
Abstract
The study of function and regulation of the phenotype of alveolar type I (AT I) epithelial cells is limited by the rareness of suitable cell lines or primary cultures of this cell type. We describe in the present study the type I-like rat epithelial cell line R3/1. This cell line displays in vitro a phenotype with several characteristic features of AT I cells. R3/1 cells were analysed for mRNA and protein content of markers related to the AT I cell type (T1α, ICAM-1, connexin-43, caveolins-1 and -2) and AT II phenotypes [surfactant proteins (SPs) A, B, C and D]. The mRNAs for SPs were found to be at a low level. Moderate protein levels for SP-A and SP-B were found, and SP-C and SP-D proteins were not detectable. R3/1 cells are positive for CD44s, E-cadherin, cytokeratin, vimentin and RAGE, and bind the lectins BPA and SBA. For demonstration of the suitability of R3/1 cells for in vitro studies on epithelial injury, the cells were treated with bleomycin. As shown by real-time RT-PCR and immunoblotting, bleomycin-treatment of R3/1 cells resulted in a decrease in mRNA and protein for both caveolin-1 and caveolin-2 in comparison with controls. The AT I-like cell line R3/1 may serve as a promising tool for the study of lung cell biology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alveolar epithelial damage is a major event of many severe pulmonary diseases such as acute respiratory distress syndrome (ARDS), chronic pulmonary inflammation and pulmonary fibrosis. The alveolar epithelium in normal adult lungs is populated by two major epithelial cell types: type I cells (AT I) and type II cells (AT II). AT I cells are large flat cells that cover more than 95% of the alveolar surface and are considered to be terminally differentiated. AT I cells contain transport proteins such as Na+-K+-ATPases, epithelial sodium channels and aquaporin (Aqp) water channels and, therefore, play an active role in regulation of lung fluid homeostasis (Johnson et al. 2002; Matthay et al. 2002). These cells also might be involved in the transport of macromolecules because of the abundance of microvesicles (caveolae; Gumbleton et al. 2003).
AT II cells, covering 2–5% of the lungs’ surface area, produce, secrete and recycle pulmonary surfactant (for review see Fehrenbach et al. 1998). They are generally considered to serve as the progenitors of AT I cells (Adamson and Bowden 1975).
Our knowledge of the general functions of AT I cells, as distinct from gas exchange, is still limited. For investigation of these functions, the correct identification of this cell type in lung tissue obtained from either animals or human lung samples, in isolated-perfused lungs or in lung explants, is necessary. The elimination of potential influences of other cell types or of extracellular matrix components on AT I cells requires the isolation and investigation of these cells in in vitro systems.
It is technically difficult to isolate and culture alveolar epithelial AT I cells. Therefore, many investigators have exploited the transformation of isolated AT II cells to AT I-like cells in vitro. In general, culturing cells on plastic in the absence of an air-liquid interface, in the absence of exogenous matrix proteins, growth factors or mechanical distension, favours the morphologic transformation of AT II cells to flattened, attenuated cells which do not contain lamellar bodies (Borok et al. 1998a, b; Campbell et al. 1999; Christensen et al. 1993; Dobbs et al. 1988; Gutierrez et al. 1998; Shannon et al. 1992). These cells express AT I cell phenotypic markers, such as ICAM-1, RTI40/T1α, caveolin-1, -2, (Cav-1, -2) and Aqp-5, but have lost phenotypic markers associated with AT II cells (for example, surfactant proteins). It has also been observed that cultured epithelial cells may coexpress both type I and type II phenotypic markers (Borok et al. 1998b; Campbell et al. 1999; Christensen et al. 1993; Dobbs et al. 1988). Currently, immortal epithelial lung cells with predominant properties of type I pneumocytes are not known, except the mouse cell line E10 which, however, also expresses some AT II cell antigens (Cao and Williams 2002; Williams 2003). The rat cell line R3/1 used in the present study was established from cells obtained from bronchopulmonary tissue of fetal Wistar rats at day 20 of gestation (Knebel et al. 1994).
The aim of the present study was the characterisation of the rat lung cell line R3/1 to demonstrate the expression of AT I and/or AT II cell-specific proteins under normal conditions and during bleomycin-induced injury. Bleomycin, a chemotherapeutic agent known to cause lung fibrosis as a side effect of its cytostatic action, is commonly used for in vitro and in vivo studies of epithelial damage in lungs (Kasper et al. 1995; Koslowski et al. 1998; Kuwano et al. 2000; Wang et al. 2000). Bleomycin-induced epithelial damage was shown to initiate a loss of specific membrane proteins from AT I cells in rat lung, for example, ICAM-1 (Kasper et al. 1995) or RTI40 (Koslowski et al. 1998).
Caveolae are small flask-shaped microvesicles found in AT I cells, but not in AT II cells (for review see Razani et al. 2001). Caveolae may be involved in the adsorption of macromolecules from the alveolar air spaces. Caveolin comprises a family of at least four proteins: Cav-1α, Cav-1β, Cav-2 and Cav-3. Cav-1, which is the major scaffolding protein of caveolae, is expressed in AT I cells, in addition to Cav-2 (Parton 1996). This protein is the most widely investigated caveolar protein and influences the structural properties of caveolae. It is involved in cell migration, lipid transport, membrane traffic and signal transduction (Liu et al. 2002; Parton 1996; Razani and Lisanti 2001).
In lung disease, for example following X-ray irradiation of animals with subsequent initiation of lung fibrosis, we noted a dramatic loss of caveolin expression in the alveolar epithelium, but an increase in expression of caveolin in the pulmonary capillary endothelium (Kasper et al. 1998). Caveolin expression seems to be a sensitive marker of acute and chronic lung injury (Kasper et al. 2004).
In the present study, therefore, we investigated the expression of the caveolae-specific proteins Cav-1 and Cav-2 in bleomycin-exposed R3/1 cells with focus on the examination of the suitability of this cell line for in vitro experiments to reproduce processes involved in lung injury.
Material and methods
Cell isolation, culture and bleomycin treatment
The rat cell line R3/1 was established from pulmonary tissue of male fetuses of Han-Wistar rats on day 20 of gestation by explant-replica techniques, as described by Knebel and co-workers (1994), without immortalisation procedures. The outgrown undifferentiated cells were expanded by Emura and co-workers (1990) using medium with low Ca2+ concentrations in combination with cis-4-hydroxy-l-proline to inhibit differentiation and proliferation of fibroblasts. Cells were passaged 12 times, cloned as described by Reid (1979) and stocked frozen at passage numbers 17–23 for morphological characterisation and further experiments.
R3/1 cells were plated into culture dishes at a density of 5×103 cells/cm2 and grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 50 IU penicillin/ml and 50 μg streptomycin/ml (medium and supplements from Biochrom, Berlin, Germany). Cells were passaged routinely and the media were changed every 3 days.
For determination of growth properties of R3/1 cells on perforated membranes, cells were plated at a density of 3.75×105/cm2 into transwell inserts (Millicell-PCF, 0.4 μm; Millipore, Eschborn, Germany) and grown in RPMI 1640 medium (supplements as described above) for at least 4 days.
For bleomycin treatment, R3/1 cells were grown as described above in culture dishes to subconfluent cultures. For estimation of an appropriate bleomycin concentration, R3/1 cells were cultured in the presence of 10, 50, 100, 200 and 500 mU bleomycin/ml (Mack, Illertissen, Germany) for 48 and 96 h. The cells were harvested, counted and the percentage of dead cells was determined using the Trypan blue exclusion test. At 100 mU bleomycin/ml cell growth was inhibited remarkably to 63% of that of untreated controls whereas the viability was at 81% after 96 h exposure to this bleomycin concentration. Above 100 mU bleomycin/ml, no cell growth was detected and the viability was reduced to 48% and to 5% at 200 and 500 mU bleomycin/ml, respectively. As a consequence, 100 mU bleomycin/ml were used for experiments. The medium was changed and the drug was added to the cultures for 48 h.
Determination of cell viability
The colorimetric MTT (3-[4,5-dimethylthiozol-2-yl]-2,5-diphenyl tetrazolium bromide) survival assay that measures the mitochondrial conversion of the tetrazolium salt to a blue formazan salt was used as previously described (Slack et al. 1996). Viability was expressed as light absorption at 570 nm with a reference wavelength of 655 nm.
Furthermore, bleomycin-treated and paraffin-embedded cells were tested for the apoptotic markers active caspase-3 (polyclonal rabbit anti-cleaved caspase-3; New England Biolabs, Frankfurt, Germany; dilution 1:50) and caspase-9 cleavage product of vimentin (rabbit anti-capase-9 vimentin split product V1, dilution 1:1,000; Nakanishi et al. 2001) by immunocytochemistry (details see below). As positive control for both caspase activities, paraffin sections from previous studies either of embedded human epithelial L132 cells which were treated with glyoxal for induction of apoptosis (Roehlecke et al. 2000) or of rat lung slices after treatment with CdCl2 (Kasper et al. 2004) were used.
Immunocytochemistry
Immunofluorescence labelling was performed on acetone–methanol-fixed (1:1; −20°C, 10 min) cells. After washing with phosphate-buffered saline, pH 7.4 (PBS), polyclonal or monoclonal antibodies (see Table 1) were added for 1 h at 25°C, followed by specific anti-rabbit, anti-goat or anti-mouse secondary antibodies conjugated to fluorescein isothiocyanate (diluted 1:80; Dianova, Hamburg, Germany) for 30 min at 25°C. As embedding medium, glycerol-PBS (9:1) supplemented with 2.5% DABCO (1,4-diazobicyclo[2,2,2]-octane; Janssen, Beerse, Belgium) was used. For controls, primary antibodies were replaced by non-specific immunoglobulins. No immunostaining of controls was detected. Immunostaining was examined with a BX60 fluorescent microscope (Olympus, Hamburg, Germany).
Immunoperoxidase staining was performed on sections of cultured cells. The culture medium was removed, cells were washed with PBS, scraped off and collected by centrifugation. Cells were carefully mixed with fibrin glue (Tissuecol Duo S; Immuno, Heidelberg, Germany) and fibrin was coagulated with thrombin solution (Immuno). After fixation with neutral-buffered formaldehyde (1 h, 4°C) and washing (three times) with PBS for 30 min, cells were embedded in paraffin. Sections of 5 μm were mounted on silane-coated slides. The sections were dewaxed, dried overnight and irradiated with microwaves in 0.01 mol/l sodium citrate buffer (pH 6.0) for 2×5 min at 850 W. After washing with PBS, the sections were treated with 0.3% hydrogen peroxide for 30 min, incubated with respective normal sera and then incubated for 1 h at 37°C with primary antibodies (Table 1). Binding of the antibodies was detected by incubation with biotinylated secondary antibodies followed by a streptavidin-biotin-peroxidase complex (Vecstatin Elite; Vector, Burlingame, CA, USA). The peroxidase activity was visualised with 3,3′-diaminobenzidine. As positive control, sections of a normal rat lung routinely fixed in 4% buffered formaldehyde solution and embedded in paraffin were used.
Lectin cytochemistry
For demonstration of lectin binding, biotinylated BPA (Bauhinia purpurea agglutinin), LEA (Lycopersicon esculentum agglutinin) and SBA (soybean agglutinin) lectins were employed. Their specificity has been tested in blocking experiments with the corresponding sugar (Kasper et al. 1993a). Paraffin sections of cells and control lungs were incubated with normal horse serum and the endogenous peroxidase was blocked for 30 min by incubation in methanol/H2O2 (0.3%). The sections were then incubated with the lectins (see Table 1). For detection, the ABC complex (Vector) was used as described (Kasper et al. 1993a).
Transmission electron microscopy
For routine electron microscopy, cells were grown on plastic dishes, harvested with a cell scraper and washed with PBS. Cells were collected by centrifugation and fixed in a solution of 4% glutaraldehyde and 2% p-formaldehyde in 0.1 mol/l cacodylate buffer (pH 7.3) for 2 h. After postfixation in 1% osmium tetroxide for 1 h at 4°C and dehydration by ethanol, the cells were embedded in epoxy resin (Epon 812). For flat embedding, R3/1 cells were grown on plastic tissue culture dishes, processed without scraping and embedded in Epon 812. Ultrathin sections were cut at 50 nm on an ultramicrotome Ultracut (Leica, Nussloch, Germany), mounted on copper grids and contrast-stained with uranyl acetate and lead citrate. The sections were examined with a Zeiss EM 10 electron microscope.
Reverse transcription-PCR (RT-PCR)
For RT-PCR, total RNA was isolated from cell lysates (RNeasy Mini kit; Qiagen, Hilden, Germany). One microgram of total RNA each was reverse transcribed using AMV-reverse transcriptase (Roche, Mannheim, Germany) with oligo-p(dT)15 primers following the manufacturer’s protocol in a final volume of 20 μl. The PCR was performed by using Taq-polymerase (Master Mix kit; Eppendorf, Hamburg, Germany) and gene-specific primers (Table 2) synthesised according to published cDNA sequences of Cav-1α, Cav-2, ICAM-1, T1α, RAGE, GAPDH and the surfactant proteins SP-A, SP-B, SP-C and SP-D [GenBank accession numbers: AF439778 (Cav-1α), NM131914 (Cav-2), NM012967 (ICAM-1), RNU07797 (T1α), NM017008 (GAPDH), primers for SP-A, SP-B, SP-C and SP-D according to Pan and co-workers (2001)]. Amplified PCR products were visualised after electrophoresis in 1.5% agarose gels containing ethidium bromide. Reactions were run on a PCR cycler under the following cycling conditions: 94°C for 4 min followed by amplification. Each cycle of amplification included 30 s denaturation at 95°C, 30 s annealing at 58°C and 45 s synthesis at 72°C. Every PCR was run with 25, 30, 35 and 40 amplification cycles for optimisation of the procedure. After optimisation, 35–40 cycles of amplification were chosen. Unspecific amplification products were not detected in any of the PCR runs. For determination of RAGE-specific mRNA, the PCR conditions were modified: after 30 s denaturation at 95°C, PCR was run for 5 cycles with annealing at 45°C (30 s) followed by 30 cycles with annealing at 52°C (30 s).
Quantification of mRNA levels by real-time PCR
For real-time PCR, we used the QuantiTect SYBR Green PCR kit (Qiagen) according to the manufacturer’s instructions. The PCR primer sequences are shown in Table 3. The gene of the eukaryotic translation elongation factor (EF-2) was used as the housekeeping gene. PCR was performed with cDNA synthesised from 50 ng of sample RNA. Reactions were run on an iCycler iQ (Biorad, Hercules, CA, USA) under the following cycling conditions: 95°C for 13 min followed by 50 cycles of amplification for Cav-1, Cav-2 and EF-2 cDNAs. Each cycle included 20 s denaturation at 95°C, 30 s annealing at 55°C and 30 s synthesis at 72°C. PCR efficiency was above 75% and linearity of the assay was proved by serial dilution of the standard.
Immunoblotting
Cells were homogenised in PBS, pH 7.4, containing 10 μmol/l E-64 (trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane), 20 μmol/l PMSF, 1 mmol/l EDTA, 100 μmol/l pepstatin A and 1 μmol/l leupeptin by sonication (all proteinase inhibitors from Sigma, Deisenhofen, Germany). Samples were centrifuged at 8,000 g at 4°C for 30 min. Protein concentrations were determined according to Bradford (1976). Proteins (25 μg/lane) were separated by SDS gel electrophoresis (5–15%) under reducing conditions and blotted onto nitrocellulose. After blocking in 20 mmol/l TRIS/HCl, pH 7.6, containing 140 mmol/l NaCl and 5% non-fat milk powder (Biorad) at 25°C for 2 h, specific proteins were detected using monoclonal mouse anti-Cav-1 and anti-Cav-2 antibodies (both: dilution 1:500; BD Biosciences, Heidelberg, Germany). Horseradish peroxidase-conjugated anti-mouse immunoglobulin (New England Biolabs) was used as secondary antibody. Protein bands were visualised by enhanced chemiluminescence (New England Biolabs) and analysed with a Genegnom chemiluminescence detection system (Synoptics, UK).
Statistical analysis
Statistical analysis was performed by analysis of variance with ANOVA.
Results
Growth properties of R3/1 cells in cell culture
Growth properties of R3/1 cells were determined for culture on plastic tissue culture dishes and on perforated PCF membranes of transwell inserts. R3/1 cells plated on plastic dishes adhere within 1 h and grow with a median population doubling time of about 38 h in RPMI 1640 medium supplemented with 10% FCS. The image of the cells is shown in Fig. 1A. The R3/1 cells cultured on plastic dishes showed a flattened morphology. Figure 1B and C demonstrate R3/1 cells grown for 4 days on perforated PCF membranes and forming a confluent monolayer.

Phase-contrast image of a subconfluent culture of R3/1 cells on a tissue culture dish. R3/1 cells were cultured for 4 days in plastic tissue culture dishes. The cells showed a flattened morphology. B, C Sections of paraffin-embedded perforated membranes covered by R3/1 cells. The cells were plated into Millicell-PCF transwell inserts and grown for at least 4 days. R3/1 cells formed monolayers on perforated membranes. B H/E staining. C Detection of RAGE by immunoperoxidase technique. ×300
Phenotype of alveolar epithelial R3/1 cells
The immunohistochemical study is aimed at the characterisation of the cell line R3/1 by means of various marker proteins such as cytoskeletal proteins, specific lung cell antigens, adhesion molecules and further structural and secretory proteins (listed in Table 1). The data of parallel immunostaining (immunofluorescence and ABC technique as well) and lectin histochemistry were summarised as follows. In formalin-fixed (paraffin sections) as well as in acetone–methanol-fixed R3/1 cells there was a strong positive staining for epithelial-type keratins numbers 7, 8, 18 and 19 as well as for vimentin (not shown). In addition, the pan-keratin antibody exhibited positive immunostaining. The proteins CD44s (not shown), connexin-43 (Cx43; Fig. 2C) and E-cadherin (Fig. 2D) were constitutively expressed by R3/1 cells. The analysis of lectin binding of embedded as well as acetone–methanol-fixed cells revealed that lectins BPA and SBA, but not LEA, which are characteristic for AT I cells in normal rat lung (Bankston et al. 1991; Kasper et al. 1993a, 1994), were bound to R3/1 cells. The binding of BPA lectin to R3/1 cells is illustrated in Fig. 2A. The expression of the AT I cell-specific protein ICAM-1 in R3/1 cells was also detected by the immunofluorescence technique (Fig. 2B).

Proof of binding of BPA lectin to R3/1 cells (A) and detection of surface expression of ICAM-1 (B), connexin-43 (Cx43; C) and E-cadherin (D) in R3/1 cells by immunofluorescence after acetone–methanol fixation. ×300
R3/1 cells expressed the AT I cell-related antigens T1α (Fig. 3B), Cav-1 (Fig. 3D) and Cav-2 (not shown). The expression was compared with that of T1α and Cav-1 proteins in normal rat lung (Fig. 3A, C). Figure 3B demonstrates that about 50% of R3/1 cells were positive for T1α protein with an estimated portion of 12–15% that were stained very strongly. The intensity of expression differed remarkably. Nearly 100% of R3/1 cells were found to be strongly positive for Cav-1 (Fig. 3D).

Expression of the marker proteins T1α and caveolin-1 (cav-1) in normal rat lung tissue (A, C) and in embedded R3/1 cells (B, D). T1α (A, B) and cav-1 (C, D) were detected using the immunoperoxidase technique. About 50% of R3/1 cells were found to be positive for the AT I-specific protein T1α protein (B) whereas nearly 100% of the cells showed intense staining for cav-1 (D). ×300
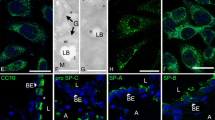
To test potential AT II cell properties of R3/1 cells, the expression of the surfactant proteins SP-A, SP-B, SP-C and SP-D in R3/1 cells was investigated and compared with that in normal rat lung tissue (Fig. 4). In normal rat lung tissue (left column of panels) and embedded R3/1 cells (right column of panels), SP-A (Fig. 4A, B), SP-B (Fig. 4C, D), SP-C (Fig. 4E, F) and SP-D (Fig. 4G, H) were detected using immunoperoxidase staining. AT II cells in lung tissue showed a strong immunostaining for all surfactant proteins (Fig. 4A, C, E, G). For SP-A and SP-B a moderate staining of R3/1 cells was observed (Fig. 4B, D). SP-C and SP-D were not found in R3/1 cells (Fig. 4F, H).

Expression of surfactant proteins (SPs) in normal rat lung tissue (A, C, E, G) and in embedded R3/1cells (B, D, F, H). Paraffin sections of lung tissue and of R3/1 cells were immunostained for SP-A (A, B), SP-B (C, D), SP-C (E, F), SP-D (G, H). ×300
Ultrastructural characteristics
Ultrastructural investigation of flat embedded cells revealed a morphology similar to AT I pneumocytes, for example, presence of caveolae, which sometimes detach from the apical cell surface, a few clathrin-coated vesicles and an abundance of filaments (Fig. 5A).

Electron micrographs of R3/1 cells. A Transmission electron micrograph of flat embedded cells for the ultrastructural localisation of caveolae (arrowheads) and a clathrin-coated vesicle (arrow). Note some detached caveolae (asterisks) and newly forming caveolae at the apical site (thick arrowhead). Scale bar 100 nm. B Ultrastructural appearance of R3/1 cells cultured on plastic surfaces. Scale bar 5 μm. C Higher magnification shows numerous vesicles, enlarged endoplasmic reticulum cisternae and abnormal lamellar bodies (arrowheads). Scale bar 1 μm
R3/1 cells grown on plastic culture dishes and harvested with a cell scraper showed polymorph nuclei, strongly enlarged cisternae of endoplasmic reticulum and numerous vesicles (Fig. 5B). Lamellar inclusions resembling abnormal lamellar bodies of AT II cells (Fig. 5C) were found in about 1–10% of cells.
mRNA expression pattern
The expression of the AT I and AT II pneumocyte-related marker genes was determined by RT-PCR based on expressed mRNA levels in R3/1 cells (Fig. 6). The expression of the housekeeping gene GAPDH was determined in parallel as a control. Figure 6A shows that the specific AT I pneumocyte-related marker genes Cav-1, Cav-2, Cx43, ICAM-1 and T1α were expressed. The expression of the RAGE-encoding gene was also detected (data not shown). The AT II-specific marker genes SP-A, SP-B, SP-C and SP-D were expressed at different levels (Fig. 6B).

RT-PCR products separated by agarose gel electrophoresis. DNA fragments were synthesised by RT-PCR with primers for: A AT I pneumocyte-specific genes Cav-1 (lane 2), Cav-2 (lane 3), ICAM-1 (lane 4), Cx43 (lane 5), T1α (lane 6) and GAPDH (lane 7); B AT II pneumocyte-specific genes SP-A (lane 2), SP-B (lane 3), SP-C (lane 4), SP-D (lane 5) and GAPDH (lane 6) with total RNA of R3/1 cells as template. Lane 1 in A and B shows DNA fragment molecular weight standards
Cav-1 and Cav-2 expression in bleomycin-exposed R3/1 cells
Bleomycin treatment of R3/1 cells was performed to investigate the expression of Cav-1 and Cav-2 under conditions known to induce epithelial injury in the lung. R3/1 cells were treated with 100 mU bleomycin/ml for 48 h. For examination of the viability of the cells in the course of bleomycin exposition, an MTT cell survival assay was performed (Table 4). This viability test reveals that the mitochondria of the bleomycin-treated R3/1 cells at all time points investigated produced lower levels of the formazan salt product of MTT compared to the untreated controls. At 48 h after bleomycin administration to the cultures, the level of this signal for the bleomycin-exposed cells was at about 85% of that of controls (Table 4). The difference between the levels of light absorption at 570 nm for bleomycin-treated cells and controls was found to be statistically significant at any time point studied (two-factorial ANOVA, P<0.05). However, this difference did not change significantly with time (P=0.102). For further confirmation of the viability data, bleomycin-treated and paraffin-embedded cells were tested for apoptotic markers by immunohistochemistry. No staining for the apoptotic markers active caspase-3 and caspase-9 cleavage product of vimentin was observed (data not shown).
For determination of the expression of Cav-1 and -2, total RNA from cell homogenates was prepared and subjected to real-time RT-PCR. The results, as shown in Table 5, demonstrate that bleomycin treatment leads to a decrease of the mRNA expression level for Cav-1 and Cav-2 to about 50% of the controls. In the case of Cav-2, the decrease of the mRNA expression level was found to be significant (Table 5).
For confirmation of the real-time PCR data, the amount of Cav-1 and Cav-2 proteins in R3/1 cells in response to bleomycin treatment was determined by immunoblotting. The time scale was extended to 72 and 96 h for reasons of time-consuming regulation at the protein level. Figure 7 shows the levels of Cav-1 and Cav-2 proteins in R3/1 cells exposed to bleomycin in comparison to untreated controls. The level of Cav-1 was slightly increased after 72 and 96 h (Fig. 7A, B). In bleomycin-treated R3/1 cells, a decrease of Cav-1 was detected at 72 and 96 h. For Cav-2, an increase in the amount of the protein was detected in control cells and a slight decrease in bleomycin-exposed R3/1 cells (Fig. 7C, D). The effects of bleomycin on the amounts of Cav-1 and Cav-2 were statistically significant at 72 and 96 h for Cav-1 and at 72 h for Cav-2. Furthermore, two-factorial ANOVA using the control value at 48 h as covariate revealed an average difference of the values spread over all three time points with P=0.005 for Cav-1 and P=0.003 for Cav-2.

Western blot analysis of R3/1 proteins for Cav-1 (A, B) and Cav-2 (C, D). R3/1 cells were treated with 100 mU bleomycin/ml and cell proteins were analysed at given times. B and D show representative blots for detection of Cav-1 and Cav-2, respectively, and A and C show the results of the densitometric evaluation of the blots (mean ± SD, n=4). Statistical analysis was performed by two-way ANOVA with two between-subject factors (treatment, time). P values for the comparison of bleomycin-treated and control cells at each time point are shown in A and C
Discussion
The cell line R3/1 derived from fetal rat lung as presented here is a new suitable tool for the in vitro study of type I cell biology. R3/1 cells are displaying a variety of proteins, which are characteristic but not exclusive for the type I cell phenotype, such as T1α, ICAM-1, Cav-1 and Cav-2 (reviewed in Williams 2003). Furthermore, two of three lectins known to bind to the surface of AT I cells displayed a positive reaction with R3/1 cells. The expression or absence of cell surface and structural antigens of cultured R3/1 cells correlated well with that seen in normal rat lung tissue. In contrast, expression of markers of the AT II phenotype in R3/1 cells was reduced: SP-C and SP-D proteins were not detectable, and SP-A and SP-B protein were at a moderate level. We detected the mRNAs specific for the surfactant proteins SP-A, SP-B, SP-C and SP-D with SP-B message at the highest level thus indicating some disturbance in the processing of surfactant proteins in R3/1 cells. We cannot exclude that R3/1 cells obtained from fetal tissue show, in part, phenotypic features of poorly differentiated cells not matching the adult AT I phenotype.
Cytokeratins of the simple epithelial type numbers 7, 8, 18 and 19, which are characteristic for rat alveolar epithelial cells in vitro and in vivo (Kasper and Singh 1995; Kasper et al. 1993b; Paine et al. 1988), were found in R3/1 cells. In addition, vimentin, the intermediate filament protein of mesenchymal cells, was detected. Vimentin is often coexpressed in epithelial cells in vitro (Ramaekers et al. 1983).
Cx43 has been detected in alveolar epithelial cells in vitro and in vivo (Abraham et al. 1999; Kasper et al. 1996; Lee et al. 1997). Alveolar epithelial cells show, however, great variability in the expression of connexins as a function of cell phenotype and cell state (Koval 2002). Under certain culture conditions, Cx43 was phenotype-specific for AT I cells (Isakson et al. 2001).
For the expression of RAGE in lung epithelial cells conflicting data exist. Katsuoka and co-workers (1997) have demonstrated the mRNA for RAGE in isolated AT II cells and by in situ hybridisation in mouse lungs. In contrast, RAGE has been selectively detected in AT I cells of human and rat lung by immunohistochemistry (Fehrenbach et al. 1998; Shirasawa et al. 2004). These data were confirmed by immunohistochemistry on paraffin sections of mouse lungs, whereas the immunoreactivity was abolished in RAGE-knockout animals (Liliensiek et al. 2004).
The R3/1 cell layer was examined after histological processing of the PCF membrane of transwell inserts. Ultrastructural analysis was carried out by transmission electron microscopy. Morphological studies showed pure and homogeneous cell cultures. The morphology of the cells growing on PCF membranes becomes reminiscent of AT I cells in that the cells were characterised by the presence of caveolae and clathrin-coated vesicles, the occurrence of very few abnormal lamellar bodies, an absence of apical microvilli and the development of highly attenuated cytoplasmic extensions. Therefore, the cells should prove a useful tool for initial studies of transepithelial transport mechanisms as well as paracellular permeability of the alveolar epithelium.
Permanent cell lines with properties of AT I pneumocytes are rare. Currently, most experiments dealing with the function of the alveolar epithelium employ primary cultures of AT II pneumocytes, immortalised lung cell lines and tumour cell lines. Primary cultures of AT I-like cells are available after isolation of AT II cells and culture on plastic surfaces (Dobbs et al. 1988). However, it is known that AT II cells in culture frequently display an intermediate AT II–I cell type, which has properties of both cell types (Campbell et al. 1999; Clegg et al. 2003; Cunningham et al. 1994; Paine et al. 1994). During culture of primary AT II cells, factors such as keratinocyte growth factor (KGF), culture conditions that facilitate maintenance of cuboidal morphology, and homologous serum support the AT II phenotype, whereas flattened shape and heterologous serum support the AT I cell phenotype (Williams 2003). The isolation of primary AT I cells has been described (Dobbs et al. 1998). Experiments investigating specific functions of AT I cells, however, have some limitations resulting from the fragility of the cells and from the limited life span of primary cultures of the terminally differentiated AT I cells.
Bleomycin exposure of R3/1 cells resulted in a decrease in mRNA levels for Cav-1 and -2 to about 50% of control values as shown by real-time RT-PCR. Immunohistochemistry and immunoblotting confirmed the loss of caveolins at the protein level. This is the first report of downregulation of caveolin expression by treatment of alveolar epithelial cells with bleomycin in vitro. The specific function of caveolins in the pulmonary epithelium is, so far, undefined (Gumbleton et al. 2003). Only sparse data exist on a modulation of caveolins in the lung in vivo. In pulmonary fibrosis induced in different experimental models, we noted a strong reduction of caveolin expression in the alveolar epithelium (Kasper 2003; Kasper et al. 1998). Mice with a null mutation in the Cav-1 gene (Drab et al. 2001; Razani and Lisanti 2001; Razani et al. 2001) and the Cav-2 gene (Razani et al. 2002) highlight the importance of caveolins in the lung, although phenotypes of the animals do not clarify the specific functions of either caveolin or caveolae. The Cav-1-knockout animals lacking caveolae are viable, fertile, do not develop lung tumours and have no embryonic lethality. In both knockout lines, the major pathology is in lung, manifest as thickened alveolar septae, abnormal cell proliferation and thickened basement membranes.
The unsolved problem is whether the loss of caveolin in bleomycin-treated R3/1 cells is related to apoptosis or not. Caveolin has been shown to be involved in the regulation of apoptosis of fibroblasts and epithelial cells via activation of caspase-3 (Liu et al. 2001) and caspase-independent in macrophages (Gargalovic and Dory 2003). Bleomycin is known to induce apoptosis in different cell types (Hamilton et al. 1995; Kuwano et al. 2000; Wang et al. 2000). Under the present experimental conditions, no signs of apoptosis were detected in the bleomycin-treated R3/1 cells. This is consistent with the fact that, to the best of our knowledge, necrotic but not apoptotic type I pneumocytes were observed in experimental models of bleomycin-induced injury. Detailed studies comparing the caveolin expression, the structural integrity and the expression of apoptosis-related proteins are necessary to determine the role of caveolins in vivo and in vitro.
Taken together, the new AT I pneumocyte-like cell line R3/1, with sustained morphological and physiological properties of AT I cells, is a promising tool for lung cell biologists. Further studies will be required to more precisely document these properties or whether this cell line remains an alveolar epithelial intermediate with properties of both alveolar epithelial cell types.
References
Abraham V, Chou ML, DeBolt KM, Koval M (1999) Phenotypic control of gap junctional communication by cultured alveolar epithelial cells. Am J Physiol 276:L825–L834
Adamson IY, Bowden DH (1975) Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab Invest 32:736–745
Bankston PW, Porter GA, Milici AJ, Palade GE (1991) Differential and specific labeling of epithelial and vascular endothelial cells of the rat lung by Lycopersicon esculentum and Griffonia simplicifolia I lectins. Eur J Cell Biol 54:187–195
Borok Z, Danto SI, Dimen LL, Zhang XL, Lubman RL (1998a) Na(+)-K(+)-ATPase expression in alveolar epithelial cells: upregulation of active ion transport by KGF. Am J Physiol 274:L149–L158
Borok Z, Danto SI, Lubman RL, Cao Y, Williams MC, Crandall ED (1998b) Modulation of t1alpha expression with alveolar epithelial cell phenotype in vitro. Am J Physiol 275:L155–L164
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Campbell L, Hollins AJ, Al-Eid A, Newman GR, von Ruhland C, Gumbleton M (1999) Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung alveolar epithelial primary cultures. Biochem Biophys Res Commun 262:744–751
Cao YX, Williams, M.C. (2002) Hyperoxia increases expression of endogenous alveolar type I cell mRNAs in the E10/C10 murine lung epithelial cell lines. Am J Respir Crit Care Med 165:A627
Christensen PJ, Kim S, Simon RH, Toews GB, Paine R III (1993) Differentiation-related expression of ICAM-1 by rat alveolar epithelial cells. Am J Respir Cell Mol Biol 8:9–15
Clegg R, Harrison DJ, McElroy MC (2003) The phenotype of cultured alveolar epithelial cells. FASEB J 17:562
Cunningham AC, Milne DS, Wilkes J, Dark JH, Tetley TD, Kirby JA (1994) Constitutive expression of MHC and adhesion molecules by alveolar epithelial cells (type II pneumocytes) isolated from human lung and comparison with immunocytochemical findings. J Cell Sci 107:443–449
Dobbs LG, Williams MC, Gonzalez R (1988) Monoclonal antibodies specific to apical surfaces of rat alveolar type I cells bind to surfaces of cultured, but not freshly isolated, type II cells. Biochim Biophys Acta 970:146–156
Dobbs LG, Gonzalez R, Matthay MA, Carter EP, Allen L, Verkman AS (1998) Highly water-permeable type I alveolar epithelial cells confer high water permeability between the airspace and vasculature in rat lung. Proc Natl Acad Sci U S A 95:2991–2996
Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293:2449–2452
Emura M, Riebe M, Ochiai A, Aufderheide M, Germann P, Mohr U (1990) New functional cell-culture approach to pulmonary carcinogenesis and toxicology. J Cancer Res Clin Oncol 116:557–562
Fehrenbach H, Kasper M, Tschernig T, Shearman MS, Schuh D, Muller M (1998) Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol (Noisy-le-grand) 44:1147–1157
Gargalovic P, Dory L (2003) Cellular apoptosis is associated with increased caveolin-1 expression in macrophages. J Lipid Res 44:1622–1632
Gumbleton M, Hollins AJ, Omidi Y, Campbell L, Taylor G (2003) Targeting caveolae for vesicular drug transport. J Control Release 87:139–151
Gutierrez JA, Gonzalez RF, Dobbs LG (1998) Mechanical distension modulates pulmonary alveolar epithelial phenotypic expression in vitro. Am J Physiol 274:L196–L202
Hamilton RF Jr, Li L, Felder TB, Holian A (1995) Bleomycin induces apoptosis in human alveolar macrophages. Am J Physiol 269:L318–L325
Isakson BE, Lubman RL, Seedorf GJ, Boitano S (2001) Modulation of pulmonary alveolar type II cell phenotype and communication by extracellular matrix and KGF. Am J Physiol Cell Physiol 281:C1291–C1299
Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG (2002) Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci U S A 99:1966–1971
Kasper M (2003) Pulmonary pathology of knockouts in mice lacking type I antigens. Ann Anat 185(suppl):144
Kasper M, Singh G (1995) Epithelial lung cell marker: current tools for cell typing. Histol Histopathol 10:155–169
Kasper M, Rudolf T, Hahn R, Peterson I, Muller M (1993a) Immuno- and lectin histochemistry of epithelial subtypes and their changes in a radiation-induced lung fibrosis model of the mini pig. Histochemistry 100:367–377
Kasper M, Rudolf T, Verhofstad AA, Schuh D, Muller M (1993b) Heterogeneity in the immunolocalization of cytokeratin-specific monoclonal antibodies in the rat lung: evaluation of three different alveolar epithelial cell types. Histochemistry 100:65–71
Kasper M, Schuh D, Muller M (1994) Bauhinia purpurea lectin (BPA) binding of rat type I pneumocytes: alveolar epithelial alterations after radiation-induced lung injury. Exp Toxicol Pathol 46:361–367
Kasper M, Koslowski R, Luther T, Schuh D, Muller M, Wenzel KW (1995) Immunohistochemical evidence for loss of ICAM-1 by alveolar epithelial cells in pulmonary fibrosis. Histochem Cell Biol 104:397–405
Kasper M, Traub O, Reimann T, Bjermer L, Grossmann H, Muller M, Wenzel KW (1996) Upregulation of gap junction protein connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis. Histochem Cell Biol 106:419–424
Kasper M, Reimann T, Hempel U, Wenzel KW, Bierhaus A, Schuh D, Dimmer V, Haroske G, Muller M (1998) Loss of caveolin expression in type I pneumocytes as an indicator of subcellular alterations during lung fibrogenesis. Histochem Cell Biol 109:41–48
Kasper M, Seidel D, Knels L, Morishima N, Neisser A, Bramke S, Koslowski R (2004) Early signs of lung fibrosis after in vitro treatment of rat lung slices with CdCl2 and TGF-beta1. Histochem Cell Biol 121:131–140
Katsuoka F, Kawakami Y, Arai T, Imuta H, Fujiwara M, Kanma H, Yamashita K (1997) Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem Biophys Res Commun 238:512–516
Knebel JW, Aufderheide M, Emura M (1994) Comparison of biological effects of different polycyclic aromatic hydrocarbons in lung cells of hamster and rat in vitro. Toxicol Lett 72:65–72
Koslowski R, Dobbs LG, Wenzel KW, Schuh D, Muller M, Kasper M (1998) Loss of immunoreactivity for RTI40, a type I cell-specific protein in the alveolar epithelium of rat lungs with bleomycin-induced fibrosis. Eur Respir J 12:1397–1403
Koval M (2002) Sharing signals: connecting lung epithelial cells with gap junction channels. Am J Physiol Lung Cell Mol Physiol 283:L875–L893
Kuwano K, Hagimoto N, Tanaka T, Kawasaki M, Kunitake R, Miyazaki H, Kaneko Y, Matsuba T, Maeyama T, Hara N (2000) Expression of apoptosis-regulatory genes in epithelial cells in pulmonary fibrosis in mice. J Pathol 190:221–229
Lee YC, Yellowley CE, Li Z, Donahue HJ, Rannels DE (1997) Expression of functional gap junctions in cultured pulmonary alveolar epithelial cells. Am J Physiol 272:L1105–L1114
Liliensiek BWM, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne H-J, Schmidt AM, Shi Du Yan, Martin E, Schleicher E, Stern DM, Hämmerling GJ, Nawroth PP, Arnold B (2004) Receptor for advanced glycation end products (RAGE) regulating sepsis but not the adaptive immune response. J Clin Invest (in press)
Liu J, Lee P, Galbiati F, Kitsis RN, Lisanti MP (2001) Caveolin-1 expression sensitizes fibroblastic and epithelial cells to apoptotic stimulation. Am J Physiol Cell Physiol 280:C823–C835
Liu P, Rudick M, Anderson RG (2002) Multiple functions of caveolin-1. J Biol Chem 277:41295–41298
Matthay MA, Folkesson HG, Clerici C (2002) Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 82:569–600
Nakanishi K, Maruyama M, Shibata T, Morishima N (2001) Identification of a caspase-9 substrate and detection of its cleavage in programmed cell death during mouse development. J Biol Chem 276:41237–41244
Paine R, Ben-Ze’ev A, Farmer SR, Brody JS (1988) The pattern of cytokeratin synthesis is a marker of type 2 cell differentiation in adult and maturing fetal lung alveolar cells. Dev Biol 129:505–515
Paine R III, Christensen P, Toews GB, Simon RH (1994) Regulation of alveolar epithelial cell ICAM-1 expression by cell shape and cell-cell interactions. Am J Physiol 266:L476–L484
Pan T, Mason RJ, Westcott JY, Shannon JM (2001) Rat alveolar type II cells inhibit lung fibroblast proliferation in vitro. Am J Respir Cell Mol Biol 25:353–361
Parton RG (1996) Caveolae and caveolins. Curr Opin Cell Biol 8:542–548
Ramaekers FC, Haag D, Kant A, Moesker O, Jap PH, Vooijs GP (1983) Coexpression of keratin- and vimentin-type intermediate filaments in human metastatic carcinoma cells. Proc Natl Acad Sci U S A 80:2618–2622
Razani B, Lisanti MP (2001) Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest 108:1553–1561
Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138
Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H Jr, Christ GJ, Edelmann W, Lisanti MP (2002) Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol 22:2329–2344
Reid LCM (1979) Cloning. In: Jacoby WBAPIH (ed) Methods in enzymology: cell culture. Academic, San Diego, pp 152–163
Roehlecke C, Kuhnt AK, Fehrenbach H, Werner C, Funk RH, Kasper M (2000) Resistance of L132 lung cell clusters to glyoxal-induced apoptosis. Histochem Cell Biol 114:283–292
Shannon JM, Jennings SD, Nielsen LD (1992) Modulation of alveolar type II cell differentiated function in vitro. Am J Physiol 262:L427–L436
Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, Hata Y (2004) Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 9:165–174
Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD (1996) Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol 135:1085–1096
Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal BD (2000) Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am J Physiol Lung Cell Mol Physiol 279:L143–L151
Williams MC (2003) Alveolar type I cells: molecular phenotype and development. Annu Rev Physiol 65:669–695
Acknowledgements
The authors thank Dr. M.C. Williams (Boston) for helpful discussion prior to the submission of the manuscript. The authors wish to thank P. Peche, B. Rost, K. Pehlke, S. Bramke and D. Streichert for their excellent technical assistance and E. Kuhlisch, Institute of Medical Informatics and Biometrics, for performing the statistical analysis. The technical support of T. Schwalm for preparation of figures is acknowledged with gratitude.
Author information
Authors and Affiliations
Corresponding author
Additional information
Roland Koslowski and Kathrin Barth contributed equally to the study
Rights and permissions
About this article
Cite this article
Koslowski, R., Barth, K., Augstein, A. et al. A new rat type I-like alveolar epithelial cell line R3/1: bleomycin effects on caveolin expression. Histochem Cell Biol 121, 509–519 (2004). https://doi.org/10.1007/s00418-004-0662-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-004-0662-4