
Abstract
Graft versus H\host disease (GVHD) can be a devastating complication following bone marrow transplantation. Acute or chronic systemic GVHD can be lethal, and severe damage of different organs and tissues can occur with both types of GVHD. Ocular involvement, either in an acute or chronic presentation, may range from mild to severe with accompanying vision loss present in 60–90 % of patients. Chronic ocular GVHD, the most common form of GVHD, affects mainly the lacrimal gland, meibomian glands, cornea and conjunctiva, mimicking other immunologically mediated inflammatory diseases of the ocular surface without specific symptoms or signs. However, dry eye disease is the main manifestation of GVHD. The long-term treatment of ocular GVHD continues to be challenging and involves a multidisciplinary approach wherein the ophthalmologist plays a major role. Besides systemic immunosuppression and ocular lubricants, topical steroids and topical cyclosporine are commonly prescribed. Newer therapeutic interventions for moderate and severe ocular GVHD include the use of serum eye drops and scleral contact lenses. In this manuscript, we review the mechanisms, clinical findings, and treatment of ocular GVHD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Allogeneic (genetically different, same species) hematopoietic stem cell transplantation (HSCT) is a curative therapy for a variety of hematological malignancies, autoimmune diseases, inherited disorders of metabolism, histiocytic disorders, and other malignant solid tumors [1–3]. The number of HSCTs continues to increase, with more than 30,000 procedures performed annually across the world [2]. The number of unrelated donors transplants, which are most commonly performed, is expected to double within the next 5 years due to improvements in techniques, including donor leukocyte infusions and isolation of umbilical cord stem cells [2, 3].
Syngeneic transplantation, between identical twins, represents the optimal form of HSCT and, unlike other allogeneic donors, does not carry risk of graft versus host disease (GVHD) [4]. Even with sibling donors, which are more likely than unrelated donors to be HLA-matched, 25–35 % of recipients develop GVHD [2, 4]. Despite HLA matching between a patient and donor (sibling or unrelated), substantial numbers of patients still develop GVHD because of differences in minor histocompatibility antigens that lie outside the matched HLA loci [2, 4].
GVHD remains the most frequent and serious complication limiting broader application of HSCT. Given the increasing number of transplant recipients, larger numbers of GVHD patients are expected in the near future. As many recipients of HSCT become long-term survivors, their quality of life and late complications have become increasingly important. Herein we review the mechanisms, clinical findings, and treatment of ocular GVHD.
Pathophysiology
Sources of hematopoetic stem cells
Peripheral blood stem cells (PBSC) have largely replaced marrow for autologous and most allogeneic transplantations; unfortunately, peripheral-blood stem cells also contain T cells that increase the incidence and prolong the course of GVHD [2]. A process called apheresis or leukapheresis is used to obtain PBSCs for transplantation [2, 3].
Another source of hematopoietic stem cells is the umbilical cord blood [5]. In cases of urgent transplantation or if donors cannot be found, umbilical cord blood can be used. The establishment of a worldwide network for umbilical cord blood cell procurement, typing, and storage has resulted in a large collection and cryopreservation that has facilitated more than 7,000 unrelated transplants. Cord blood as a source of stem cells has several advantages: its transplantation requires less-stringent HLA matching than is required for that of peripheral blood or marrow, and mismatched cord-blood cells are less likely to cause GVHD [6, 7]. A recent study has reported that the incidence of dry eye was significantly higher in the recipients of peripheral blood stem cells than those receiving bone marrow or cord blood [8].
Preparation of donor
For 4 or 5 days before apheresis, the donor may be given granulocyte colony-stimulating factor (GCSF) to increase the number of stem cells in circulation. The stem cells are isolated from circulation based on the cell membrane expression of CD34+, a hematopoietic stem cell marker. These peripheral blood CD34+ stem cells are capable of forming colonies of granulocytes/macrophages, erythrocytes, and other multipotential or immature progenitors. The CD34+ stem cells are frozen until they are infused to the recipient.
Preparation of recipient
Recipient first receives a conditioning regimen consisting of chemotherapy, which is often combined with radiotherapy and T-cell-depleting antibody designed to immunosuppress the host in order to decrease the possibility of graft rejection, and, when used to treat cancer, to reduce the number of malignant cells. This is followed by the infusion of donor cells [2].
Basis for tissue damage
Whereas bone-marrow cells and GCSF-mobilized PBSCs are both enriched with hematopoietic progenitors, they also contain mature T cells that are responsible for graft rejection. [2] Three main strategies to deplete T cells and decrease the incidence of GVHD have been proposed: (1) selection of T cells ex-vivo before transplantation; (2) positive selection of CD34+ stem cells ex vivo by immunomagnetic separation [9]; and (3) antibodies against T cells in vivo [10]. These approaches showed substantial reduction of both acute and chronic GVHD. Unfortunately, reduced frequency of severe GVHD is offset by high rates of graft failure, relapse of malignant disease, infections, and Epstein-Barr virus-associated lymphoproliferative disorders. Moreover, overall survival has not significantly improved as compared with non-T-cell-depleted bone marrow [9, 10].
In contrast with acute GVHD (aGVHD), the pathophysiology of chronic GVHD (cGVHD) is not well understood. Current data suggests that Th1 subpopulation of cells play a critical role in the pathogenesis of aGVHD, whereas Th2 cells may be central in pathogenesis of cGVHD [11].
Classification of GVHD
Graft versus host disease presents in an acute or chronic form. Historically, the acute and chronic forms were arbitrarily defined based on the time of onset since transplant (less than or more than 100 days, respectively) [2]. A clear distinction between acute and chronic forms of GVHD as originally described can no longer be delineated, given the alterations in the recipient’s immunosuppression [2, 12]. Mindful of factors that produce clinical variability among transplant recipients, in 2005, a National Institutes of Health working group sought to standardize the definitions of acute and chronic GVHD (Table 1). Currently, the diagnosis of cGVHD is based on specific signs, degree of organ involvement (mild, moderate, severe), laboratory data, or histopathological confirmation, rather than time of onset since transplant (Table 2) [13].
aGVHD
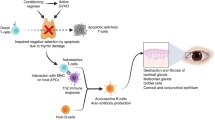
Despite prophylactic measures, the incidence of aGVHD is estimated to be 40–60 % among patients receiving transplants from HLA-identical sibling donors and 75 % in patients receiving HLA matched unrelated donors [14]. The sine qua non of aGVHD is selective epithelial damage of target organs [15] such as skin, liver, gastrointestinal tract within 14–42 days of infusion [12] (Fig. 1).

A 28-year-old male patient developed generalized skin pain and pruritus a month after allogeneic PBSC transplantation. Evaluation of periocular skin showed erythema with some areas of skin desquamation and bullae formation. Evaluation of lid margins showed areas of severe keratinization. A clinical diagnosis of acute graft versus host disease (aGVHD) was done based on the clinical findings
A “hyperacute” form of GVHD may occur within 14 days of infusion, in patients with severe HLA mismatched donor or in those that have received inadequate GVHD prophylaxis [12]. Hyperacute GVHD is manifested by high fever, severe cutaneous component (generalized erythema with desquamation), in addition to hepatitis and intestinal symptoms; this form of GVHD may be rapidly fatal [13].
cGVHD
The chronic form of graft versus host disease (cGVHD) has features resembling autoimmune disorders such as scleroderma, Sjögren syndrome, primary biliary cirrhosis, wasting syndrome, bronchiolitis obliterans, immune cytopenias, and chronic immunodeficiency. Patients who have received stem cells/bone marrow from an HLA (human leukocyte antigen) mismatched related donor or from an HLA matched unrelated donor are at an increased risk of cGVHD [2]. Other factors that increase the risk of cGVHD include older recipients and those who have already experienced aGVHD [2].
Symptoms usually present within 3 years after allogeneic HSCT and are often preceded by a history of aGVHD [2, 4]. Manifestations of cGVHD may be restricted to a single organ or tissue or may be widespread. The diagnostic criteria are listed in Table 3. Dry eye is the most frequent ocular complication usually occurring approximately 6 months post-transplantation [16]. Chronic GVHD can lead to debilitating sequelae such as joint contractures, loss of sight, end-stage lung disease or mortality from profound chronic immune suppression induced life-threatening infections [2, 4].
Ocular surface manifestations
Symptoms and signs
Ocular cGVHD mimics other immunologically mediated inflammatory diseases of the ocular surface and there are no specific symptoms or clinical signs. Ocular manifestations, present in 60–90 % of patients with cGVHD, primarily affect structures of the anterior segment, mainly the lacrimal gland, meibomian glands, and conjunctiva [17] (Fig. 2). Typical symptoms of cGVHD are dry eye, photophobia, foreign body sensation, irritation, burning, epiphora, redness and blurriness (Table 4) [18, 19].

Slit lamp examination in two different patients affected with chronic (c)GVHD following allogeneic bone marrow transplantation. a and b a mild form of corneal staining is noted, fluorescein staining is mostly localized to the interpalpebral area. A normal tear meniscus is noted. c and d a more severe form is shown. More diffuse and confluent staining in the interpalpebral zone is present. Tear meniscus is absent and mucus discharge noted
Target tissues
Lacrimal gland
The lacrimal gland is an important ocular target for the pathogenesis of GVHD [17, 20, 21]. Fibrotic processes often affect the lacrimal gland reducing its secretory capacity or even causing complete stasis with distended ductules and obliteration of ducts lumen, similar to bile duct damage seen in liver cGVHD [20]. Histological studies also showed extensive destruction, tissue atrophy and fibrosis of the tubuloalveolar glands and ducts in the lacrimal gland with an increase in CD34+ stromal fibroblasts accompanied by mild lymphocytic infiltration [21].
Meibomian glands
Besides aqueous tear deficiency, progressive decline of conjunctival goblet cells and the dysfunction of meibomian glands contribute to the overall breakdown of the ocular tear film causing severe keratoconjunctivitis sicca [17, 22]. In vivo confocal microscopy shows destruction of the ductal epithelia due to lymphocyte infiltration, sloughing of epithelial cells, pseudomembrane formation, and subsequent excessive fibrosis around the orifice, ducts, ductules, and acini of the meibomian gland, all these findings together may explain the development of meibomian gland disease [23].
Conjunctiva
Pseudomembranous conjunctivitis can rarely occur in cGVHD and is partly considered as an acute variant of GVHD [22]. Sterile inflammatory conjunctival involvement is a common finding, which can be accompanied by formation of pseudomembranes, loss of lashes and stenosis or closure of the lacrimal punctum. Palpebral and subtarsal conjunctival scarring is seen in a number of patients, sometimes resulting in the formation of cicatricial lagophthalmos [22, 24] (Figs. 3 and 5).

Slit lamp examination in a patient affected with cGVHD following allogeneic bone marrow transplantation. a and b a mild form of lissamine green staining in the cornea and conjunctiva is localized to the interpalpebral area. A descreased tear meniscus is noted. c and d evaluation of the tarsal conjunctiva shows subconjunctival fibrosis and abundant scarring in both eyes
Cornea
Corneal findings include punctate keratopathy, formation of mucus filaments, painful erosions and eventually secondary corneal infections [22, 24]. Figure 2 less frequently, sterile corneal stromal necrosis and perforations may occur [22, 24, 25]. Superior limbic keratoconjunctivitis in the setting of ocular cGVHD is believed to be more common in peripheral stem cell transplantation patients compared with bone marrow recipients [17] (Figs. 4 and 5).

A 65-year-old African American patient status post bone marrow transplant and a diagnosis of cGVHD complains of severe discomfort, dryness and decreased vision. a an absent tear meniscus is noted. b areas of subepithelial haze in the interpalpebral cornea and mucus filaments are noted on theright cornea. c Staining of the superior limbal area suggestive of superior limbal keratitis is noted

A 61-year-old white female patient status post bone marrow transplant and a history of cGVHD complains of severe decreased vision, discomfort and dryness. a and b slit lamp photos show prominent lissamine green staining in the upper limbal region of both eye. c and d everted upper lids show punctate staining of the tarsal conjunctiva and prominent subconjunctival scarring
Diagnostic evaluation
Vital staining using fluorescein, rose bengal or lissamine green needs to be performed. In the clinical setting, the staining profile of lissamine green is nearly identical to that of rose Bengal and they are considered to be interchangeable [26]. Fluorescein staining is usually used for cornea staining and rose Bengal or lissamine green for conjunctival staining. Additional tests for ocular surface examination include: tear break up time, schirmer test, tear evaporimetry, meibomian gland examination, conjunctival and corneal impression cytology, cornea sensitivity and other tests [26]. Schirmer score of less than 5 mm or new onset of dryness with a Schirmer of 6–10 mm is sufficient for a diagnosis of cGVHD if another organ is affected [13].
Wang et al. [27]. evaluated the baseline profiles of ocular surface and tear function alterations using tear evaporimetry, meibomian gland examination, impression cytology, and cornea sensitivity for a detailed ocular surface evaluation in patients with cGVHD, post-HSCT patients without dry eye disease, and healthy controls. They found decreased corneal sensitivity and an increased rate of meibomian gland obstruction in all post-HSCT patients. They also found that cGVHD-related mild and severe dry eyes showed significantly decreased goblet cell density compared with normal controls and post-HSCT without dry eye specimens. Moreover, high grades of squamous metaplasia and increased number of inflammatory cells with a decrease in number of goblet cell were found in the severe versus mild dry eye patients. as a form of posterior blepharitis, is the most common cause of evaporative-type dry eye disease. The authors speculated that the extent of the inflammatory process has a pivotal role in the outcome of cGVHD related dry eye disease with changes in tear evaporation, cornea sensitivity and goblet cell density acting as determinants of the ocular surface status, with MGD as a form of posterior blepharitis being the most common cause of evaporative-type dry eye disease [27].
Tabbara et al. reported the largest series of ocular involvement following allogeneic HSCT [24]. They retrospectively evaluated 620 patients that underwent allogeneic HSCT from bone marrow, peripheral blood and umbilical cord. Although only 34 of the 620 developed cGVHD, 80 of 620 patients (13 % of the total) developed some form of eye involvement. The most common ocular complication was dry eye syndrome with or without cGVHD. Twenty-nine of 34 patients with cGVHD had keratoconjunctivitis sicca. Patients with keratoconjunctivitis sicca who did not have cGVHD may have developed lacrimal deficiency secondary to immunosuppression, total body irradiation, or both. Four patients developed vernal or atopic keratoconjunctivitis after allogeneic HSCT from atopic donors.
Fifteen patients had corneal ulcers including bacterial corneal ulcers (10), herpetic keratitis (1), and sterile epithelial defects (4) [24]. Five patients with cGVHD developed ocular cicatricial pemphigoid-like clinical picture with evidence of conjunctival cicatrization, fornices obliteration, symblepharon, conjunctival keratinization and punctal occlusion [24]. It is important to realize that severe immunosuppression from treating cGVHD can lead to opportunistic ocular infections, such as CMV retinitis or bacterial corneal ulcers [24].
In a prospective study of 101 patients [18], ocular GVHD developed in 54 patients manifesting combination of dry eyes in 42 %, conjunctivitis in 28 %, blepharitis in 26 %, and uveitis in 4 %. Chronic systemic GVHD developed in 45 of 101 patients and was strongly associated with the occurrence of ocular GVHD, specifically with dry eye. The severity of dry eyes and pseudomembranous conjunctivitis increased over time, although most patients were already treated for their ocular and systemic GVHD. It was interesting that the highest prevalence of ocular GVHD was observed in related donors (60 %) compared to matched unrelated donors (45 %). This might reflect the effect of antithymocyte globulin in the conditioning regimen for patients with matched unrelated donors, which causes T-cell depletion and consequently prevents GVHD.
Interestingly, Inamoto et al. reported that Schirmer test correlated poorly with both clinician-reported and patient-reported changes in cGVHD, demonstrating that Schirmer test is a poor diagnostic test of dry eye [28].
Treatment of systemic GVHD
Prevention of GVHD
Prevention of aGVHD by the use of pharmacologic prophylaxis is an integral component to the management of patients undergoing allogeneic HSCT [4, 12, 22]. A regimen based on methotrexate with a calcineurin inhibitor, a cytoplasmic enzyme important for activation of T cells, is standard practice and is recommended in different studies [14]. The most widely used regimen includes a combination of either cyclosporine or tacrolimus with a brief course of methotrexate [12, 22]. For higher risk groups (such as mismatched donors, older patients) more intensive immunosuppression is often required [2, 4, 22].
Treatment of established GVHD
Although many therapeutic options have been used in the management of ocular GVHD, adequate treatment remains a challenge. The management is guided by a multidisciplinary approach, including adjustment of immunosuppression and aggressive supportive care. The treatment approach should include multiple strategies (topical and oral medications, surgery, environmental control, and systemic immunosuppression). Communication with the transplantation team is crucial in the optimal management of GVHD patients. Symptomatic mild cGVHD may often be treated with local therapies alone (e.g., artificial tears, topical steroid,serum drops). However, in patients with cGVHD that involves three or more organs or severe damage in any single organ, systemic immunosuppressive therapy may be considered.
Use of steroids (with or without a calcineurin inhibitor) is the standard of care, but findings of a randomized trial of more than 300 patients with cGVHD observed no difference between cyclosporine plus prednisone versus prednisone alone [29].
Treatment of ocular GVHD
Patients with ocular symptoms need close ocular supportive care focused on improving ocular surface moisture and decreasing ocular surface inflammation.
Topical lubricants
The traditional treatment for dry eye symptoms consists of topical lubricants [26]. Many brands are available and there is a large individual variability in symptoms relief efficiency and tolerance. No data is available on the efficacy of specific artificial tears medications in ocular GVHD. Although artificial tears and lubricants are needed to lubricate the ocular surface, ocular cGVHD is a complex problem of the ocular surface with multiple dysfunctional tear components.
Topical corticosteroids
Corticosteroids remain essential for controlling active chronic graft-versus-host disease. Systemic steroids represent the mainstay in the treatment of acute (exacerbations of) cGVHD but not enough data are available on the efficacy of topical steroids in ocular GVHD. In a small study of seven patients with conjunctival GVHD, resolution or improvement of the conjunctival signs was achieved using topical corticosteroids but the signs of keratoconjunctivitis sicca remained unchanged [30]. Patients receiving topical corticosteroids should be monitored for adverse effects. In presence of corneal epithelial defects, stromal thinning, or infiltrates, topical corticosteroids are contraindicated.
Topical cyclosporine A
Cycloporine A is a cyclic polypeptide produced by the fungus Tolypocladium inflatum Gams. Cyclosporine 0.05 % ophthalmic emulsion (Restasis; Allergan, Inc., Irvine, CA) has been FDA-approved for treatment of dry eye disease since 2003 [31]. Cylosporine A emulsion has shown to decrease the number of activated T cells in the ocular surface, increases the goblet cell density of the conjunctiva, decreases epithelial cell apoptosis and reduce proinflammatory citokines [32]. Treatment with CsA, 0.05 % or 0.1 %, gave significantly greater improvement than vehicle in two objective signs of dry eye disease (corneal staining and categorized Schirmer values) in patients with moderate to severe dry eye not related to GVHD [33]. In a small study ofeight cGVHD patients treated with cyclosporine 0.05 %, ophthalmic suspension twice a day for at least 3 months. Researchers noted mean Schirmer scores increases, tear breakup time improvement and subjective symptoms improvement [34]. In another study of only 16 patients (32 eyes) with GVHD, dry eye symptoms improved in 62.5 % of patients and corneal fluorescein staining improved in all eyes after a mean follow-up of 90 days [35]. Baptista Malta reported a retrospective study with 105 patients, of whom 43 patients developed cGVHD. All patients were initially started on topical cyclosporine before the HSCT. They conclude that cyclosporine is helpful in decreasing the incidence and severity of dry eyes in patients who are under topical cyclosporine before the HSCT [36].
Although beneficial effect of topical cyclosporine on ocular GVHD has been documented in several studies comprised of small number of cases, there is at present, no large randomized study that clearly suggests its usefulness as a consequence of the rare incidence of ocular GVHD.
Tacrolimus
FK506 (tacrolimus) is a macrolide antibiotic extracted from the soil fungus Streptomyces tsukubaensis and its mechanisms of action and pharmacokinetics are similar to CsA [37], although the immunosuppressive potency of tacrolimus in vitro is 50–200 times greater. Although the beneficial effect of systemic tacrolimus on ocular GVHD has been observed [14, 37], topical administration might form a better treatment option because of the adverse effects associated with its long-term systemic administration. Except for two case reports in which ocular GVHD was successfully treated, not enough data is available on the use of topical tacrolimus [38, 39].
Autologous serum eye drops
Fox et al. in described serum drops as a tear substitute free of preservatives in 1984 [40]. Autologous serum contains vitamin A, epidermal growth factor, fibronectin, transforming growth factor beta, which all are need for a healthy ocular surface epithelium [41]. The efficacy and safety of autologous serum drops were investigated in a small study of 14 patients with ocular GVHD and severe cGVHD not responsive to conventional artificial tears therapy. After 4 weeks of treatment, significant improvement was observed in dryness symptoms and fluorescein scores, and also in rose bengal staining and tear break-up time [42]. The improvement was noted in all patients at the 4-week follow-up [42]. The risk of contamination and subsequent infection forms a possible complication of autologous serum drops.
Contact scleral lenses
In patients with cGVHD affecting the ocular surface, two different types of lenses can be used, the bandage soft lens and the scleral rigid lens. The fluid-ventilated, gas-permeable scleral lens has been effective in mitigating symptoms and resurfacing corneal erosions in the treatment of moderate and severe ocular surface disorders of multiple etiologies. The fluid-filled reservoir shields the cornea from blink trauma, noxious environmental stimuli, and inflammatory mediators in the tears. The body-temperature saline reservoir also prevents corneal cooling and nerve firing that occurs during the inter-blink intervals [43].
One of the scleral lens used (Boston Scleral Lens Prosthetic Device) was approved for the management of corneal disorders by the Food and Drug Administration in 1994. Takahide published a retrospective review on nine patients fitted for refractory ocular surface disease secondary to cGVHD [44]. Contact lens fitting was prompted by debilitating ocular discomfort, visual impairment, or keratopathy. Some of the patients evaluated were using artificial tears, cyclosporine eye drops, punctal plugs, autologous serum tears, and moisture chamber eye wear. All patients reported improvement of ocular symptoms, reduced use of topical lubricants after fitting and improvement in the ocular surface disease index [44].
The same group published results of 33 consecutive patients with severe dry eye from cGVHD, unresponsive to conventional therapy. Ninety-four percent of patients reported improvement in photophobia in the worse eye. Ninety seven percent of patients reported improvement in life quality with no complications noted during the follow-up period [45].
Schornack reported the successful use of the Jupiter scleral contact lens (Medlens Innovations, Front Royal, VA or Essilor Contact Lens, Inc., Dallas, TX) in the management of ten eyes of five patients with cGVHD. All patients had improvement in symptoms and some improved visual acuity. Jupiter lenses are commercially available in the US, and may therefore be more accessible and affordable to patients who could potentially benefit from this treatment [46].
Although scleral lenses are maybe the most important tool in the armamentarium, their use is not widespread. Published data suggests that scleral rigid gas permeable lenses are an important therapeutic option for patients with recalcitrant ocular surface compromise and debilitating symptoms. In our experience, high cost, inadequate fitting, poor tolerance by some patients, and discomfort with blinking in presence of severe meibomian gland disease and keratinization are some of the drawbacks. To our knowledge, no comparative prospective study has evaluated these two different types of available scleral lenses.
Prognosis of GVHD
Systemic GVHD
Cahn and colleagues recently reported on a multicenter study comparing the IBMTR (International Bone Marrow Transplant Registry) and the Glucksberg scales. In general, patients with grade C (IBMTR) [47] or grade III (Glucksberg) [48] acute GVHD have about a 30 % probability of long-term survival. Those with grade D/grade IV acute GVHD, have under 5 % long-term survival. Patients without GVHD or with grade A-B/grade I-II acute GVHD have above 80 % probability of long-term survival [49].
Ocular GVHD
Few reports have studied the long-term prognosis in patients affected with ocular cGVHD. Sales et al., in a case series of 49 patients, report that in the long-term, many patients with cGVHD may experience improved dry eye symptoms as a result of effective treatment. Although only nine patients completed this 3-year prospective case series, stable visual acuity, tear production, and lissamine green staining and a statistically insignificant improvement in dry eye symptoms was reported [50].
Another retrospective cohort study reported that of 56 patients with cGVHD, 39 % developed symptoms of photophobia, irritation and foreign body sensation [51]. Over time, patients required fewer topical medications to control their symptoms; only 5 % of patients required more than two medications for dry eye disease management at the end of follow-up [51].
In contrast, Ogawa et al.'s series of 53 patients suggested that the symptoms of dry eye, including ocular fatigue, foreign body sensation, pain, blurring, photophobia, and epiphora, were almost universally worse among 22 participants at 30 months after HSCT in comparison to before HSCT [16].
Conclusions
As the number of stem-cell transplants performed worldwide increases, it is important that ophthalmologists taking care of these patients are able to understand the basics of HSCT, as well as to recognize and treat the ophthalmic manifestations and complications of GVHD. Early recognition and treatment of ocular GVHD may be important to halt the progress of this sight threatening disease. A multidisciplinary approach is needed. An individualized assessment of each patient and the combined use of the different medications and devices available is needed to help these patients deal with a chronic, and in some cases, very difficult to treat debilitating disease.
Methods of literature search
A PubMed literature search was conducted for manuscripts related to GVHD and ocular GVHD. Key words included GVHD, acute GVHD, chronic GVHD, ocular GVHD, scleral contact lenses, serum drops and cyclosporine.
References
Copelan EA (2006) Hematopoietic stem-cell transplantation. N Engl J Med 354:1813–1826
Ferrara JL, Levine JE, Reddy P, Holler E (2009) Graft-versus-host disease. Lancet 373:1550–1561
de la Morena MT, Gatti RA (2010) A history of bone marrow transplantation. Immunol Allergy Clin North Am 30:1–15
Choi SW, Levine JE, Ferrara JL (2010) Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am 30:75–101
Broxmeyer HE, Douglas GW, Hangoc G, Cooper S, Bard J, English D, Arny M, Thomas L, Boyse EA (1989) Human umbilical cord blood as a potential source of transplantable hematopoietic stem/progenitor cells. Proc Natl Acad Sci U S A 86:3828–3832
Barker JN, Krepski TP, DeFor TE, Davies SM, Wagner JE, Weisdorf DJ (2002) Searching for unrelated donor hematopoietic stem cells: availability and speed of umbilical cord blood versus bone marrow. Biol Blood Marrow Transplant 8:257–260
Wagner JE, Barker JN, DeFor TE, Baker KS, Blazar BR, Eide C, Goldman A, Kersey J, Krivit W, MacMillan ML, Orchard PJ, Peters C, Weisdorf DJ, Ramsay NK, Davies SM (2002) Transplantation of unrelated donor umbilical cord blood in 102 patients with malignant and nonmalignant diseases: influence of CD34 cell dose and HLA disparity on treatment-related mortality and survival. Blood 100:1611–1618
Uchino M, Ogawa Y, Uchino Y, Mori T, Okamoto S, Tsubota K (2012) Comparison of stem cell sources in the severity of dry eye after allogeneic haematopoietic stem cell transplantation. Br J Ophthalmol 96:34–37
Platzbecker U, Ehninger G, Bornhauser M (2004) Allogeneic transplantation of CD34+ selected hematopoietic cells—clinical problems and current challenges. Leuk Lymphoma 45:447–453
Wagner JE, Thompson JS, Carter SL, Kernan NA (2005) Effect of graft-versus-host disease prophylaxis on 3-year disease-free survival in recipients of unrelated donor bone marrow (T-cell Depletion Trial): a multi-centre, randomised phase II-III trial. Lancet 366:733–741
Coghill JM, Sarantopoulos S, Moran TP, Murphy WJ, Blazar BR, Serody JS (2011) Effector CD4+ T cells, the cytokines they generate, and GVHD: something old and something new. Blood 117:3268–3276
Socie G, Blazar BR (2009) Acute graft-versus-host disease: from the bench to the bedside. Blood 114:4327–4336
Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, Martin P, Chien J, Przepiorka D, Couriel D, Cowen EW, Dinndorf P, Farrell A, Hartzman R, Henslee-Downey J, Jacobsohn D, McDonald G, Mittleman B, Rizzo JD, Robinson M, Schubert M, Schultz K, Shulman H, Turner M, Vogelsang G, Flowers ME (2005) Biol Blood Marrow Transplant 11:945–956
Ram R, Gafter-Gvili A, Yeshurun M, Paul M, Raanani P, Shpilberg O (2009) Prophylaxis regimens for GVHD: systematic review and meta-analysis. Bone Marrow Transplant 43:643–653
Sale GE (1996) Does graft-versus-host disease attack epithelial stem cells? Mol Med Today 2:114–119
Ogawa Y, Okamoto S, Wakui M, Watanabe R, Yamada M, Yoshino M, Ono M, Yang HY, Mashima Y, Oguchi Y, Ikeda Y, Tsubota K (1999) Dry eye after haematopoietic stem cell transplantation. Br J Ophthalmol 83:1125–1130
Kim SK (2006) Update on ocular graft versus host disease. Curr Opin Ophthalmol 17:344–348
Westeneng AC, Hettinga Y, Lokhorst H, Verdonck L, van Dorp S, Rothova A (2010) Ocular graft-versus-host disease after allogeneic stem cell transplantation. Cornea 29:758–763
Dietrich-Ntoukas T, Cursiefen C, Westekemper H, Eberwein P, Reinhard T, Bertz H, Nepp J, Lawitschka A, Heiligenhaus A, Seitz B, Messmer EM, Meyer-ter-Vehn T, Basara N, Greinix H, Datiles MB, Lee SJ, Pavletic SZ, Wolff D (2012) Diagnosis and treatment of ocular chronic graft-versus-host disease: report from the German-Austrian-Swiss Consensus Conference on Clinical Practice in chronic GVHD. Cornea 31:299–310
West RH, Szer J, Pedersen JS (1991) Ocular surface and lacrimal disturbances in chronic graft-versus-host disease: the role of conjunctival biopsy. Aust N Z J Ophthalmol 19:187–191
Ogawa Y, Kuwana M (2003) Dry eye as a major complication associated with chronic graft-versus-host disease after hematopoietic stem cell transplantation. Cornea 22:S19–S27
Riemens A, Te BL, Imhof S, Kuball J, Rothova A (2010) Current insights into ocular graft-versus-host disease. Curr Opin Ophthalmol 21:485–494
Ban Y, Ogawa Y, Ibrahim OM, Tatematsu Y, Kamoi M, Uchino M, Yaguchi S, Dogru M, Tsubota K (2011) Morphologic evaluation of meibomian glands in chronic graft-versus-host disease using in vivo laser confocal microscopy. Mol Vis 17:2533–2543
Tabbara KF, Al-Ghamdi A, Al-Mohareb F, Ayas M, Chaudhri N, Al-Sharif F, Al-Zahrani H, Mohammed SY, Nassar A, Aljurf M (2009) Ocular findings after allogeneic hematopoietic stem cell transplantation. Ophthalmology 116:1624–1629
Koch KR, Joussen AM, Huber KK (2011) Ocular involvement in chronic graft-versus-host disease: therapeutic approaches to complicated courses. Cornea 30:107–113
Savini G, Prabhawasat P, Kojima T, Espana E (2008) The challenge of dry eye diagnosis. Clin Ophthalmol 2:31–55
Wang Y, Ogawa Y, Dogru M, Tatematsu Y, Uchino M, Kamoi M, Okada N, Okamoto S, Tsubota K (2010) Baseline profiles of ocular surface and tear dynamics after allogeneic hematopoietic stem cell transplantation in patients with or without chronic GVHD-related dry eye. Bone Marrow Transplant 45:1077–1083
Inamoto Y, Chai X, Kurland BF, Cutler C, Flowers ME, Palmer JM, Carpenter PA, Heffernan MJ, Jacobsohn D, Jagasia MH, Pidala J, Khera N, Vogelsang GB, Weisdorf D, Martin PJ, Pavletic SZ, Lee SJ, Chronic GVHD Consortium (2012) Validation of measurement scales in ocular graft-versus-host disease. Ophthalmology 119:487–493
Koc S, Leisenring W, Flowers ME, Anasetti C, Deeg HJ, Nash RA, Sanders JE, Witherspoon RP, Storb R, Appelbaum FR, Martin PJ (2002) Therapy for chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus prednisone versus prednisone alone. Blood 100:48–51
Robinson MR, Lee SS, Rubin BI (2004) Topical corticosteroid therapy for cicatricial conjunctivitis associated with chronic graft-versus-host disease. Bone Marrow Transplant 33:1031–1035
Stevenson D, Tauber J, Reis BL (2000) Efficacy and safety of cyclosporin A ophthalmic emulsion in the treatment of moderate-to-severe dry eye disease: a dose-ranging, randomized trial. The Cyclosporin A Phase 2 Study Group. Ophthalmology 107:967–974
Donnenfeld E, Pflugfelder SC (2009) Topical ophthalmic cyclosporine: pharmacology and clinical uses. Surv Ophthalmol 54:321–338
Sall K, Stevenson OD, Mundorf TK, Reis BL (2000) Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology 107:631–639
Rao SN, Rao RD (2006) Efficacy of topical cyclosporine 0.05 % in the treatment of dry eye associated with graft versus host disease. Cornea 25:674–678
Lelli GJ Jr, Musch DC, Gupta A (2006) Ophthalmic cyclosporine use in ocular GVHD. Cornea 25:635–638
Malta JB, Soong HK, Shtein RM, Musch DC, Rhoades W, Sugar A, Mian SI (2010) Treatment of ocular graft-versus-host disease with topical cyclosporine 0.05 %. Cornea 29:1392–1396
Simpson D (2000) Drug therapy for acute graft-versus-host disease prophylaxis. J Hematother Stem Cell Res 9:317–325
Ogawa Y, Okamoto S, Kuwana M, Tsubota K (2001) Successful treatment of dry eye in two patients with chronic graft-versus-host disease with systemic administration of FK506 and corticosteroids. Cornea 20:430–434
Tam PM, Young AL, Cheng LL, Lam PT (2010) Topical 0.03 % tacrolimus ointment in the management of ocular surface inflammation in chronic GVHD. Bone Marrow Transplant 45:957–958
Fox RI, Chan R, Michelson JB, Belmont JB, Michelson PE (1984) Beneficial effect of artificial tears made with autologous serum in patients with keratoconjunctivitis sicca. Arthritis Rheum 27:459–461
Tsubota K, Goto E, Fujita H (1999) Treatment of dry eye by autologous serum application in Sjogren’s syndrome. Br J Ophthalmol 83:390–395
Ogawa Y, Okamoto S, Mori T, Tsubota K (2003) Autologous serum eye drops for the treatment of severe dry eye in patients with chronic graft-versus-host disease. Bone Marrow Transplant 31:579–583
Pflugfelder SC (2011) Tear dysfunction and the cornea: LXVIII Edward Jackson Memorial Lecture. Am J Ophthalmol 152:900–909
Takahide K, Parker PM, Wu M, Hwang WY, Carpenter PA, Moravec C, Stehr B, Martin PJ, Rosenthal P, Forman SJ, Flowers ME (2007) Use of fluid-ventilated, gas-permeable scleral lens for management of severe keratoconjunctivitis sicca secondary to chronic graft-versus-host disease. Biol Blood Marrow Transplant 13:1016–1021
Jacobs DS, Rosenthal P (2007) Boston scleral lens prosthetic device for treatment of severe dry eye in chronic graft-versus-host disease. Cornea 26:1195–1199
Schornack MM, Baratz KH, Patel SV, Maguire LJ (2008) Jupiter scleral lenses in the management of chronic graft versus host disease. Eye Contact Lens 34:302–305
Rowlings PA, Agahan AL, Perez-Simon JA, López A, Caballero D, Hernández E, Barrientos-Gutierrez T, Calonge M (2011) IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 97:855–864
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, Lerner KG, Thomas ED (1974) Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 18:295–304
Cahn JY, Klein JP, Lee SJ, Milpied N, Blaise D, Antin JH, Leblond V, Ifrah N, Jouet JP, Loberiza F, Ringden O, Barrett AJ, Horowitz MM, Socie G (2005) Prospective evaluation of 2 acute graft-versus-host (GVHD) grading systems: a joint Societe Francaise de Greffe de Moelle et Therapie Cellulaire (SFGM-TC), Dana Farber Cancer Institute (DFCI), and International Bone Marrow Transplant Registry (IBMTR) prospective study. Blood 106:1495–1500
Sales CS, Johnston LJ, Ta CN (2011) Long-term clinical course of dry eye in patients with chronic graft-versus-host disease referred for eye examination. Cornea 30:143–149
Parra-Colin P, Agahan AL, Perez-Simon JA, Lopez A, Caballero D, Hernandez E, Barrientos-Gutierrez T, Calonge M (2011) Dry eye disease in chronic graft-versus-host disease: results from a Spanish retrospective cohort study. Transplant Proc 43:1934–1938
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Espana, E.M., Shah, S., Santhiago, M.R. et al. Graft versus host disease: clinical evaluation, diagnosis and management. Graefes Arch Clin Exp Ophthalmol 251, 1257–1266 (2013). https://doi.org/10.1007/s00417-013-2301-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-013-2301-z