
Abstract
Background
Since Wiethe first described the clinical presentation of two optic disc depressions in a 62-year-old woman in 1882, there have been many studies addressing what later become known as the “optic disc pit.” The main complication of this condition, termed optic disc pit maculopathy, is associated with visual deterioration. Treatment of optic disc pit maculopathy remains challenging.
Methods
Here we review the body of literature that documents the clinical findings, pathophysiology, histology, main complications, treatment options, special features and presentations, and differential diagnosis of optic disc pit.
Results
The source of the intraretinal fluid in optic disc pit maculopathy remains controversial. Four possible sources of this fluid have been proposed: fluid from the vitreous cavity; cerebrospinal fluid originating from the subarachnoid space; fluid from leaky blood vessels at the base of the pit; and fluid from the orbital space surrounding the dura.
Conclusions
Optic disc pits are a very rare clinical entity, affecting approximately one in 11,000 people. Patients with congenital optic disc pit sometimes remain asymptomatic, but 25% to 75% present with visual deterioration in their 30s or 40s after developing macular schisis and detachment. The most widely accepted treatment for such patients is a surgical approach involving pars plana vitrectomy with or without internal limiting membrane peeling, with or without endolaser photocoagulation and C3F8 endotamponade.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since 1882, when Wiethe first reported and described the clinical presentation of two optic disc depressions in a 62-year-old woman [1], there have been many reports concerning what later become known as the “optic disc pit” (ODP). ODPs can be congenital or acquired. A congenital ODP is usually a standalone, oval, gray-white depression that is most commonly located in the inferotemporal segment of the optic disc and that is frequently associated with macular involvement, either as a serous retinal detachment or as a cystoid retinal edema [2–7].
ODPs are considered extremely rare, with an incidence of about one in 11,000 people for congenital ODPs [6]. Although ODPs are bilateral in 15% of the affected individuals, they typically present unilaterally in patients with larger optic discs [9, 10]. In the majority of cases, there is one pit per disc, although two or three pits occasionally occur [11]. ODPs occur equally in women and men; although they usually appear sporadically, it has been suggested that unilateral pits may be inherited in an autosomal dominant fashion [12].
ODP clinical findings
Clinical symptoms
ODPs are usually asymptomatic and diagnosed as incidental findings during fundus examination. Visual deterioration occurs only when congenital ODPs are complicated by macular lesions such as macular edema ,schisis-related macular detachment, or changes in macular pigment (see ODP maculopathy (ODP-M) section). On some occasions, optic disc pits may result in an arcuate scotoma or an enlarged blind spot [9].
Clinical appearance
An ODP appears as a localized grey (60%), white/yellow (30%), or black (10%) round or oval depression in the optic disc (Figs. 1 and 2). This color variation is attributed to the presence of glial tissue [9, 13]. ODPs are most commonly located at the inferotemporal segment of the optic disc, with 20% occurring centrally and 10% located in other regions of the optic disc. The size of the pit varies from 0.1 to 0.7 disc diameters, and the average depth measures ∼0.3 diopters with a maximum value of 0.5 diopters [14].

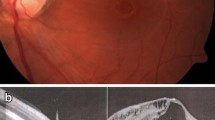
Color fundus photography of the left eye of a patient with optic disc pit maculopathy. Please note that the pit is located temporally

Color fundus photography of an eye of with optic disc pit maculopathy
ODPs may be associated with other abnormalities, such as nerve head coloboma and nerve head enlargement. Atrophy of the pigment epithelium along the temporal edge of the disc may also occur as a symptom related to the chronicity of the subretinal fluid [15]. The latter occurs when the pit is located temporally. Centrally located pits are associated with temporal disc pallor [16]. In up to 60% of patients with ODP, one or two cilioretinal arteries can be seen emerging from the pit base [9]. Unlike optic disc coloboma, an ODP does not affect the disc margin, and the physiological optic cup remains distinct.
Pathophysiology of ODP
Although ODP represents a “mature” clinical entity, its pathophysiological background remains unclear and somewhat controversial. For years, ODPs have been considered a more benign variant of optic disc coloboma [6, 11, 15, 17, 18]. Specifically, it has been suggested that ODPs occur as a result of incomplete closure of the optic fissure during development, due to anomalous differentiation of the primitive epithelial papilla that allows abnormal microcommunication between the subarachnoid space surrounding the nerve and the pit [14]. In addition, a lack or loss of retinal nerve fibers has been observed at the site of the ODP [19].
Nonetheless, in their review of 75 cases, Brown et al. [11] concluded that the lack of inferonasal pits “casts doubts as to whether the pits are truly colobomas resulting from incomplete closure of the embryonic fissure.” Moreover, Brodsky et al. [9] argue against this widely accepted hypothesis of the common pathophysiology between pits and colobomas as follows. First, the usually unilateral and sporadic nature of ODPs, as well as the lack of systemic association, contrast with colobomas, which are as often unilateral as bilateral, with a strong systemic association and a commonly autosomal dominant inheritance pattern. Second, ODPS are rarely found with iris or retinochoroidal colobomas. Third, ODPs are not usually located near the optic fissure.
Histology of ODP
An ODP is a herniation of a dysplastic retina into a collagen-rich excavation that often extends into the subarachnoid space through a defect in the lamina cribrosa [20]. Moreover, strands of condensed vitreous terminate at the margin of the pit, and nerve axons enter the optic disc and cross over the pit [20, 21].
ODP maculopathy (ODP-M)
Although uncomplicated congenital ODPs do not cause visual deterioration, Halbertsma [22] described the association of pits with macular lesions and decreased visual acuity as early as 1927. Moreover, some years later, Kranenburg [6] reported that approximately 65% of his patients demonstrated significant macular changes resulting in non-reversible central visual field defects and reduced central visual acuity. These macular changes consisted of serous detachment, cystic degeneration, and degenerative pigment changes (Figs. 1 and 2). Of course, it is possible that all of these are remnants of longstanding ODP-M [6, 23, 24].
Petersen [25] was the first to report the relationship between ODPs and serous retinal detachment, which is usually confined to the macular area and rarely exceeds 1.5 mm in height. Serous macular elevations develop in approximately 25% to 75% of eyes with pits [6, 11, 24–26]. The great majority of ODP-Ms occur in eyes with temporally located pits, and they usually become symptomatic in the 3rd or 4th decade of life [9]. However, there have been some reports of serous detachments in children [24, 27–30].
Although spontaneous resolution of ODP-M with recovery of excellent visual acuity has been reported [23, 24, 30] and is estimated to occur in 25% of such cases, the overall prognosis remains poor, particularly when there is longstanding serous detachment.
Pathophysiology of ODP-M
There are two aspects of the pathophysiological background of ODP-M that remain unclear, namely the origin of the fluid and the mechanism by which the fluid causes detachment of the sensory retina.
Origin of the fluid
The source of the intraretinal fluid in ODP-M remains controversial. Four possible sources/mechanisms have been proposed.
-
1.
The fluid is from the vitreous cavity. This hypothesis is supported by the work of Brown et al. [31] who conducted experiments on Collie dogs. Specifically, India ink injected into the vitreous cavity was later observed in the subretinal space. However, direct communication between the liquefied vitreous and the subretinal space has not been demonstrated conclusively using optical coherence tomography (OCT). In addition, the Collie dog study failed to identify any glycosaminoglycans, which are an important vitreous component, in the subretinal space [31].
-
2.
The fluid is cerebrospinal fluid (CSF) originating from the subarachnoid space. In 1996, an OCT-based study by Krivoy et al. [13] demonstrated that there was direct communication between the schisis cavity/subretinal space and the subarachnoid space. The study found no vitreous cavity–subretinal space communication. More recently, Kuhn et al. [32] reported intracranial migration of silicon oil from an eye with an ODP that had been treated previously with vitrectomy. This is strong supporting evidence that CSF can move towards the submacular space via the subarachnoid space.
-
3.
The fluid source is leaky blood vessels at the base of the pit [15, 33–35]. Although flourescein angiography shows early hypofluorescent staining followed in most cases by late hyperfluorescent staining, pits do not demonstrate leakage, and there is no extension of fluorescein into the subretinal space.
-
4.
The fluid originates from the orbital space surrounding the dura [10, 26, 36].
Mechanism of ODP-M
Until 1988, most investigators believed that all ODP-associated macular elevations represented serous detachments. In 1988, Lincoff et al. [37] carried out a stereoscopic imaging study of 15 eyes in conjunction with kinetic perimetry, which documented the progression of events in the development of ODP-M. They proposed the following sequence of events:
-
1.
Initially, fluid from the pit causes an elevation of the nerve fiber layer leading to a schisis-like inner layer separation. This produces a mild centrocecal scotoma.
-
2.
An outer layer macular hole develops beneath the inner layer. This causes a dense central scotoma.
-
3.
Subsequently, an outer layer retinal detachment occurs around the macular hole, possibly due to the movement of fluid towards the outer segment. Clinically, this presents as a retinal pigment epithelium detachment.
-
4.
Finally, the outer layer detachment increases. At this point, the detachment is indistinguishable from a serous detachment.
Since ODPs are usually congenital, and ODP-M represents a late phenomenon, it is reasonable to assume that changes later in life promote fluid movement from the pit towards the retina. In order to understand the initial events in the formation of ODP-Ms, it is important to determine the microanatomy of the pits and the overlying tissue. Brockhurst [38] noted the presence of a small hole overlying the pit in two of his cases. He suggested that this hole might serve as a passage for fluid. Bonnet [26] and Postel et al. [39] reported similar observations. In addition, Theodossiadis [40, 41] noted the disappearance of the membrane overlying the ODPs in patients that were observed for more than 6 years.
Based on this evidence, Postel et al. [39] proposed that there may be a pocket of liquefied vitreous over the abnormally developed optic nerve, and that as tractional forces develop over the years, a tear or hole might form in the overlying diaphanous tissue that allows liquefied vitreous to spread under and/or into the retina. Extending this hypothesis, Postel et al. supported this rhegmatogenous element in the triggering mechanism for ODP-M formation. Moreover, this group suggested that the delayed onset of macular detachment, its occasional spontaneous resolution, and the apparent response to vitrectomy comprise supporting evidence for this hypothesis. In addition, Doyle et al. proposed that the membrane spanning the optic disc cup may act as a protective factor against the development of maculopathy [42].
Irrespective of the fluid origin or the triggering event for ODP-M, a very recent study of 16 patients with ODP using high-resolution optical coherence tomography [43] showed that fluid can move directly from the optic pit to the subinternal limiting membrane space, ganglion cell layer, inner and outer nuclear layer, or subretinal space. The outer nuclear layer was the one most commonly affected. Interestingly, outer layer holes were not a common finding.
Diagnosis of ODP-M
ODP-M generally presents as visual acuity worse than 20/70 in the affected eye. Of the eyes with ODP-M, 80% will end up with visual acuity of 20/200 or worse and a generally poor prognosis [14, 36, 37]. The diagnosis is based mainly on fundus examination and OCT imaging. Fundus examination demonstrates an ODP that is usually located temporally (with or without an overlying diaphanous membrane that may have small holes) with a coexistent macular elevation (Figs. 1 and 2). In addition, as suggested previously, it may be possible to observe cystic retinal degeneration or a macular hole located in the outer retinal layer.
OCT is the most helpful diagnostic tool for ODP-M, since it reveals the pattern unique to ODP-M (Figs. 3 and 4). Specifically, OCT reveals the dual morphology of the detachment with a schisis cavity (in the majority of patients) and with a coexisting outer layer detachment from the retinal pigment epithelium (Figs. 5 and 6) [13, 44–46]. OCT may also reveal retinal edema in the inner layers, with cystic degeneration overlying a more central detachment [14], although the most prominent edema is located in the outer layers. Also, OCT examination of the ODP reveals a hypo-reflective area that corresponds to the edge of the pit where there is communication with the schisis cavity [14].

Time-domain optical coherence tomography of the optic nerve in an eye with an optic disc pit maculopathy

Time-domain optical coherence tomography of the macular area in an eye with optic disc pit maculopathy. Please note the intraretinal and subretinal fluid

Color fundus photography of the left eye of a patient with an optic disc pit. Please note that the intraretinal and subretinal fluid is barely visible

Spectral domain optical coherence tomography of the macular area of a patient with intraretinal and subretinal fluid in the macular area
A recent study [47] showed that OCT can detect vitreous abnormalities in patients with ODP-M. Specifically, vitreomacular traction, vitreous strands over the optic disc, and complete or partial posterior vitreous detachment were detected with OCT.
As noted previously, a recent high-resolution optical coherence tomography study [43] showed that the outer nuclear layer was the one most commonly affected. Interestingly, outer layer holes were not a common finding.
Treatment of ODP-M
There is no single ODP-M treatment that is universally accepted, since none have been shown to be clearly better than the others. This is partly due to the rarity of this clinical entity, and partly due to the challenging nature of the retinal detachment.
Conservative management used to be the initial recommendation, as 25% of ODP-Ms resolve spontaneously. However, the poor final outcome of the natural course of these spontaneously-resolved detachments [23, 24, 30] has made a more aggressive surgical approach the preferred treatment modality for most of the retinal specialists. The treatment proposed initially for ODP-M, which included bed rest with bilateral patching and oral corticosteroids, did not prove beneficial [14]. Gass [15] used Xenon photocoagulation along the temporal disc margin in two patients to create a chorioretinal adhesion at the disc edge that would minimize fluid movement from the pit to the subretinal space. The results were not very promising, since the retinal detachment became worse in the first patient and remained stable in the other. In 1972, Mustonen [48] reported the use of Argon laser photocoagulation to treat three patients with ODP-M. The retina became re-attached after 1 or 2 years in all three cases, but visual acuity improved in only one patient. In 1975, Brockhurst [38] reported the results of Argon laser photocoagulation along the disc margin in the area of the retinal detachment in six patients. In five cases, re-attachment of the retina occurred. Overall, laser treatment remains questionable given how long it takes for fluid reabsorption [14].
Other proposed therapeutic modalities include intravitreal gas injection alone [49] and laser therapy in combination with gas injection [50]. Theodossiadis [41] proposed a novel technique that used macular buckling surgery as primary treatment for serous macular detachment. In this technique, a scleral 7.5 × 5.5-mm sponge is placed on the area corresponding to the macula, with no application of additional gas, laser, or cryotherapy. The results look very promising.
Ten years ago, most of the retinal specialists performed laser photocoagulation if there was no resolution of the detachment within 3 months [14]. If the fluid did not resolve within 3 months of treatment, the treatment of choice was a pars plana vitrectomy with posterior vitreous detachment, or gas exchange with or without laser photocoagulation [14, 51]. In recent years, the release of traction [either vitreous or internal limiting membrane- (ILM)-induced] has been considered a very important factor in the management of patients with ODP-M. Therefore, several investigators have performed vitrectomy alone or with ILM-peel, in combination with laser photocoagulation in the peripapillary area [52–55]. The results have been very promising both for retinal re-attachment as well as for visual improvement. Very recently, Georgalas et al. [29, 56] reported favorable results in three patients with ODP-M (one case was a 5-year-old boy) treated with vitrectomy, posterior vitreous detachment (PVD) induction, and ILM peel so as to completely eliminate vitreoretinal traction in the macular area and facilitate absorption of intraretinal and subretinal fluid. Laser photocoagulation was not performed, firstly to avoid any adverse effects of the laser treatment in the papillomacular region, and secondly because the authors thought that the rationale for laser photocoagulation prevention of fluid transportation within the inner retinal layers of the macula was weak.
Although Spaide et al. [57] recently proposed a new technique suggesting surgery of the inner retinal fenestration as a possible treatment approach for ODP-M, the current management trend for these cases appear to be a pars plana vitrectomy with PVD induction, with or without ILM peeling (although the latter further eliminates traction), with or without endolaser photocoagulation (although there have been favorable results for cases without laser application).
The same vitreoretinal approach has been reported for pediatric ODP-M cases. Snead et al. [58] reported a case of a 9-year-old boy with ODP-M successfully treated with vitrectomy, endolaser, and SF6 tamponade without ILM peeling. Hirakata et al. [59] treated an 8-year-old girl with vitrectomy and gas, while an unusual posterior hyaloid strand connected with the ODP was removed with forceps. Laser photocoagulation followed by vitrectomy, ILM peeling, and gas tamponade was performed with favorable results by Ishikawa et al. [60] in a 7-year-old girl with ODP-M. Recently, Ghosh et al. [55] retrospectively reviewed the outcome of vitrectomy, laser photocoagulation, and gas tamponade in seven patients; among them, two were children, 7 and 11 years old. In both patients, the macula was flattened postoperatively; however, one required further surgery.
Georgalas et al. [29] reported that PVD seemed impossible intraoperatively in the case of a 5-year-old boy with ODP-M, and therefore the posterior vitreous was peeled off from the macular area within the retinal arcades. Air was used as an endotamponade agent in order to avoid the possible interactions of the gas bubble with the remaining attached vitreous. Endolaser photocoagulation was not performed.
Very recently, the use of autologous platelet treatment has been reported for persistent ODP-related macular detachment. Specifically, autologous platelets were injected over the ODP after a three-port pars plana vitrectomy. This surgical approach demonstrated promising anatomical and functional results in a 44-year-old woman [61].
Special features and presentations
ODP-M and trauma
A few reports in the literature support the association between ocular trauma and ODP-M. Specifically, the conversion from an asymptomatic optic pit to an ODP-M could occur as a result of antero-posterior vitreous traction that allowed vitreous fluid to move into the sub- and intra-retinal space. A different pathophysiological mechanism has been also postulated: following a closed head trauma, cerebrospinal fluid could be transmitted into the subretinal space, causing macular elevation [62–64]. Similarly, the mechanical stress on the vitreoretinal interface that is generated during laser emission in a patient subjected to laser in-situ keratomileusis (LASIK) has been proposed as a possible etiological factor in the development of ODP-M [65].
ODP and systemic associations
Basal encephalocele is a rare midline bony defect of the base of the skull that may allow protrusion of the meninges and their contents [66]. Optic nerve anomalies, including ODP as well as coloboma, aplasia, hypoplasia, megalopapilla, and morning glory anomaly, have been associated with basal encephalocele [67].
In addition, OPDs have been reported in Aicardi syndrome [14], and Alagille syndrome [68, 69]. The latter is an autosomal dominant disorder caused by mutations in the JAG1 gene. The JAG1 gene encodes a ligand for the Notch receptor, and thus is part of a critical signalling pathway during development [69]. OPDs have also been reported to be associated with bilateral renal hypoplasia [70] and with some cases of midline neurological developmental malformations [71]. An unusual case involving the coexistence of bilateral keratoconus and ODP has been reported, although a pathogenetic correlation can not be established until further investigations confirm this association [72].
Accessory evaluation tools
Visual fields
Although the results of visual field tests in patients with ODP are variable and often do not correspond to the location of the pit, paracentral arcuate scotoma and an enlarged blind spot are the most common defects [6, 11].
Fluorescein angiography (FA) and indocyanine green angiography (ICGA)
FA shows early hypofluorescence of the pit, followed in many cases by late hyperfluorescent staining [7, 11, 26, 73]. ODPs do not generally leak fluorescein, and there is no extension of the dye towards the macula (Fig. 7) [36].

Color fundus photography and fluorescein angiography of an eye with an optic disc pit
In a study of 17 patients with ODP-M, absolute hypofluorescence of the ODP was noted in all eyes using ICGA [73]. In addition, all 17 eyes showed a delineated late hyperfluorescence that corresponded to the area of macular elevation using both ICGA and FA [73].
Infrared (IR) and fundus autofluorescence (FAF) imaging
In a very recent retrospective study analyzing the IR and FAF features of eyes with ODP-M, the areas of serous retinal detachment and inner retinal schisis were dark but changed to brighter images following re-attachment after vitrectomy. Moreover, there was an increase in the granular hyperfluorescence in the FAF images that was accompanied by an increase in the number of subretinal precipitates [74].
Differential diagnosis
Acquired ODP (AODP) and glaucoma
Glaucoma may present with various patterns of progressive optic nerve damage. An AODP is an acquired focal structural abnormality with a distinct type of progressive glaucomatous optic nerve defect. This clinical entity was first described in the late 1970s by Smith [75] and by Lichter et al. [76] in patients without glaucoma. Further reports followed that described a similar type of damage in glaucoma patients [77–79].
AODPs may not be morphologically distinguishable from congenital ODPs. They are more frequent in women than in men, with the ratio ranging from 3:1 to 2:1, and higher percentages of inferior location have been reported compared to congenital ODPs [80, 81]. In addition, AODPs have been reported to present bilaterally in 21% [81] to 48% [8] of cases, and are associated with deep, sharply demarcated scotomas [75–81]. The prevalence of AODP has been reported to be higher in patients with low-tension glaucoma (74%) than in those with high-tension glaucoma (15%) [8, 79]. In one study [8], the frequency of disk hemorrhage was 40% in patients with AODPs, which was significantly higher than in those without (8%). In addition, disc progression and glaucomatous visual field loss were more frequent in patients with AODPs [8]. A 2007 population-based study that included 3,654 participants [82] concluded that ODPs were primarily associated with glaucoma and some of its features (disc hemorrhage and peripapillary atrophy). Interestingly, classic temporal ODP was the rarest subtype in the studied population; thus, the association with low-tension glaucoma may represent acquisition bias [82].
Usually ODP-M is not a complication of AODP, although one report suggested that in cases with untreated glaucoma, progression of glaucomatous optic disc damage with subsequent asymmetrical increase in the cup depth and width might result in a localized defect of the rim that would allow leakage from the choroid into the subretinal space [83].
ODP and other congenital anomalies of the optic disc
The main features of the differential diagnosis of ODP from other congenital anomalies of the optic nerve head are presented in Table 1 [36, 84, 85].
Conclusion
ODPs are a very rare clinical entity, affecting approximately one in 11,000 people. Patients with congenital ODP sometimes remain asymptomatic, but 25% to 75% present with visual deterioration in their 30s or 40s after developing macular schisis and detachment. The most widely accepted treatment for such patients is a surgical approach involving pars plana vitrectomy with or without internal limiting membrane peeling, with or without endolaser photocoagulation and C3F8 endotamponade
References
Wiethe T (1882) Ein Fall von angeborener Deformität der Sehnervenpapille. Arch Augenheilkd 11:14–19
Reis W (1908) Eine wenig bekannte typische Missbildung am Sehnerveneintritt: umschriebene Grubenbildung auf der Papilla r optici. Arch Augenheilkd 19:505–528
James R (1913) Crater-like hole in the disc associated with changes at the macula. Ophthalmol Rev 32:38–40
Halbertsma K (1927) Crater-like hole and coloboma of the disc associated with changes at the macula. Br J Ophthalmol 11:11–17
Rosen E (1948) Crater-like holes in the optic disc. Br J Ophthalmol 32:465–478
Kranenburg E (1960) (1960) Crater-like holes in the optic disc and central serous retinopathy. Arch Ophthalmol 64:912–924
Chang M (1976) Pits and crater-like holes of the optic disc. Ophthalmol Sem 1:21–61
Ugurlu S, Weitzman M, Nduaguba C, Caprioli J (1998) Acquired pit of the optic nerve: a risk factor for progression of glaucoma. Am J Ophthalmol 125(4):457–464
Brodsky MC (1994) Congenital optic disc anomalies. Surv Ophthalmol 39:89–112
Brown G, Tasman W (1983) Congenital anomalies of the optic disc. Grune & Stratton, New York, pp 31–215
Brown GS, Shieds JA, Goldberg RE (1980) Congenital pits of the optic nerve head II. Clinical studies in humans. Ophthalmology 87:51–65
Stefko ST, Campochiaro P, Wang P, Li Y, Zhu D, Traboulsi EI (1997) Dominant inheritance of optic pits. Am J Ophthalmol 124(1):112–113
Krivoy D, Gentile R, Liebmann JM, Stegman Z, Rosen R, Walsh JB, Ritch R (1996) Imaging congenital optic disc pits and associated maculopathy using optical coherence tomography. Arch Ophthalmol 114(2):165–170
Reed D (1999) Congenital pits of the optic nerve. Clin Eye Vis Care 11(2):75–80
Gass JDM (1969) Serous detachment of the macula secondary to congenital pit of the opticnervehead. Am J Ophthalmol 67:821–841
Chan JW (2007) Optic nerve disorders: diagnosis and management. Chapter 8. Springer, New York, pp 208–209
Irvine AR, Crawford JB, Sullivan JH (1986) The pathogenesis of retinal detachment with morning glory disc and optic pit. Retina 6(3):146–150
Lin CC, Tso MO, Vygantas CM (1984) Coloboma of optic nerve associated with serous maculopathy. A clinicopathologic correlative study. Arch Ophthalmol 102(11):1651–1654
Meyer CH, Rodrigues EB, Schmidt JC (2003) Congenital optic nerve head pit associated with reduced retinal nerve fibre thickness at the papillomacular bundle. Br J Ophthalmol 87(10):1300–1301
Ferry AP (1963) Macular detachment associated with congenital pit of the optic nerve head. Arch Ophthalmol 70:106–117
Akiba J, Kakehashi A, Hikichi T, Trempe CL (1993) Vitreous findings in cases of optic nerve pits and serous macular detachment. Am J Ophthalmol 116(1):38–41
Halbertsma KTA (1927) Craterlike hole and coloboma of disc associated with changes at macula. Br J Ophthalmol 11:11
Sugar HS (1964) Congenital pits of the optic disc. Am J Ophthalmol 57:833–835
Sugar HS (1967) Congenital pits of the optic disc. Am J Ophthalmol 63:298–307
Petersen HP (1958) Pits or crater-like holes in the optic disc. Acta Ophthalmol (Copenh) 36:453–43
Bonnet M (1991) Serous macular detachment associated with optic nerve pits. Arch Clin Exp Ophthalmol 229:526–532
Snead MP, James N, Jacobs PM (1991) Vitrectomy, argon laser, and gas tamponade for serous retinal detachment associated with an optic disc pit: a case report. Br J Ophthalmol 75(6):381–382
Schatz H, McDonald HR (1988) Treatment of sensory retinal detachment associated with optic nerve pit or coloboma. Ophthalmology 95(2):178–186
Georgalas I, Kouri A, Ladas I, Gotzaridis E (2010) Optic disc pit maculopathy treated with vitrectomy, internal limiting membrane peeling, and air in a 5-year-old boy. Can J Ophthalmol 45(2):189–191
Yuen CH, Kaye SB (2002) Spontaneous resolution of serous maculopathy associated with optic disc pit in a child: a case report. J AAPOS 6(5):330–331
Brown GC, Shields JA, Patty BE, Goldberg RE (1979) Congenital pits of the optic nerve head. I. Experimental studies in collie dogs. Arch Ophthalmol 97(7):1341–1344
Kuhn F, Kover F, Szabo I, Mester V (2006) Intracranial migration of silicone oil from an eye with optic pit. Graefes Arch Clin Exp Ophthalmol 244(10):1360–1362
Regenbogen L, Stein R, Lazar M (1964) Macular and juxtapapillary serous retinal detachment associated with pit of the optic disc. Ophthalmologica 148:247–251
Gordon R, Chatfield RK (1969) Pits in the disc associated with macular degeneration. Br J Ophthalmol 53:481–489
Wise G, Dollery C, Henkind P (1971) Disciform macular disease. In: Wise G, Dollery C, Henkind P (eds) The retinal circulation. Harper & Row, New York, p 471
Apple DJ, Rabb MF, Walsh PM (1988) Congenital anomalies of the optic disc. Surv Ophthalmol 27:3–41
Lincoff H, Lopez R, Kreissig I, Yannuzzi L, Cox M, Burton T (1988) Retinoschisis associated with optic nerve pits. Arch Ophthalmol 106:61–67
Brockhurst RJ (1975) Optic pits and posterior retinal detachment. Trans Am Ophthalmol Soc 73:264–291
Postel EA, Pulido JS, McNamara JA, Johnson MW (1998) The etiology and treatment of macular detachment associated with optic nerve pits and related anomalies. Trans Am Ophthalmol Soc 96:73–88
Theodossiadis G (1977) Evolution of congenital pit of the optic disc with macular detachment in photocoagulated and non photocoagulated eyes. Am J Ophthalmol 84:620–631
Theodossiadis GP (1996) Treatment of maculopathy associated with optic disc pit by sponge explant. Am J Ophthalmol 121:630–637
Doyle E, Trivedi D, Good P, Scott RA, Kirkby GR (2009) High-resolution optical coherence tomography demonstration of membranes spanning optic disc pits and colobomas. Br J Ophthalmol 93(3):360–365
Imamura Y, Zweifel SA, Fujiwara T, Freund KB, Spaide RF (2010) High-resolution optical coherence tomography findings in optic pit maculopathy. Retina 30(7):1104–1112
Chiu YT, Chen HY, Tsai YY, Lin JM, Chiang CC (2006) Stratus optical coherence tomography for evaluating optic disc pits associated with maculopathy before and after vitrectomy: two case reports. Kaohsiung J Med Sci 22(5):229–234
Lincoff H, Schiff W, Krivoy D, Ritch R (1996) Optic coherence tomography of optic disk pit maculopathy. Am J Ophthalmol 122(2):264–266
Brasil OF, Brasil MV, Brasil OM (2006) Different presentations of intraretinal fluid collections in optic disc pits: OCT study of 3 cases. Arq Bras Oftalmol 69(5):745–747
Theodossiadis PG, Grigoropoulos VG, Emfietzoglou J, Theodossiadis GP (2007) Vitreous findings in optic disc pit maculopathy based on optical coherence tomography. Graefes Arch Clin Exp Ophthalmol 245(9):1311–1318
Mustonen E, Varonen T (1972) Congenital pit of the optic nerve head associated with serous detachment of the macula. Acta Ophthalmol (Kbh) 50:689–698
Lincoff H, Yannuzzi L, Singerman L, Kreissig I, Fisher Y (1993) Improvement in visual function after displacement of the retinal elevations emanating from optic pits. Arch Ophthalmol 111(8):1071–1079
Rosa AAM, Primiano Júnior HP, Nakashima Y (2006) Retinopexia pneumática e fotocooagulação a laser para tratamento de descolamento secundário à fosseta de disco óptico: relato de caso. Arq Bras Oftalmol 69(1):101–105
Cox MS, Witherspoon CD, Morris RE, Flynn HW (1988) Evolving techniques in the treatment of macular detachment caused by optic nerve pits. Ophthalmology 95(7):889–896
Hirakata A, Okada AA, Hida T (2005) Long-term results of vitrectomy without laser treatment for macular detachment associated with an optic disc pit. Ophthalmology 112(8):1430–1435
Dai S, Polkinghorne P (2003) Peeling the internal limiting membrane in serous macular detachment associated with congenital optic disc pit. Clin Exp Ophthalmol 31(3):272–275
Snead MP, James N, Jacobs PM (1991) Vitrectomy, argon laser, and gas tamponade for serous retinal detachment associated with an optic disc pit: a case report. Br J Ophthalmol 75(6):381–382
Ghosh YK, Banerjee S, Konstantinidis A, Athanasiadis I, Kirkby GR, Tyagi AK (2008) Surgical management of optic disc pit associated maculopathy. Eur J Ophthalmol 18(1):142–146
Georgalas I, Petrou P, Koutsandrea C, Papaconstadinou D, Ladas I, Gotzaridis E (2009) Optic disc pit maculopathy treated with vitrectomy, internal limiting membrane peeling, and gas tamponade: a report of two cases. Eur J Ophthalmol 19(2):324–326
Spaide RF, Fisher Y, Ober M, Stoller G (2006) Surgical hypothesis: inner retinal fenestration as a treatment for optic disc pit maculopathy. Retina 26(1):89–91
Snead MP, James N, Jacobs PM (1991) Vitrectomy, argon laser, and gas tamponade for serous retinal detachment associated with an optic disc pit: a case report. Br J Ophthalmol 75:381–382
Hirakata A, Hida T, Wakabayashi T, Fukuda M (2005) Unusual posterior hyaloid strand in a young child with optic disc pit maculopathy: intraoperative and histopathological findings. Jpn J Ophthalmol 49:264–266
Ishikawa K, Terasaki H, Mori M, Sugita K, Miyake Y (2005) Optical coherence tomography before and after vitrectomy with internal limiting membrane removal in a child with optic disc pit maculopathy. Jpn J Ophthalmol 49:411–413
Rosenthal G, Bartz-Schmidt KU, Walter P, Heimann K (1998) Autologous platelet treatment for optic disc pit associated with persistent macular detachment. Graefes Arch Clin Exp Ophthalmol 236(2):151–153
Meyer CH, Rodrigues EB (2004) Optic disc pit maculopathy after blunt trauma. Eur J Ophthalmol 14(1):71–73
Colyer MH, Weichel ED, Ward TP (2007) Blast injury-associated optic disc pit maculopathy. Br J Ophthalmol 91(4):558
Hirakata A, Hida T, Wakabayashi T, Fukuda M (2005) Unusual posterior hyaloid strand in a young child with optic disc pit maculopathy: intraoperative and histopathological findings. Jpn J Ophthalmol 49(3):264–266
Rodriguez-Coleman H, Schiff WM, Hwang JC, Speaker MG (2007) Optic pit maculopathy after laser-assisted in situ keratomileusis. Can J Ophthalmol 42(1):123–124
Pollock JA, Newton TH, Hoyt WF (1968) Transsphenoidal and transethmoidal encephaloceles. Radiology 90:442–453
Caprioli J, Lesser R (1983) Basal encephalocele and morning glory syndrome. Br J Ophthalmol 67:349–351
Fea A, Grosso A, Rabbione M, Grignolo F (2007) Alagille syndrome and optic pit. Graefes Arch Clin Exp Ophthalmol 245(2):315–317
Kim BJ, Fulton AB (2007) The genetics and ocular findings of Alagille syndrome. Semin Ophthalmol 22(4):205–210
Asensio Sánchez VM, Corral Azor A, Bartolomé Aragón A, De Paz García M (2002) Renal-coloboma syndrome. Arch Soc Esp Oftalmol 77(11):635–638
Corbett JJ, Savino PJ, Schatz NJ, Orr LS (1980) Cavitary developmental defects of the optic disc. Visual loss associated with optic pits and colobomas. Arch Neurol 37(4):210–213
Fasciani R, Mosca L, Giannico ML, Legrottaglie EF, Balestrazzi E (2008) Unusual coexistence of bilateral keratoconus and optic disc pit: a case report. Eur J Ophthalmol 18(1):134–137
Theodossiadis GP, Ladas ID, Panagiotidis DN, Kollia AC, Voudouri AN, Theodossiadis PG (1999) Fluorescein and indocyanine green angiographic findings in congenital optic disk pit associated with macular detachment. Retina 19(1):6–11
Hiraoka T, Inoue M, Ninomiya Y, Hirakata A (2010) Infrared and fundus autofluorescence imaging in eyes with optic pit maculopathy. Clin Exp Ophthalmol 38(7):669–677
Smith JL (1977) The optic nerve work up. Trans Am Acad Ophthalmol Otolaryngol 83:778–785
Lichter PR, Henderson JW (1977) Optic nerve infarction. Trans Am Ophthalmol Soc 75:103–121
Radius RL, Maumenee AE, Green WR (1978) Pit-like changes of the optic nerve head in open-angle glaucoma. Br J Ophthalmol 62(6):389–393
Spaeth GL (1980) Low-tension glaucoma: its diagnosis and management. In: Greve EL (ed) Glaucoma symposium: diagnosis and therapy, Amsterdam, 1979. Doc Ophthalmol Proc Ser 22. Dr W. Junk, The Hague, pp 263–287
Javitt JC, Spaeth GL, Katz LJ, Poryzees E, Addiego R (1990) Acquired pits of the optic nerve. Increased prevalence in patients with low-tension glaucoma. Ophthalmology 97(8):1038–1043, discussion 1043–1034
Spaeth GL (1994) A new classification of glaucoma including focal glaucoma. Surv Ophthalmol 38:S9–S17
Cashwell LF, Ford JG (1995) Central visual field changes associated with acquired pits of the optic nerve. Ophthalmology 102(9):1270–1278
Healey PR, Mitchell P (2008) The prevalence of optic disc pits and their relationship to glaucoma. J Glaucoma 17(1):11–14
Rath EZ, Rumelt S (2007) Acute visual loss due to serous retinal detachment from acquired optic pit may be a rare presentation of primary open-angle glaucoma. Can J Ophthalmol 42(2):339–340
Yamakiri K, Uemura A, Sakamoto T (2004) Retinal detachment caused by a slitlike break within the excavated disc in morning glory syndrome. Retina 24(4):652–653
Coll GE, Chang S, Flynn TE, Brown GC (1995) Communication between the subretinal space and the vitreous cavity in the morning glory syndrome. Graefes Arch Clin Exp Ophthalmol 233(7):441–443
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 28.5 kb)
Rights and permissions
About this article
Cite this article
Georgalas, I., Ladas, I., Georgopoulos, G. et al. Optic disc pit: a review. Graefes Arch Clin Exp Ophthalmol 249, 1113–1122 (2011). https://doi.org/10.1007/s00417-011-1698-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-011-1698-5