific redox status; instead it requires a precise pH gradient. However, oxidative stress does affect this organelle, by inducing fragmentation of the Golgi network, as well as specific changes in the transport of different proteins from Golgi vesicles to the cell surface. The mechanism of these changes is not yet known.
Life and death decisions: mitochondria and the apoptosome
Mitochondria play a major role in both life and death of cells. They are the site of fatty acid oxidation and the citric acid cycle, which produce NADH and FADH2. These molecules, in turn, transfer electrons to the respiratory chain, and finally to oxygen, a process that generates ATP. Mitochondria are also the site of several components which, when released into the cytosol, trigger the apoptotic events. In certain circumstances, the very process of respiration generates the ROS that elicit, directly or indirectly, cell death (Duchen 2004).
It has long been recognized that the mitochondrial electron transport chain is a site of free radical generation (Das et al. 1989). The two sites where this happens are complex I (NADH-coQ reductase) and complex III (cytochrome c oxidase), especially when electron transport becomes slow. In such conditions, the half-life of reduced electron transport chain components increases, resulting in an increased likelihood of one electron reduction of O2 to O2·− (Sadek et al. 2003). A pathological condition where this mechanism occurs is ischemia, defined by decreased levels of the terminal electron acceptor O2 (Baker and Kalyanaraman 1989; Grill et al. 1992). However, the normal functioning of mitochondria is also characterized by electron leaks and formation of O2·−. Although in an aqueous environment O2·− is only moderately reactive, it can generate other more potent ROS: H2O2 by direct dismutation, and indirectly the highly oxidative and cytotoxic radical OH·, through reductive homolytic cleavage of H2O2 (Fleury et al. 2002). O2·−, H2O2, and OH· all inactivate components of the respiratory chain, as well as some enzymes of the Krebs cycle and other mitochondrial proteins. To prevent oxidative damage to their own proteins, mitochondria contain a complex antioxidant system that includes antioxidants (GSH, NADH, and thioredoxin) and enzymatic activities: MnSOD, catalase, glutathione reductase, and glutathione peroxidase (Bai and Cederbaum 2001; Fleury et al. 2002; Tanaka et al. 2002). The importance of these systems is even more evident in dysfunctional states. Such is the case of MnSOD: in a mouse model of oxidative stress due to lack of SOD2 (sod2−/− mice), proteomic and functional analyses revealed a high and specific sensitivity of some enzymes from the Krebs cycle, glutathione S-transferase, peroxiredoxin 5, and electron transport complexes I, II, III, and IV (but not complex V), to mitochondrial oxidative stress (Hinerfeld et al. 2004).
A consequence of oxidative stress in cells is the rise in cytosolic Ca2+, due to influx from the extracellular space through the PM, or release from intracellular stores such as the endoplasmic/sarcoplasmic reticulum. Intracellular calcium signaling responds in specific ways, with respect to intracellular location, amplitude, and duration, to a variety of stimuli (Ermak and Davies 2002). Mitochondria, too, have a load of Ca2+ that depends on the cytosolic Ca2+ concentration (Gunter et al. 1998). Physiological increases of intramitochondrial Ca2+ activate metabolism. However, under oxidative stress this process switches to become a death signal (Ermak and Davies 2002; for a review of Ca2+ effects on mitochondria, see Duchen 2000). The major mechanism is considered to be the formation of mitochondrial permeability transition pores (mPTPs). The mPTP is a large conductance pathway in the inner mitochondrial membrane, thought to be generated by a conformational alteration of adenine nucleotide translocase (ANT). mPTPs appear mostly at contact sites between the inner and outer mitochondrial membranes, where ANT as well as the voltage-dependent anion channel (VDAC; an outer mitochondrial membrane protein) are found (Duchen 2004). The mPTP is opened not only by high cytosolic Ca2+, but also by oxidative stress (by way of sulfhydryl oxidation; McStay et al. 2002) and by ATP depletion (Hagen et al. 2003).
There is not yet a consensus for what kind of cell death is triggered by the opening of mPTPs: mostly necrotic (Duchen 2004), or apoptotic as well (Newmeyer and Ferguson-Miller 2003), and not even on the role of ANT (Kokoszka et al. 2004). The hallmark of apoptosis (“programmed cell death”), as opposed to necrosis, is its active character, in that it requires the presence of a functional mitochondrial respiratory chain (Fleury et al. 2002). Apoptosis is executed by a set of cystein proteases (caspases), activated by signaling pathways initiated either by ligation of cell surface death receptors (the extrinsic pathway) or by mitochondria perturbation by different stressors (intrinsic pathway) (Fumarola and Guidotti 2004).
Perturbation of mitochondrial membrane integrity is followed by release in the cytosol of a number of pro-apoptotic proteins: cytochrome c, the flavoprotein apoptosis-inducing factor (AIF), endonuclease G, and others (Fumarola and Guidotti 2004; Lorenzo et al. 1999; Susin et al. 1999). In the cytosol, cytochrome c interacts with the adaptor protein Apaf-1 (apoptosis protease activating factor). As a result, a conformational change of Apaf-1 is induced that allows stable binding of dATP/ATP and formation of a complex (the “apoptosome”), and recruitment and activation of procaspase-9 to caspase-9. This, in turn, activates the downstream caspases-3, -6, and -7 (Acehan et al. 2002; Hengartner 1997; Li et al. 1997). It is worth noting that almost every molecule and step involved in this process is regulated by microenvironmental redox status, more specifically thiol redox (Sato et al. 1995; Ueda et al. 2002), as shown, for instance, by experiments with cells deficient in mitochondrial thioredoxin-2 gene (Tanaka et al. 2002). ANT may be activated by thiol crosslinkers and lead to mitochondrial membrane permeabilization independent of any other control or interactions (Costantini et al. 2000). AIF is a flavoprotein that bears homology, within FAD and NAD binding sites, to human glutathione reductase. This suggests a bifunctional protein with an electron acceptor/donor (oxidoreductase), as well as an apoptogenic function (Lorenzo et al. 1999). The activity of caspases is also profoundly influenced by cytosolic redox state: oxidative stress shifts apoptosis to necrosis (Hirsch et al. 1997), while maintenance of a reducing environment by thioredoxin and glutathione is required for caspase-3 activity (Ueda et al. 1998). Finally, oxidative stress induces molecules on signaling pathways leading to apoptosis: ASK1, JNK, p38 MAP kinase, and p53 (Ueda et al. 2002).
The question is how is it possible that the same organelle that provides cells with critical energy is also the source of death-triggering components? An interesting hypothesis was put forward by Punj and Chakrabarty (2003). It starts from the believed prokaryotic ancestry of mitochondria, and the fact that some present-day prokaryotes release redox proteins that induce apoptosis in mammalian cells, through stabilization of the tumor suppressor protein p53. It is conceivable that before mitochondria became endosymbionts, their ancestors released redox proteins with the same final effect, and that during their evolution the mitochondria maintained this function, i.e., to program their own host cell death (Punj and Chakrabarty 2003), to the benefit of the organism.
In summary, mitochondria are essential organelles that provide cells with ATP, the “energy quanta”, through the metabolic pathways and the respiratory chain they harbor. However, these very processes may also result in oxidative stress that attacks mitochondrial self-proteins. Whether intrinsic or extrinsic, oxidative stress may trigger opening of mPTP and release into the cytosol of several different molecules that form a macromolecular complex, the apoptosome. This, in turn, activates a caspase cascade, which eventually leads to programmed cell death. To protect themselves from this byproduct of their own activity, mitochondria contain a battery of antioxidant components. Defects in the antioxidant capacity of mitochondria are involved in ageing and many pathological situations defined by increased, mitochondria-derived, oxidative stress.
The degradation pathways: lysosomes and peroxisomes
Lysosomes were until recently viewed as sites for terminal degradation of macromolecules derived either from the extracellular space, through some form of endocytosis, or from the cell itself, through autophagocytic recycling (Sun-Wada et al. 2003). Lysosomes are involved in many other functions, from bone remodeling (Goto et al. 2003; Li et al. 2004) to antigen presentation (Lizee et al. 2003; for recent reviews of lysosomal organization and function, see Eskelinen et al. 2003; Sun-Wada et al. 2003). It was also found that some redox-active molecules have roles in lysosomal function. A consequence of this feature is that lysosomes may have, in certain circumstances, a significant role in the cellular redox homeostasis and free radical-induced processes.
More than a decade ago flavins, ubiquinone, and a b-type cytochrome were discovered as components of purified lysosomal membranes (Arai et al. 1991). These proteins actually form a functional electron transport system, starting with the donor NADH, in the direction NADH→FAD→cytochrome b→ubiquinone→O2 (Gille and Nohl 2000). The role of this redox chain is to support proton accumulation within lysosomes (Gille and Nohl 2000), in addition to the vacuolar-type proton ATPase, thus maintaining the optimal pH for the acidic hydrolases (Sun-Wada et al. 2003). Interestingly, a direct correlation was found between lysosomal oxidative activity (increase in O2·− and H2O2) and pH rise (due to consumption of H+) (Chen 2002).
A biochemical characteristic of this electron transport chain is the promotion of a three-electron reduction of O2, thus giving rise to the extremely reactive radical hydroxyl (HO·; Nohl and Gille 2002). Although the physiological relevance of this process is not yet understood, it is thought to interfere with the redox-active iron pool that accumulates as a result of lysosomal digestion (Yu et al. 2003b). Inhibition of intralysosomal degradation (through the raising of pH with NH4Cl) had comparable effects with iron chelation by desferrioxamine or alpha lipoic acid-plus, in terms of H2O2-induced formation of HO· (Persson et al. 2003; Yu et al. 2003b). The Fenton reaction, in which Fe (II) reacts with H2O2 to yield Fe (III) + HO·, was proposed to be at the origin of this process (Fridovich 1997), although there is not yet a consensus on its relevance in vivo (Saran et al. 2000).
A consequence of oxidative stress upon lysosomes is destabilization of their membranes, due to peroxidation (Nilsson et al. 1997). H2O2, an agent that induces oxidative stress, also promotes lysosomal membrane rupture (Antunes et al. 2001; Yu et al. 2003b), and so does radiation (Ogawa et al. 2004). Moreover, lysosomal membrane permeabilization per se, induced by different chemicals such as the synthetic retinoid CD437 (Zang et al. 2001), the antibiotics ciprofloxacin and norfloxacin (Boya et al. 2003), 3-aminopropanal (Yu et al. 2003a), or the lysosomotropic detergent O-methyl-serine dodecylamide HCl (Zhao et al. 2003), triggers intracellular formation of ROS. Some of the above processes were mediated by the action of lysosomal proteases that leaked into the cytosol, notably cathepsins B and D. These enzymes affected mitochondria, further inducing cytochrome c release and activation of caspase-mediated cell death (Boya et al. 2003; Johansson et al. 2003; Yu et al. 2003a; Zhao et al. 2003). An alternative pathway, unveiled by blocking mPTPs by cyclosporin A, consists in a concomitant generation of superoxide that leads to lysosomal damage and a regulated necrotic cell death, different from apoptosis (Raymond et al. 2003).
Thus it became apparent that lysosomal proteases are involved in both apoptotic and necrotic cell death, via either intrinsic or extrinsic oxidative perturbations in this organelle’s membrane. The process is even more prominent with age, when a new player enters the stage: lipofuscin, formed of protein residues crosslinked due to iron-catalyzed oxidative processes (Brunk and Terman 2002). Several phenomena associated with lipofuscin predispose the cells in which it is formed to even more damage. Lipofuscin has as strong autofluorescence, and as such it may sensitize cells to visible light, a process that implicates it in cell dysfunction such as occurs in ageing and retinal diseases (Davies et al. 2001). Lipofuscin may interfere with proteolytic processes, both intralysosomal (Brunk and Terman 2002) as well as proteasome-mediated (Szweda et al. 2003).
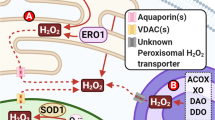
Peroxisomes are other organelles involved in not only catabolic but also synthetic biochemical pathways. Peroxisomes have an essential role in cellular metabolism, and their functions include fatty acid beta- and alpha-oxidation, amino acid metabolism, glyoxylate metabolism, as well as the synthesis of various lipidic compounds (plasmalogens, cholesterol, and dolichol) (Singh 1996, 1997). It is beyond our scope to review the rich literature that refers to peroxisome biogenesis, functions, signaling, genetic defects, or the role of peroxisome proliferators and their receptors in the control of peroxisome functions and fate, for there are many available reviews (Brown and Baker 2003; Masters 1996; Masters and Crane 1995a; Michalik et al. 2004; Singh 1996, 1997; van der Klei and Veenhuis 2002). Instead, we will focus on the fact that these organelles also have a key role in both the production and scavenging of ROS, in particular H2O2.
Most of the metabolic reactions hosted by peroxisomes consume oxygen, in amounts that are estimated between 10% and 30% of the total oxygen consumption by the liver (De Duve and Baudhuin 1966). Among the ROS-producing enzymes found in peroxisomes are cytochrome B5 (Gutierrez et al. 1988), a cytochrome P-450-dependent hydroxylation system (Singh et al. 1993), and numerous oxidases (del Rio et al. 1992), of which we will discuss in more detail xanthine oxidase (Angermuller et al. 1987). Substrates of these enzymes are long-chain fatty acids, hydroxyacids, amino acids, purines and pyrimidines, and uric acid (Masters and Crane 1995b). The result of their activity is mostly the production of H2O2, but also O2·− and HO·. If uncontrolled, these ROS are obviously very damaging. Therefore, peroxisomes also contain the “antidote”, i.e., antioxidant enzymes: catalase (De Duve and Baudhuin 1966; Moreno et al. 1995), Cu,Zn-SOD (Wanders and Denis 1992), and MnSOD (Singh et al. 1999), as well as glutathione peroxidase (Muse et al. 1994; Singh et al. 1994) and members of the peroxiredoxin family (Fujii and Ikeda 2002).
As with all the other organelles discussed so far, the peroxisome displays mechanisms to maintain the equilibrium between production and scavenging of ROS, which are inevitable byproducts of the oxidative-type metabolic activity that takes place within this organelle. There are, however, situations when the capacity of the antioxidant machinery is overwhelmed. One such situation is the sharp increase in peroxisome numbers stimulated by a heterogeneous class of chemicals, collectively known as peroxisome proliferators. Their effects are mediated by peroxisome proliferator-activated receptors (PPARs). Their endogenous ligands are fatty acids and fatty acid derivatives. Through the pathways they activate, PPARs can regulate cell proliferation, differentiation, and survival, consequently controlling carcinogenesis in various tissues (Michalik et al. 2004). It is considered that one trigger of peroxisome proliferator-induced carcinogenesis is the peroxisome-induced oxidative stress, although in humans this process is different from rodents, which are much more susceptible to this process (Dzhekova-Stojkova et al. 2001; Jiao and Zhao 2002).
Xanthine dehydrogenase (XD) is a molybdenum hydroxylase flavoprotein located mostly in peroxisomes (Frederiks and Vreeling-Sindelarova 2002), and the rate-limiting enzyme in purine catabolism (Pritsos 2000). In particular situations (most significant being hypoxia) it is converted to an oxidase (XO; Terada et al. 1997; Xia and Zweier 1995; Zweier et al. 1994). Both forms catalyze the hydroxylation of hypoxanthine to xanthine, and of xanthine to urate, the difference being that XD used NAD+ as electron acceptor, while XO uses O2, a reaction that generates O2·− (McManaman and Bain 2002). The pathological significance of this finding consists in explaining ROS generation that occurs during reperfusion after ischemia in various organs, and which mediates extensive, often irreversible, tissue damage (Canas 1999; Frederiks and Bosch 1995; Hotter et al. 1995; Nilsson et al. 1994; Tabuchi et al. 2001; Zhang et al. 1998; Zweier et al. 1994).
Ageing is another situation where higher amounts of peroxisome-derived ROS, especially H2O2, are detected. This is due not mainly to their increased generation, as in hypoxia-reperfusion, but rather to the decreased antioxidant capacity of the peroxisomal milieu. Several defects may account for this failure of antioxidant defense. First, a progressive alteration of the catalase import system was found (Legakis et al. 2002). Second, decrease of PPARα availability (Iemitsu et al. 2002), and diminished levels of retinoid X receptor (RXR) alpha, with which PPARα forms heterodimers, were detected. These heterodimers control expression of genes involved in oxidative stress, which is decreased especially as a consequence of the decline in availability of RXR alpha receptor (Chao et al. 2002).
Thus, lysosomes and peroxisomes are involved mostly in catabolic processes, however both of them also have other functions within cells. The enzymatic activities accommodated by these organelles depend on pH and/or the redox environment, and are mostly oxidative reactions. The result of lysosomal and peroxisomal functioning includes damaging chemical species, either directly (H2O2) or indirectly (iron stores and lipofuscin). Therefore, both organelles have powerful defense mechanisms, but when these mechanisms are overcome (for example, lysosomal membrane destabilization or decrease in the antioxidant potential of peroxisomes), oxidant stress results that leads to apoptotic or necrotic cell death.
The headquarters: nucleus
The effect of ROS and oxidative stress on gene expression and on DNA has been frequently and extensively reviewed (see, for example, Cadet et al. 2003; Izumi et al. 2003; Martinez et al. 2003; Slupphaug et al. 2003 for redox damage of DNA; Mikkelsen and Wardman 2003; Nguyen et al. 2003; Saran 2003 for ROS signaling and proliferation). We will, therefore, not address these issues and instead we will consider some other less-analyzed roles and effects of the redox environment on the nucleus as an organelle.
The nuclear content is separated from the cytoplasm by a double membrane, the nuclear envelope. Nucleocytoplasmic exchanges take place through specialized “gateways”, the nuclear pore complexes. Passage of molecules in and out of nucleus through nuclear pores is not a passive event, but an actively controlled process. A mechanism of control is the activity of the enzyme NTPase. This enzyme is modulated by the nuclear membrane content in cholesterol, and it was found that oxidation of cholesterol, especially in the internal leaflet of the envelope, inhibits NTPase (Ramjiawan et al. 1997). Thus, oxidative stress, which may certainly lead to cholesterol oxidation, could also induce decreased nucleocytoplasmic transport.
This raises the question whether there is an intranuclear defense mechanism against changes of redox status in the vicinity of chromatin. At least two mechanisms were found, namely glutathione and thioredoxin. The cytosolic and nuclear concentrations of glutathione are at near equilibrium, and protein-glutathione mixed disulfides were present in highest concentrations in the nuclear area and in the perinuclear region (Soderdahl et al. 2003). Monochlorbimane fluorescence microscopy revealed a similar distribution of GSH (Bellomo et al. 1997). Moreover, these authors found a GSH-stimulated ATP hydrolysis and an ATP-stimulated GSH accumulation in isolated nuclei. They consider that this active process may modulate the thiol/disulfide redox status of nuclear proteins and control chromatin compacting and decondensation (Bellomo et al. 1997). In fact, it has previously been shown that about 50% of nuclear matrix proteins had oxidized sulfhydryl groups, possibly stabilizing the nuclear matrix by disulfide bridges (Stuurman et al. 1992). Even DNA–nuclear matrix anchoring may be achieved in the same way, since DTT decreased and diamide increased DNA association with the nuclear matrix, an effect mediated by a nuclear PDI (VanderWaal et al. 2002). In addition to this distinct pool of GSH, a nuclear thioredoxin pool was found as well, more reduced than the cytosolic thioredoxin (Watson and Jones 2003).
It seems, thus, that despite the abundant knowledge accumulated on redox control of specific transcription factors and of gene expression, much less is known about ROS and nuclear matrix redox status. The presence of disulfide bridges suggests an ER-like environment, but this is not clearly defined, nor is the control of nuclear redox homeostasis or the physiological meaning of intranuclear redox changes.
ROS and animal cell movement
Actin cytoskeleton, a “distributed” organelle
Cellular motility is a complex process, with numerous points of control and feedback. Its central mechanism is the actin cytoskeleton reorganization that has both regulator and effector roles (Moldovan and Goldschmidt-Clermont 1998). Through its interactions with the extracellular matrix, via integrins and focal adhesions, actin actually controls cell shape, motility, and proliferation (Flusberg et al. 2001; Ingber 2002; Moldovan et al. 1997). On the other hand, signaling to the numerous actin-binding proteins induces changes in the rate, extent, and site of actin polymerization/depolymerization, thus controlling when, where, and how fast the cells move (Condeelis 1998; Defilippi et al. 1999). Comprising up to 10% of the total cellular protein, there is no wonder that actin is one of the most studied proteins. About half of the actin pool in any non-muscle cell is unpolymerized (G-actin). Upon polymerization actin monomers associate head-to-tail and form filaments (F-actin), which associate in various higher-level structures (Pollard and Cooper 1986). Many quiescent cells in culture, such as endothelial cells and fibroblasts, display bundles of actin filaments associated with actin-binding proteins, called stress fibers (Byers and Fujiwara 1982; Gabbiani et al. 1983; Kreis and Birchmeier 1980; Wong et al. 1983). At one or both ends stress fibers come in tight contact with specialized domains of the PM, named focal adhesions, through which cells adhere to the extracellular matrix (Kano et al. 1996).
A dramatic change in the organization of actin cytoskeleton occurs when cells are induced to migrate. A redistribution of cortical actin filaments occurs, resulting in an intense membrane activity known as “ruffling”, in changes of cell shape, and in directional locomotion (Small et al. 2002). Actin fibers are continuously assembled at the PM, while depolymerization occurs at the opposite end, toward the cytosol. A battery of actin-binding proteins (ABPs) contribute to the organization of F-actin, both in resting and motile cells; they differ according to the cellular activity and to the intracellular location (cell body, lamellipodia, filopodia, and membrane ruffles; Small et al. 2002). Although the term “actin cytoskeleton” has a connotation of rigidity, suggesting that it mainly contributes to the mechanical support of the cell, this is not entirely correct. The actin cytoskeleton is highly dynamic, since only such dynamic interactions between actin and binding proteins can support cellular motile responses on time scales ranging from subseconds to hours. The different steps of actin fiber assembly and disassembly are controlled by ABPs through weak interactions (Kd in the range of 10−5 M; Pollard 1986). An obvious advantage of such weak interactions resides in the rate with which they form and dissociate, which, in turn, is essential for the dynamic properties of the actin cytoskeleton and for the promptitude with which the cells respond to stimuli. As a corollary, one would expect that signaling pathways exert control upon the interactions between actin and its regulatory proteins (Pollard 1988). Capping, severing, and sequestering proteins regulate the size of the pools of F-actin and unassembled G-actin by affecting the steady-state concentration of ATP-G-actin (Carlier and Pantaloni 1997).
The signaling transduction pathways that trigger cellular motility operate by regulating actin reorganization. A major role was ascribed to the small GTP-binding proteins from the Rho family: Rho, Rac, and Cdc42. Each has a particular role in specific motility states of cells. Rho proteins mediate the lysophosphatidic acid and bombesin-induced formation of focal adhesions and actin stress fibers in quiescent cells (Ridley and Hall 1992). Rac proteins are required for the platelet-derived growth factor-, insulin-, bombesin-, and phorbol ester-stimulated actin polymerization at the PM that results in membrane ruffling in motile cells (Ridley et al. 1992). Cdc42 controls the formation of filopodia induced, for example, by bradykinin receptor activation (Kozma et al. 1995; Nobes and Hall 1995). Moreover, in these cells, a sequential activation of Cdc42→Rac→Rho was described (Nobes and Hall 1995). Each of these mediators have activators and inhibitors, and interacting proteins that act upstream or downstream on the respective pathways, in a complex network of interactions and with multiple points of control (Aspenstrom et al. 2004).
Role of ROS in cellular motility
Necessary components of the NAD(P)H oxidase functional complex are two small GTPase isoforms also involved in actin reorganization: Rac1 in macrophages (Abo et al. 1991) and Rac2 in neutrophils (Knaus et al. 1991). Interestingly, although both roles of Rac have been known for a decade and intensely studied, the two processes—actin reorganization and NAD(P)H oxidase activation—were regarded until recently independently. In fact, there might be a crosstalk between O2·− generation by the activated NAD(P)H oxidase and actin cytoskeleton remodeling, Rac1 being the “common denominator” (Goldschmidt-Clermont and Moldovan 1999). In posthypoxic endothelial cells (EC), a condition known to induce XO-mediated production of O2·− (Zweier et al. 1994), the pool of filamentous actin increased, and this effect was reversed by overexpression of Cu,Zn-SOD (Crawford et al. 1996). This suggested that O2·− could be involved in actin control. To further test this hypothesis, we used a replication-deficient adenoviral vector to overexpress the constitutively active form of Rac1, RacV12 in EC. RacV12 induced an increase in ROS production by EC, as well as actin reorganization and ruffles formation. Both effects could be counteracted by either overexpressing Cu,Zn-SOD or by other antioxidants (Moldovan et al. 1999).
One possible pathway for this effect was recently described. Rac-mediated activation of NAD(P)H oxidase and ROS generation would inhibit a low molecular weight protein tyrosine phosphatase, which as a consequence does not dephosphorylate p190Rho-GAP and therefore maintains it in an activated state (Nimnual et al. 2003). p190Rho-GAP is a Rho inhibitor, which means that Rac downregulates Rho activity in a redox-dependent manner, inhibits stress fibers formation, and thus allows actin remodeling (Nimnual et al. 2003). Another connection was recently reported: the human immunodeficiency virus type 1 Tat induces alterations in the actin cytoskeleton mediated by Pak1 and downstream activation of NAD(P)H oxidase (Wu et al. 2004). Pak1 is a serine/threonine kinase and a Rac-interacting protein, and although the cited report does not focus on Rac, the cytoskeletal changes observed in the presence of Tat match those specifically induced by Rac activation.
The physiological significance of these findings could be the following: since (1) rapid actin reorganization controls cellular migration and (2) intracellular O2·− generation is needed for Rac to exert its control on actin, therefore, (3) O2·− production, most probably due to Rac-mediated NAD(P)H oxidase activation, may regulate cell migration. Indeed, in an in vitro EC wounding model, we found that either preventing the formation of O2·− with the NAD(P)H oxidase inhibitor diphenylene iodonium or scavenging O2·− with the SOD mimetic manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) consistently reduced speed and directionality of otherwise unstimulated EC migration into the wound (Moldovan et al. 2000). These findings were confirmed by the fact that inhibition of Rac-mediated ROS production blocked EC VE-cadherin-mediated cell–cell adhesion and migration (van Wetering et al. 2002).
In several other instances intracellular ROS were involved in endothelial cell motility: ethanol-induced in vitro angiogenesis (Qian et al. 2003), vascular endothelial growth factor-induced migration of EC (Abid et al. 2000; Ushio-Fukai et al. 2002) and of smooth muscle cells (Wang et al. 2001), in vivo wound healing (Gordillo and Sen 2003), and hypoxia/reoxygenation-induced Matrigel invasion by EC (Lelkes et al. 1998). Likewise, ROS were demonstrated to be necessary in other actin-based cellular activities: human sperm acrosomal reaction (de Lamirande et al. 1998), low-density lipoprotein-induced stress fiber formation (Holland et al. 2001), VCAM-1 signaling upon engagement during lymphocyte migration (Cook-Mills 2002), integrin engagement during cell adhesion (Chiarugi et al. 2003), or migration of human fibrosarcoma cells (Liu et al. 2003).
Although the actin cytoskeleton, unlike the organelles discussed so far, does not seem to contain bound O2·−, H2O2, or other ROS-generating systems, it is profoundly affected by the redox environment. For years, the regulation of the actin cytoskeleton has been reconstituted in vitro in privileged redox conditions, in which a large excess of antioxidants, such as DTT, is added to protein preparations (Moldovan et al. 2000; Pardee and Spudich 1982). The reason for this redox bias is that in the absence of reducing agents, actin preparations are too unstable for studies of protein–protein interaction (Xu et al. 1998). However, until recently this redox sensitivity was not investigated in terms of actin regulation, but in terms of what damage ROS, and especially H2O2, may inflict upon actin (see, for example, Milzani et al. 1997). This view has changed as it became apparent that cytosolic redox conditions do not just alter actin, but alter it in a physiologically meaningful way (Goldschmidt-Clermont and Moldovan 1999).
Another line of thinking on the actin–redox connection started with the finding that glutathionylated actin is present in vivo in various conditions of oxidative stress (Fratelli et al. 2002; Pastore et al. 2003). Most importantly, actin glutathionylation is reversible, and so is its decreased polymerizability in this condition (Dalle-Donne et al. 2003). These authors suggested that S-thiolation of actin may prevent irreversible organization of microfilaments within cells, also preventing excessive actin polymerization and crosslinking, thereby preserving actin dynamics under oxidative stress (Dalle-Donne et al. 2001).
The above findings do not explain yet the sequence of events by which NAD(P)H oxidase-derived O2·− controls actin dynamics and cell motility. It could be either an indirect mechanism (Nimnual et al. 2003), or by modulating the activity of proteins that directly bind actin. An example is cofilin, which was found to form reversible disulfide-linked dimers and oligomers, with reduced severing activity and instead increased actin bundling (Pfannstiel et al. 2001). Alternatively, it could be a direct interaction of ROS, O2·− or H2O2, with actin. Although no proof of such an interaction in vivo exists yet, it is not inconceivable, because in both phagocytic (Tamura et al. 2000) and non-phagocytic cells (Gu et al. 2002; Li and Shah 2002; Wu et al. 2003) NAD(P)H oxidase was found associated with actin or with the F-actin binding protein moesin (Wientjes et al. 2001). Moreover, this association would not be a permanent one, but inducible, as suggested by the fact that vascular endothelial growth factor stimulation of EC triggers translocation of p47phox, together with WAVE1, Rac, and Pak1, to the membrane ruffles (Wu et al. 2003). This is exactly where the most active actin reorganization takes place.
In summary, ROS are “new” players in the physiological control of actin cytoskeleton dynamics, besides the 30+ families of ABPs. Actin is a very redox sensitive protein, a fact that may be important for its control through the cytosolic redox environment. Despite the fact that more evidence is accumulating that links ROS generation with cellular motility, this process is not yet understood in detail.
Conclusions
In this review, we documented the likely presence and roles of O2·− and other ROS, as well as the redox environment in membrane-bound compartments and membrane-free structures of cells. We consider that organelles, as well as other molecules and/or supramolecular ensembles, need a redox “activation”, which would increase their biochemical and possibly their biomechanical reactivity, making them suitable for the various processes to which they contribute. In this respect, of particular relevance is the highly dynamic actin cytoskeleton. On a larger scale, any protein may benefit from the redox milieu in adopting an optimal functional three-dimensional structure. While O2·− might be a “hidden variable”, other partners of the redox system, such as the concentrations of thiols, are also involved in the maintenance of an optimal redox (micro)environment.
The question remains whether it is possible to reconcile the apparently vital production of ROS with the also largely documented deleterious effects of “oxidative stress” (in other words: are ROS good or bad for cells?). Probably there is no simple answer, but in any case this is clearly a matter of what kind, where, when, and how much ROS are produced. The intracellular oxidative stress of significant consequences represents large quantities of ROS, usually derived from mitochondria, either acutely in specific pathophysiological reactions, or as chronic, cumulative leakage. Given the evolutionary symbiotic origin of mitochondria, these ROS might still be viewed as “exogenous” to the host cell. Therefore, the various types of ROS, namely those derived from NAD(P)H oxidase and from mitochondria, as well as those derived from or associated with the functioning of lysosomes, peroxisomes, Fenton reactions, etc., would represent different “physiological” entities, even though chemically similar.
References
Abid MR, Kachra Z, Spokes KC, Aird WC (2000) NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett 486:252–256
Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW (1991) Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353:668–670
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW (2002) Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9:423–432
Al Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB (1998) Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res 83:730–737
Angermuller S, Bruder G, Volkl A, Wesch H, Fahimi HD (1987) Localization of xanthine oxidase in crystalline cores of peroxisomes. A cytochemical and biochemical study. Eur J Cell Biol 45:137–144
Antunes F, Cadenas E, Brunk UT (2001) Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J 356:549–555
Arai K, Kanaseki T, Ohkuma S (1991) Isolation of highly purified lysosomes from rat liver: identification of electron carrier components on lysosomal membranes. J Biochem (Tokyo) 110:541–547
Aridor M, Balch WE (1999) Integration of endoplasmic reticulum signaling in health and disease. Nat Med 5:745–751
Aslan M, Ozben T (2003) Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal 5:781–788
Aspenstrom P, Fransson A, Saras J (2004) Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J 377:327–337
Babior BM (1982) The production of superoxide by neutrophils. In: Karnovsky ML, Bolis L (eds) Phagocytosis: past and future. Academic, New York, pp 157–166
Babior BM (1999) NADPH oxidase: an update. Blood 93:1464–1476
Bai J, Cederbaum AI (2001) Mitochondrial catalase and oxidative injury. Biol Signals Recept 10:189–199
Baker JE, Kalyanaraman B (1989) Ischemia-induced changes in myocardial paramagnetic metabolites: implications for intracellular oxy-radical generation. FEBS Lett 244:311–314
Baker MA, Lawen A (2000) Plasma membrane NADH-oxidoreductase system: a critical review of the structural and functional data. Antioxid Redox Signal 2:197–212
Banfi B, Clark RA, Steger K, Krause KH (2003) Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278:3510–3513
Banhegyi G, Lusini L, Puskas F, Rossi R, Fulceri R, Braun L, Mile V, di Simplicio P, Mandl J, Benedetti A (1999) Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J Biol Chem 274:12213–12216
Bass R, Ruddock LW, Klappa P, Freedman RB (2004) A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J Biol Chem 279:5257–5262
Bayraktutan U, Blayney L, Shah AM (2000) Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler Thromb Vasc Biol 20:1903–1911
Bellomo G, Palladini G, Vairetti M (1997) Intranuclear distribution, function and fate of glutathione and glutathione-S-conjugate in living rat hepatocytes studied by fluorescence microscopy. Microsc Res Tech 36:243–252
Benedetti A, Comporti M, Esterbauer H (1980) Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta 620:281–296
Berridge MV, Tan AS (2000) Cell-surface NAD(P)H-oxidase: relationship to trans-plasma membrane NADH-oxidoreductase and a potential source of circulating NADH-oxidase. Antioxid Redox Signal 2:277–288
Bhatnagar A, Srivastava SK, Szabo G (1990) Oxidative stress alters specific membrane currents in isolated cardiac myocytes. Circ Res 67:535–549
Blanc EM, Keller JN, Fernandez S, Mattson MP (1998) 4-hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia 22:149–160
Bokoch GM, Knaus UG (2003) NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci 28:502–508
Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, Ojcius DM, Jaattela M, Kroemer G (2003) Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med 197:1323–1334
Braakman I, Helenius J, Helenius A (1992) Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J 11:1717–1722
Brar SS, Kennedy TP, Whorton AR, Sturrock AB, Huecksteadt TP, Ghio AJ, Hoidal JR (2001) Reactive oxygen species from NAD(P)H:quinone oxidoreductase constitutively activate NF-kappaB in malignant melanoma cells. Am J Physiol Cell Physiol 280:C659–C676
Brown LA, Baker A (2003) Peroxisome biogenesis and the role of protein import. J Cell Mol Med 7:388–400
Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100:391–398
Brunk UT, Terman A (2002) Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med 33:611–619
Byers HR, Fujiwara K (1982) Stress fibers in cells in situ: immunofluorescence visualization with antiactin, antimyosin, and anti-alpha-actinin. J Cell Biol 93:804–811
Cadet J, Douki T, Gasparutto D, Ravanat JL (2003) Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res 531:5–23
Canas PE (1999) The role of xanthine oxidase and the effects of antioxidants in ischemia reperfusion cell injury. Acta Physiol Pharmacol Ther Latinoam 49:13–20
Carlier MF, Pantaloni D (1997) Control of actin dynamics in cell motility. J Mol Biol 269:459–467
Carmel-Harel O, Storz G (2000) Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol 54:439–461
Chao C, Youssef J, Rezaiekhaleigh M, Birnbaum LS, Badr M (2002) Senescence-associated decline in hepatic peroxisomal enzyme activities corresponds with diminished levels of retinoid X receptor alpha, but not peroxisome proliferator-activated receptor alpha. Mech Ageing Dev 123:1469–1476
Chen CS (2002) Phorbol ester induces elevated oxidative activity and alkalization in a subset of lysosomes. BMC Cell Biol 3:21
Cheng G, Cao Z, Xu X, Van Meir EG, Lambeth JD (2001) Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269:131–140
Chiarugi P (2003) Reactive oxygen species as mediators of cell adhesion. Ital J Biochem 52:28–32
Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G (2003) Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161:933–944
Condeelis J (1998) The biochemistry of animal cell crawling. In: Soll DR, Wessels D (eds) Motion analysis of living cells. Wiley-Liss, New York, pp 85–100
Cook-Mills JM (2002) VCAM-1 signals during lymphocyte migration: role of reactive oxygen species. Mol Immunol 39:499–508
Costantini P, Belzacq AS, Vieira HL, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G (2000) Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 19:307–314
Crane FL, Sun IL, Sun EE (1993) The essential functions of coenzyme Q. Clin Invest 71:S55–S59
Crawford LE, Milliken EE, Irani K, Zweier JL, Becker LC, Johnson TM, Eissa NT, Crystal RG, Finkel T, Goldschmidt-Clermont PJ (1996) Superoxide-mediated actin response in post-hypoxic endothelial cells. J Biol Chem 271:26863–26867
Csala M, Fulceri R, Mandl J, Benedetti A, Banhegyi G (2001) Ryanodine receptor channel-dependent glutathione transport in the sarcoplasmic reticulum of skeletal muscle. Biochem Biophys Res Commun 287:696–700
Csala M, Fulceri R, Mandl J, Benedetti A, Banhegyi G (2003) Glutathione transport in the endo/sarcoplasmic reticulum. Biofactors 17:27–35
Cuozzo JW, Kaiser CA (1999) Competition between glutathione and protein thiols for disulphide-bond formation. Nat Cell Biol 1:130–135
Dalle-Donne I, Rossi R, Milzani A, di Simplicio P, Colombo R (2001) The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med 31:1624–1632
Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A (2003) Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med 34:23–32
Dancis A, Klausner RD, Hinnebusch AG, Barriocanal JG (1990) Genetic evidence that ferric reductase is required for iron uptake in Saccharomyces cerevisiae. Mol Cell Biol 10:2294–2301
Das DK, George A, Liu XK, Rao PS (1989) Detection of hydroxyl radical in the mitochondria of ischemic-reperfused myocardium by trapping with salicylate. Biochem Biophys Res Commun 165:1004–1009
Davies S, Elliott MH, Floor E, Truscott TG, Zareba M, Sarna T, Shamsi FA, Boulton ME (2001) Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic Biol Med 31:256–265
DeCoursey TE (2003) Interactions between NADPH oxidase and voltage-gated proton channels: why electron transport depends on proton transport. FEBS Lett 555:57–61
De Duve C, Baudhuin P (1966) Peroxisomes (microbodies and related particles). Physiol Rev 46:323–357
Defilippi P, Olivo C, Venturino M, Dolce L, Silengo L, Tarone G (1999) Actin cytoskeleton organization in response to integrin-mediated adhesion. Microsc Res Tech 47:67–78
Dekroon RM, Armati PJ (2002) The effects of oxidative stress and altered intracellular calcium levels on vesicular transport of apoE-EGFP. Cell Biol Int 26:407–420
de Lamirande E, Tsai C, Harakat A, Gagnon C (1998) Involvement of reactive oxygen species in human sperm acrosome reaction induced by A23187, lysophosphatidylcholine, and biological fluid ultrafiltrates. J Androl 19:585–594
del Rio LA, Sandalio LM, Palma JM, Bueno P, Corpas FJ (1992) Metabolism of oxygen radicals in peroxisomes and cellular implications. Free Radic Biol Med 13:557–580
Droge W (2003) Oxidative stress and aging. Adv Exp Med Biol 543:191–200
Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J Physiol 529:57–68
Duchen MR (2004) Roles of mitochondria in health and disease. Diabetes 53(suppl 1):S96–S102
Dzhekova-Stojkova S, Bogdanska J, Stojkova Z (2001) Peroxisome proliferators: their biological and toxicological effects. Clin Chem Lab Med 39:468–474
Ermak G, Davies KJ (2002) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38:713–721
Eskelinen EL, Tanaka Y, Saftig P (2003) At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol 13:137–145
Feranchak AP, Kilic G, Wojtaszek PA, Qadri I, Fitz JG (2003) Volume-sensitive tyrosine kinases regulate liver cell volume through effects on vesicular trafficking and membrane Na+ permeability. J Biol Chem 278:44632–44638
Fleury C, Mignotte B, Vayssiere JL (2002) Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84:131–141
Flusberg DA, Numaguchi Y, Ingber DE (2001) Cooperative control of Akt phosphorylation, bcl-2 expression, and apoptosis by cytoskeletal microfilaments and microtubules in capillary endothelial cells. Mol Biol Cell 12:3087–3094
Frand AR, Kaiser CA (1998) The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol Cell 1:161–170
Frand AR, Kaiser CA (1999) Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell 4:469–477
Frand AR, Cuozzo JW, Kaiser CA (2000) Pathways for protein disulphide bond formation. Trends Cell Biol 10:203–210
Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A 99:3505–3510
Frederiks WM, Bosch KS (1995) The role of xanthine oxidase in ischemia/reperfusion damage of rat liver. Histol Histopathol 10:111–116
Frederiks WM, Vreeling-Sindelarova H (2002) Ultrastructural localization of xanthine oxidoreductase activity in isolated rat liver cells. Acta Histochem 104:29–37
Fridovich I (1997) Superoxide anion radical (O2−.), superoxide dismutases, and related matters. J Biol Chem 272:18515–18517
Fujii J, Ikeda Y (2002) Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep 7:123–130
Fujita Y, Okamoto K, Sakurai A, Amari M, Nakazato Y, Gonatas NK (1999) Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J Neurol Sci 163:81–85
Fujita Y, Okamoto K, Sakurai A, Gonatas NK, Hirano A (2000) Fragmentation of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J Neurol Sci 174:137–140
Fujita Y, Okamoto K, Sakurai A, Kusaka H, Aizawa H, Mihara B, Gonatas NK (2002) The Golgi apparatus is fragmented in spinal cord motor neurons of amyotrophic lateral sclerosis with basophilic inclusions. Acta Neuropathol (Berl) 103:243–247
Fumarola C, Guidotti GG (2004) Stress-induced apoptosis: toward a symmetry with receptor-mediated cell death. Apoptosis 9:77–82
Gabbiani G, Gabbiani F, Lombardi D, Schwartz SM (1983) Organization of actin cytoskeleton in normal and regenerating arterial endothelial cells. Proc Natl Acad Sci U S A 80:2361–2364
Geiszt M, Kopp JB, Varnai P, Leto TL (2000) Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A 97:8010–8014
Geiszt M, Lekstrom K, Brenner S, Hewitt SM, Dana R, Malech HL, Leto TL (2003a) NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol 171:299–306
Geiszt M, Lekstrom K, Witta J, Leto TL (2003b) Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J Biol Chem 278:20006–20012
Georgatsou E, Alexandraki D (1999) Regulated expression of the Saccharomyces cerevisiae Fre1p/Fre2p Fe/Cu reductase related genes. Yeast 15:573–584
Gess B, Hofbauer KH, Wenger RH, Lohaus C, Meyer HE, Kurtz A (2003) The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur J Biochem 270:2228–2235
Gething MJ, Sambrook J (1992) Protein folding in the cell. Nature 355:33–45
Ghyczy M, Boros M (2001) Electrophilic methyl groups present in the diet ameliorate pathological states induced by reductive and oxidative stress: a hypothesis. Br J Nutr 85:409–414
Gilbert HF (1990) Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol 63:69–172
Gille L, Nohl H (2000) The existence of a lysosomal redox chain and the role of ubiquinone. Arch Biochem Biophys 375:347–354
Goldschmidt-Clermont PJ, Moldovan L (1999) Stress, superoxide, and signal transduction. Gene Expr 7:255–260
Gordillo GM, Sen CK (2003) Revisiting the essential role of oxygen in wound healing. Am J Surg 186:259–263
Goto T, Yamaza T, Tanaka T (2003) Cathepsins in the osteoclast. J Electron Microsc (Tokyo) 52:551–558
Grill HP, Zweier JL, Kuppusamy P, Weisfeldt ML, Flaherty JT (1992) Direct measurement of myocardial free radical generation in an in vivo model: effects of postischemic reperfusion and treatment with human recombinant superoxide dismutase. J Am Coll Cardiol 20:1604–1611
Gu Y, Xu YC, Wu RF, Souza RF, Nwariaku FE, Terada LS (2002) TNFalpha activates c-Jun amino terminal kinase through p47(phox). Exp Cell Res 272:62–74
Gunter TE, Buntinas L, Sparagna GC, Gunter KK (1998) The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochim Biophys Acta 1366:5–15
Gutierrez C, Okita R, Krisans S (1988) Demonstration of cytochrome reductases in rat liver peroxisomes: biochemical and immunochemical analyses. J Lipid Res 29:613–628
Hagen T, Lagace CJ, Modica-Napolitano JS, Aprille JR (2003) Permeability transition in rat liver mitochondria is modulated by the ATP-Mg/Pi carrier. Am J Physiol Gastrointest Liver Physiol 285:G274–G281
Halliwell B (1991) Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med 91:14S–22S
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Hengartner MO (1997) Apoptosis. CED-4 is a stranger no more. Nature 388:714–715
Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S (2004) Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem 88:657–667
Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G (1997) The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15:1573–1581
Hohler B, Holzapfel B, Kummer W (2000) NADPH oxidase subunits and superoxide production in porcine pulmonary artery endothelial cells. Histochem Cell Biol 114:29–37
Holland JA, Goss RA, O’Donnell RW, Chang MM, Johnson DK, Ziegler LM (2001) Low-density lipoprotein induced actin cytoskeleton reorganization in endothelial cells: mechanisms of action. Endothelium 8:117–135
Hotter G, Closa D, Gelpi E, Prats N, Rosello-Catafau J (1995) Role of xanthine oxidase and eicosanoids in development of pancreatic ischemia-reperfusion injury. Inflammation 19:469–478
Hoyal CR, Thomas AP, Forman HJ (1996) Hydroperoxide-induced increases in intracellular calcium due to annexin VI translocation and inactivation of plasma membrane Ca2+-ATPase. J Biol Chem 271:29205–29210
Huschenbett J, Zaidi A, Michaelis ML (1998) Sensitivity of the synaptic membrane Na+/Ca2+ exchanger and the expressed NCX1 isoform to reactive oxygen species. Biochim Biophys Acta 1374:34–46
Hwang C, Sinskey AJ, Lodish HF (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257:1496–1502
Iemitsu M, Miyauchi T, Maeda S, Tanabe T, Takanashi M, Irukayama-Tomobe Y, Sakai S, Ohmori H, Matsuda M, Yamaguchi I (2002) Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol 283:H1750–H1760
Ingber DE (2002) Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ Res 91:877–887
Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S, Hazra TK (2003) Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology 193:43–65
Jiao HL, Zhao BL (2002) Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol Appl Pharmacol 185:172–179
Johansson AC, Steen H, Ollinger K, Roberg K (2003) Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ 10:1253–1259
Kano Y, Katoh K, Masuda M, Fujiwara K (1996) Macromolecular composition of stress fiber-plasma membrane attachment sites in endothelial cells in situ. Circ Res 79:1000–1006
Kersteen EA, Raines RT (2003) Catalysis of protein folding by protein disulfide isomerase and small-molecule mimics. Antioxid Redox Signal 5:413–424
Kikuchi H, Hikage M, Miyashita H, Fukumoto M (2000) NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene 254:237–243
Knaus UG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM (1991) Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 254:1512–1515
Kohno K, Normington K, Sambrook J, Gething MJ, Mori K (1993) The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol Cell Biol 13:877–890
Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–465
Kozma R, Ahmed S, Best A, Lim L (1995) The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol 15:1942–1952
Kreis TE, Birchmeier W (1980) Stress fiber sarcomeres of fibroblasts are contractile. Cell 22:555–561
Kudo T, Katayama T, Imaizumi K, Yasuda Y, Yatera M, Okochi M, Tohyama M, Takeda M (2002) The unfolded protein response is involved in the pathology of Alzheimer’s disease. Ann N Y Acad Sci 977:349–355
Lambeth JD (2002) Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol 9:11–17
Lane N (2003) A unifying view of ageing and disease: the double-agent theory. J Theor Biol 225:531–540
Legakis JE, Koepke JI, Jedeszko C, Barlaskar F, Terlecky LJ, Edwards HJ, Walton PA, Terlecky SR (2002) Peroxisome senescence in human fibroblasts. Mol Biol Cell 13:4243–4255
Lelkes PI, Hahn KL, Sukovich DA, Karmiol S, Schmidt DH (1998) On the possible role of reactive oxygen species in angiogenesis. Adv Exp Med Biol 454:295–310
Li JM, Shah AM (2002) Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277:19952–19960
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489
Li Z, Yasuda Y, Li W, Bogyo M, Katz N, Gordon RE, Fields GB, Bromme D (2004) Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J Biol Chem 279:5470–5479
Lipinski B (2002) Evidence in support of a concept of reductive stress. Br J Nutr 87:93–94
Liu JW, Kayasuga A, Nagao N, Masatsuji-Kato E, Tuzuki T, Miwa N (2003) Repressions of actin assembly and RhoA localization are involved in inhibition of tumor cell motility by lipophilic ascorbyl phosphate. Int J Oncol 23:1561–1567
Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA (2003) Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol 4:1065–1073
Lorenzo HK, Susin SA, Penninger J, Kroemer G (1999) Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ 6:516–524
Losch A, Koch-Brandt C (1995) Dithiothreitol treatment of Madin-Darby canine kidney cells reversibly blocks export from the endoplasmic reticulum but does not affect vectorial targeting of secretory proteins. J Biol Chem 270:11543–11548
Machen TE, Leigh MJ, Taylor C, Kimura T, Asano S, Moore HP (2003) pH of TGN and recycling endosomes of H+/K+-ATPase-transfected HEK-293 cells: implications for pH regulation in the secretory pathway. Am J Physiol Cell Physiol 285:C205–C214
Malorni W, Iosi F, Santini MT, Testa U (1993) Menadione-induced oxidative stress leads to a rapid down-modulation of transferrin receptor recycling. J Cell Sci 106:309–318
Malorni W, Testa U, Rainaldi G, Tritarelli E, Peschle C (1998) Oxidative stress leads to a rapid alteration of transferrin receptor intravesicular trafficking. Exp Cell Res 241:102–116
Martinez GR, Loureiro AP, Marques SA, Miyamoto S, Yamaguchi LF, Onuki J, Almeida EA, Garcia CC, Barbosa LF, Medeiros MH, Di Mascio P (2003) Oxidative and alkylating damage in DNA. Mutat Res 544:115–127
Masters CJ (1996) Cellular signalling: the role of the peroxisome. Cell Signal 8:197–208
Masters CJ, Crane DI (1995a) On the role of the peroxisome in ontogeny, ageing and degenerative disease. Mech Ageing Dev 80:69–83
Masters CJ, Crane D (1995b) The peroxisome: a vital organelle. Cambridge University Press, Cambridge, MA
McManaman JL, Bain DL (2002) Structural and conformational analysis of the oxidase to dehydrogenase conversion of xanthine oxidoreductase. J Biol Chem 277:21261–21268
McStay GP, Clarke SJ, Halestrap AP (2002) Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J 367:541–548
Meyer JW, Holland JA, Ziegler LM, Chang MM, Beebe G, Schmitt ME (1999) Identification of a functional leukocyte-type NADPH oxidase in human endothelial cells: a potential atherogenic source of reactive oxygen species. Endothelium 7:11–22
Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R (2001) Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J 20:6288–6296
Michalik L, Desvergne B, Wahli W (2004) Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer 4:61–70
Mikkelsen RB, Wardman P (2003) Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 22:5734–5754
Milzani A, DalleDonne I, Colombo R (1997) Prolonged oxidative stress on actin. Arch Biochem Biophys 339:267–274
Moldovan L, Goldschmidt-Clermont PJ (1998) Of proteins, redox states and living things. In: Beysens D, Forgacs G (eds) Dynamical networks in physics and biology. Springer, Berlin Heidelberg New York, pp 51–66
Moldovan NI, Milliken EE, Irani K, Chen J, Sohn RH, Finkel T, Goldschmidt-Clermont PJ (1997) Regulation of endothelial cell adhesion by profilin. Curr Biol 7:24–30
Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ (1999) The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal 1:29–43
Moldovan L, Moldovan NI, Sohn RH, Parikh SA, Goldschmidt-Clermont PJ (2000) Redox changes of cultured endothelial cells and actin dynamics. Circ Res 86:549–557
Moreno S, Mugnaini E, Ceru MP (1995) Immunocytochemical localization of catalase in the central nervous system of the rat. J Histochem Cytochem 43:1253–1267
Mori K (2003) Frame switch splicing and regulated intramembrane proteolysis: key words to understand the unfolded protein response. Traffic 4:519–528
Morre DJ, Morre DM (2003) Cell surface NADH oxidases (ECTO-NOX proteins) with roles in cancer, cellular time-keeping, growth, aging and neurodegenerative diseases. Free Radic Res 37:795–808
Murshid A, Presley JF (2004) ER-to-Golgi transport and cytoskeletal interactions in animal cells. Cell Mol Life Sci 61:133–145
Muse KE, Oberley TD, Sempf JM, Oberley LW (1994) Immunolocalization of antioxidant enzymes in adult hamster kidney. Histochem J 26:734–753
Nathan C (2003) Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest 111:769–778
Navas P, Fernandez-Ayala DM, Martin SF, Lopez-Lluch G, De Caboa R, Rodriguez-Aguilera JC, Villalba JM (2002) Ceramide-dependent caspase 3 activation is prevented by coenzyme Q from plasma membrane in serum-deprived cells. Free Radic Res 36:369–374
Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112:481–490
Nguyen T, Sherratt PJ, Pickett CB (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 43:233–260
Nilsson UA, Schoenberg MH, Aneman A, Poch B, Magadum S, Beger HG, Lundgren O (1994) Free radicals and pathogenesis during ischemia and reperfusion of the cat small intestine. Gastroenterology 106:629–636
Nilsson E, Ghassemifar R, Brunk UT (1997) Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J 29:857–865
Nimnual AS, Taylor LJ, Bar-Sagi D (2003) Redox-dependent downregulation of Rho by Rac. Nat Cell Biol 5:236–241
Nobes CD, Hall A (1995) Rho, Rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53–62
Nohl H, Gille L (2002) The bifunctional activity of ubiquinone in lysosomal membranes. Biogerontology 3:125–131
Ogawa Y, Kobayashi T, Nishioka A, Kariya S, Ohnishi T, Hamasato S, Seguchi H, Yoshida S (2004) Reactive oxygen species-producing site in radiation-induced apoptosis of human peripheral T cells: involvement of lysosomal membrane destabilization. Int J Mol Med 13:69–73
Palade G (1975) Intracellular aspects of the process of protein synthesis. Science 189:347–358
Pardee JD, Spudich JA (1982) Purification of muscle actin. Methods Cell Biol 24:271–289
Parinandi NL, Kleinberg MA, Usatyuk PV, Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG, Natarajan V (2003) Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am J Physiol Lung Cell Mol Physiol 284:L26–L38
Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, Di Cesare S, Bonetto V, Casoni F, Carrozzo R, Federici G, Piemonte F (2003) Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: a potential role in the pathogenesis of the disease. J Biol Chem 278:42588–42595
Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT (2003) Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med 34:1295–1305
Pfannstiel J, Cyrklaff M, Habermann A, Stoeva S, Griffiths G, Shoeman R, Faulstich H (2001) Human cofilin forms oligomers exhibiting actin bundling activity. J Biol Chem 276:49476–49484
Polishchuk RS, Mironov AA (2004) Structural aspects of Golgi function. Cell Mol Life Sci 61:146–158
Pollard TD (1986) Assembly and dynamics of the actin filament system in nonmuscle cells. J Cell Biochem 31:87–95
Pollard T (1988) Macromolecular motion in the actin cytoskeleton depends on dynamic, low-affinity interactions. In: Satir P, Condeelis J, Lazarides E (eds) Signal transduction in cytoplasmic organization and cell motility. Liss, New York, pp 263–269
Pollard TD, Cooper JA (1986) Actin and actin-binding proteins. A critical evaluation of mechanisms and functions. Annu Rev Biochem 55:987–1035
Pollard MG, Travers KJ, Weissman JS (1998) Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol Cell 1:171–182
Pritsos CA (2000) Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem Biol Interact 129:195–208
Punj V, Chakrabarty AM (2003) Redox proteins in mammalian cell death: an evolutionarily conserved function in mitochondria and prokaryotes. Cell Microbiol 5:225–231
Qian Y, Luo J, Leonard SS, Harris GK, Millecchia L, Flynn DC, Shi X (2003) Hydrogen peroxide formation and actin filament reorganization by Cdc42 are essential for ethanol-induced in vitro angiogenesis. J Biol Chem 278:16189–16197
Rabek JP, Boylston WH, III, Papaconstantinou J (2003) Carbonylation of ER chaperone proteins in aged mouse liver. Biochem Biophys Res Commun 305:566–572
Ramjiawan B, Czubryt MP, Massaeli H, Gilchrist JS, Pierce GN (1997) Oxidation of nuclear membrane cholesterol inhibits nucleoside triphosphatase activity. Free Radic Biol Med 23:556–562
Raymond MA, Mollica L, Vigneault N, Desormeaux A, Chan JS, Filep JG, Hebert MJ (2003) Blockade of the apoptotic machinery by cyclosporin A redirects cell death toward necrosis in arterial endothelial cells: regulation by reactive oxygen species and cathepsin D. FASEB J 17:515–517
Ridley AJ, Hall A (1992) The small GTP-binding protein Rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399
Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A (1992) The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 70:401–410
Roberts LJ, Morrow JD (2002) Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci 59:808–820
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14:20–28
Sadek HA, Nulton-Persson AC, Szweda PA, Szweda LI (2003) Cardiac ischemia/reperfusion, aging, and redox-dependent alterations in mitochondrial function. Arch Biochem Biophys 420:201–208
Saran M (2003) To what end does nature produce superoxide? NADPH oxidase as an autocrine modifier of membrane phospholipids generating paracrine lipid messengers. Free Radic Res 37:1045–1059
Saran M, Michel C, Stettmaier K, Bors W (2000) Arguments against the significance of the Fenton reaction contributing to signal pathways under in vivo conditions. Free Radic Res 33:567–579
Sato N, Iwata S, Nakamura K, Hori T, Mori K, Yodoi J (1995) Thiol-mediated redox regulation of apoptosis. Possible roles of cellular thiols other than glutathione in T cell apoptosis. J Immunol 154:3194–3203
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30:1191–1212
Sevier CS, Cuozzo JW, Vala A, Aslund F, Kaiser CA (2001) A flavoprotein oxidase defines a new endoplasmic reticulum pathway for biosynthetic disulphide bond formation. Nat Cell Biol 3:874–882
Shamu CE, Cox JS, Walter P (1994) The unfolded-protein-response pathway in yeast. Trends Cell Biol 4:56–60
Siems W, Capuozzo E, Lucano A, Salerno C, Crifo C (2003) High sensitivity of plasma membrane ion transport ATPases from human neutrophils towards 4-hydroxy-2,3-trans-nonenal. Life Sci 73:2583–2590
Singh I (1996) Mammalian peroxisomes: metabolism of oxygen and reactive oxygen species. Ann N Y Acad Sci 804:612–627
Singh I (1997) Biochemistry of peroxisomes in health and disease. Mol Cell Biochem 167:1–29
Singh I, Pahan K, Dhaunsi GS, Lazo O, Ozand P (1993) Phytanic acid alpha-oxidation. Differential subcellular localization in rat and human tissues and its inhibition by nycodenz. J Biol Chem 268:9972–9979
Singh AK, Dhaunsi GS, Gupta MP, Orak JK, Asayama K, Singh I (1994) Demonstration of glutathione peroxidase in rat liver peroxisomes and its intraorganellar distribution. Arch Biochem Biophys 315:331–338
Singh AK, Dobashi K, Gupta MP, Asayama K, Singh I, Orak JK (1999) Manganese superoxide dismutase in rat liver peroxisomes: biochemical and immunochemical evidence. Mol Cell Biochem 197:7–12
Skulachev VP (1996) Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett 397:7–10
Slupphaug G, Kavli B, Krokan HE (2003) The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res 531:231–251
Small JV, Stradal T, Vignal E, Rottner K (2002) The lamellipodium: where motility begins. Trends Cell Biol 12:112–120
Soderdahl T, Enoksson M, Lundberg M, Holmgren A, Ottersen OP, Orrenius S, Bolcsfoldi G, Cotgreave IA (2003) Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J 17:124–126
Souza HP, Liu X, Samouilov A, Kuppusamy P, Laurindo FR, Zweier JL (2002) Quantitation of superoxide generation and substrate utilization by vascular NAD(P)H oxidase. Am J Physiol Heart Circ Physiol 282:H466–H474
Stieber A, Chen Y, Wei S, Mourelatos Z, Gonatas J, Okamoto K, Gonatas NK (1998) The fragmented neuronal Golgi apparatus in amyotrophic lateral sclerosis includes the trans-Golgi-network: functional implications. Acta Neuropathol (Berl) 95:245–253
Stieber A, Gonatas JO, Collard J, Meier J, Julien J, Schweitzer P, Gonatas NK (2000) The neuronal Golgi apparatus is fragmented in transgenic mice expressing a mutant human SOD1, but not in mice expressing the human NF-H gene. J Neurol Sci 173:63–72
Stuurman N, Floore A, Colen A, de Jong L, van Driel R (1992) Stabilization of the nuclear matrix by disulfide bridges: identification of matrix polypeptides that form disulfides. Exp Cell Res 200:285–294
Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD (1999) Cell transformation by the superoxide-generating oxidase Mox1. Nature 401:79–82
Sun-Wada GH, Wada Y, Futai M (2003) Lysosome and lysosome-related organelles responsible for specialized functions in higher organisms, with special emphasis on vacuolar-type proton ATPase. Cell Struct Funct 28:455–463
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–446
Szweda PA, Camouse M, Lundberg KC, Oberley TD, Szweda LI (2003) Aging, lipofuscin formation, and free radical-mediated inhibition of cellular proteolytic systems. Ageing Res Rev 2:383–405
Tabuchi K, Tsuji S, Ito Z, Hara A, Kusakari J (2001) Does xanthine oxidase contribute to the hydroxyl radical generation in ischemia and reperfusion of the cochlea? Hear Res 153:1–6
Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H (2003) Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem 278:25234–25246
Tamura M, Kai T, Tsunawaki S, Lambeth JD, Kameda K (2000) Direct interaction of actin with p47(phox) of neutrophil NADPH oxidase. Biochem Biophys Res Commun 276:1186–1190
Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J (2002) Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J 21:1695–1703
Terada LS, Piermattei D, Shibao GN, McManaman JL, Wright RM (1997) Hypoxia regulates xanthine dehydrogenase activity at pre- and posttranslational levels. Arch Biochem Biophys 348:163–168
Trotter EW, Grant CM (2002) Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol Microbiol 46:869–878
Tu BP, Weissman JS (2002) The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell 10:983–994
Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164:341–346
Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS (2000) Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290:1571–1574
Tyurina YY, Shvedova AA, Kawai K, Tyurin VA, Kommineni C, Quinn PJ, Schor NF, Fabisiak JP, Kagan VE (2000) Phospholipid signaling in apoptosis: peroxidation and externalization of phosphatidylserine. Toxicology 148:93–101
Ueda S, Nakamura H, Masutani H, Sasada T, Yonehara S, Takabayashi A, Yamaoka Y, Yodoi J (1998) Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J Immunol 161:6689–6695
Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J (2002) Redox control of cell death. Antioxid Redox Signal 4:405–414
Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW (2002) Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 91:1160–1167
van der Klei I, Veenhuis M (2002) Peroxisomes: flexible and dynamic organelles. Curr Opin Cell Biol 14:500–505
van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, Post JA (2003a) Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal 5:381–387
van der Vlies D, Woudenberg J, Post JA (2003b) Protein oxidation in aging: endoplasmic reticulum as a target. Amino Acids 25:397–407
VanderWaal RP, Spitz DR, Griffith CL, Higashikubo R, Roti Roti JL (2002) Evidence that protein disulfide isomerase (PDI) is involved in DNA-nuclear matrix anchoring. J Cell Biochem 85:689–702
van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL (2002) Reactive oxygen species mediate Rac-induced loss of cell–cell adhesion in primary human endothelial cells. J Cell Sci 115:1837–1846
Vignais PV (2002) The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59:1428–1459
Villalba JM, Navas P (2000) Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal 2:213–230
Voeltz GK, Rolls MM, Rapoport TA (2002) Structural organization of the endoplasmic reticulum. EMBO Rep 3:944–950
Wanders RJ, Denis S (1992) Identification of superoxide dismutase in rat liver peroxisomes. Biochim Biophys Acta 1115:259–262
Wang Z, Castresana MR, Newman WH (2001) Reactive oxygen and NF-kappaB in VEGF-induced migration of human vascular smooth muscle cells. Biochem Biophys Res Commun 285:669–674
Ward TH, Brandizzi F (2004) Dynamics of proteins in Golgi membranes: comparisons between mammalian and plant cells highlighted by photobleaching techniques. Cell Mol Life Sci 61:172–185
Watson WH, Jones DP (2003) Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett 543:144–147
Wientjes FB, Reeves EP, Soskic V, Furthmayr H, Segal AW (2001) The NADPH oxidase components p47(phox) and p40(phox) bind to moesin through their PX domain. Biochem Biophys Res Commun 289:382–388
Wong AJ, Pollard TD, Herman IM (1983) Actin filament stress fibers in vascular endothelial cells in vivo. Science 219:867–869
Wu RF, Gu Y, Xu YC, Nwariaku FE, Terada LS (2003) Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem 278:36830–36840
Wu RF, Gu Y, Xu YC, Mitola S, Bussolino F, Terada LS (2004) Human immunodeficiency virus type 1 Tat regulates endothelial cell actin cytoskeletal dynamics through PAK1 activation and oxidant production. J Virol 78:779–789
Xia Y, Zweier JL (1995) Substrate control of free radical generation from xanthine oxidase in the postischemic heart. J Biol Chem 270:18797–18803
Xu J, Schwarz WH, Kas JA, Stossel TP, Janmey PA, Pollard TD (1998) Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys J 74:2731–2740
Young J, Kane LP, Exley M, Wileman T (1993) Regulation of selective protein degradation in the endoplasmic reticulum by redox potential. J Biol Chem 268:19810–19818
Yu Z, Li W, Brunk UT (2003a) 3-Aminopropanal is a lysosomotropic aldehyde that causes oxidative stress and apoptosis by rupturing lysosomes. APMIS 111:643–652
Yu Z, Persson HL, Eaton JW, Brunk UT (2003b) Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med 34:1243–1252
Zang Y, Beard RL, Chandraratna RA, Kang JX (2001) Evidence of a lysosomal pathway for apoptosis induced by the synthetic retinoid CD437 in human leukemia HL-60 cells. Cell Death Differ 8:477–485
Zhang Z, Blake DR, Stevens CR, Kanczler JM, Winyard PG, Symons MC, Benboubetra M, Harrison R (1998) A reappraisal of xanthine dehydrogenase and oxidase in hypoxic reperfusion injury: the role of NADH as an electron donor. Free Radic Res 28:151–164
Zhao M, Antunes F, Eaton JW, Brunk UT (2003) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem 270:3778–3786
Zweier JL, Broderick R, Kuppusamy P, Thompson-Gorman S, Lutty GA (1994) Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J Biol Chem 269:24156–24162
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moldovan, L., Moldovan, N.I. Oxygen free radicals and redox biology of organelles. Histochem Cell Biol 122, 395–412 (2004). https://doi.org/10.1007/s00418-004-0676-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-004-0676-y