
Abstract
Huntington’s disease (HD) is a fatal neurodegenerative disease caused by mutant HTT gene expansions of CAG triplet repeat numbers that are inherited in an autosomal dominant manner. HD patients display multiple clinical features that are correlated with HTT CAG repeat numbers that include age of disease onset, motor dysfunction, cognitive deficits, compromised daily living capacity, and brain neurodegeneration. It is important to understand the significant relationships of the multiple HD clinical deficits correlated with the number of mutant HTT CAG expansions that are the genetic basis for HD disabilities. Therefore, this review highlights the significant correlations of the HD clinical features of age of onset, motor and cognitive disabilities, decline in living capabilities, weight loss, risk of death, and brain neurodegeneration with respect to their associations with CAG repeat lengths of the HTT gene. Quantitative HTT gene expression patterns analyzed in normal adult human brain regions demonstrated its distribution in areas known to undergo neurodegeneration in HD, as well as in other brain regions. Future investigation of the relationships of the spectrum of clinical HD features with mutant HTT molecular mechanisms will be important to gain understanding of how mutant CAG expansions of the HTT gene result in the devastating disabilities of HD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease (HD) is an inherited neurodegenerative disease that progresses to multiple behavioral dysfunctions in motor and cognitive abilities, which eventually result in death 15–20 years after onset of symptoms [1,2,3,4]. Expansion of the CAG triplet repeat domain in the mutant HTT gene is responsible for causing HD that is inherited in an autosomal dominant manner. The disease progression to increasing severity of these clinical features involves brain neurodegeneration in HD. The purpose of this review is to describe the associations of significant clinical features of HD with CAG repeat lengths of the mutant HTT gene, and to gain understanding of the extent of clinical disabling severities with HTT CAG expansions. This topic has not been recently covered among the many HD reviews in the literature. Understanding data about the extent of HTT CAG expansions with clinical features will benefit investigations of HTT gene and cellular mechanisms of the multiple human HD disabilities suffered by patients.
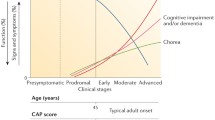
The mutant HTT gene with expansion of CAG repeats is responsible for HD [3,4,5,6]. Normal healthy individuals have fewer than 35 CAG repeats (35Q), while affected HD individuals have 40 or more CAG repeats (Fig. 1). Most normal individuals have 15–21Q, while the majority of HD patients have adult-onset HD with repeats in the range of 40–50Q. The disease has reduced penetrance in the range of 35–39 repeats, while > 60 repeats usually results in juvenile HD with symptoms present in children before age 20. Importantly, these genetic features of the HTT gene illustrate that HD results from a spectrum of expanded CAG repeat lengths that initiate progressive dysfunctions of numerous HD clinical features. Thus, there is no single mutation identical in all patients, but rather a disease threshold followed by a spectrum of polyQ lengths which regulate HD disease outcomes. The mutant CAG repeat lengths of the HTT gene result in an age-dependent onset of HD motor symptoms and chorea, cognitive decline, loss of capacity for independent daily life, and brain tissue loss in HD that are more severe with increasing CAG repeat numbers. These dysfunctions of HD are significantly associated with HTT CAG repeat length, discussed in this review.

HTT gene CAG triplet repeat expansions in Huntington’s disease (HD) corresponding to phenotypic dysfunctions in HD patients. The HD causative CAG triplet repeat mutations in the HTT gene and corresponding ages of onset for clinical symptoms are illustrated for normal CAG repeat lengths of < 35, 36–39 repeat lengths that may or may not result in HD, adult HD repeat lengths of 40 to ~ 50, and juvenile HD repeat lengths of ~ > 60. The clinical dysfunction features for each of these three ranges of HTT CAG repeats are displayed
HD age of onset correlates with CAG repeat length of the HTT gene
The age of onset of HD motor dysfunction correlates with the CAG repeat lengths of the mutant HTT gene. Because the HTT disease allele is dominant, affected individuals are usually heterozygotes, with one wild-type allele. With the mutant allele, longer CAG repeats of > 60Q cause juvenile HD. These high expansion lengths have the strongest association with age of motor symptom onset, with repeat length accounting for about 70% of variance [7]. Studies examining a wide range of CAG repeat lengths have provided estimates of about 40–60% of variance in age of HD onset explained by repeat length for the whole spectrum of HD (summarized in Table 1) [7,8,9,10,11,12,13,14,15,16]. Also, lower correlation in individuals closest to the normal repeat range was observed [17]. A large study of more than 3500 participants found that CAG repeat length accounted for 66% of variation in onset age in the range of 40–53 repeats, representing most HD cases [13]. These findings demonstrate the strength of the correlation of CAG repeat numbers with age of HD onset.
Further analyses of the change in onset age for each CAG repeat were carried out by Andresen and coworkers, who found in two separate large HD populations that there was an inflection point in the regression curve at approximately 50Q [18, 19]. Interestingly, there was a steeper slope in the 40–50Q range compared to > 50Q, suggesting that CAG repeat number actually has a stronger effect on age of onset in adult HD patients compared to juvenile HD patients [18, 19]. Further evidence for the greater variance at shorter CAG repeat lengths has been provided by the PREDICT-HD study of pre-symptomatic HD patients [20, 21] with greater than 35 CAG repeats. It is unknown as to why greater variance in age-at-onset occurs for lower CAG repeat lengths, compared to the decreasing variance with higher CAG repeats. Further, it is noted that the rare HD individuals who carry two copies of mutant HTT gene alleles show similarities in age of HD onset compared to heterozygotes when adjusted for the longest HTT CAG repeat length [7, 13, 22,23,24].
These findings demonstrate the significant relationships for correlation of CAG repeat numbers of the HTT gene with the age of onset of HD disease. Further, data show greater variance for age of onset at lower CAG repeat lengths, compared to lower variance with high CAG repeats. These findings suggest some differences in how the different CAG repeat lengths of mutant HTT regulate the age of onset of HD clinical features.
Motor dysfunction correlates with CAG repeat length of mutant HTT in HD
HD progression results in characteristic extrapyramidal motor signs of chorea and incoordination early in the disease, progressing to bradykinesia, dystonia, saccadic eye movements, and impaired voluntary movement [3, 4, 25]. Several studies have examined the relationship of CAG repeat length and progression of chorea and motor dysfunctions (summarized in Table 2), measured by the Quantified Neurological Examination (QNE) or the total motor score from the Unified Huntington’s Disease Rating Scale (UHDRS) [10, 11, 26,27,28]. QNE and UHDRS motor examination consist of tests quantifying ocular, speech, walking, and movement dysfunctions. As shown in Table 2, inverse correlation of the progression of motor dysfunction per CAG repeat number was found to be significantly correlated by Pearson correlation coefficient (r) values and p values. Higher scores indicate deteriorating motor function. Investigation of chorea alone showed that progression was significantly related to CAG repeat length in the study by Brandt and colleagues [10] with 46 subjects. On the other hand, two other studies with larger patient group sizes found trends towards correlation of chorea progression with CAG repeat number, but were not statistically significant [26, 28]. Rosenblatt et al. [27] conducted a mathematical correction for the age at onset of motor symptoms, and this factor increased the effect of CAG repeat numbers on motor impairment scores by 76%. The varying age at onset of motor symptoms for the same CAG repeat number may reflect genetic and environmental factors and the aging process [27].
Overall, studies of the QNE and UHDSRS for neurological motor impairment and total motor scores showed significant correlation of motor dysfunction progression with changes in CAG repeat numbers for subjects with 36–109 CAG repeats in the HTT gene.
Cognitive deficits correlate with CAG repeat lengths of HTT
HD patients display cognitive changes before and during motor dysfunction. Development of cognitive deficits in HD often progresses to severe behavioral disturbances [3, 25, 29]. Studies examining the relationship of CAG repeat length with changes in cognitive symptoms, as measured by scores related to psychomotor function, emotional recognition, inhibition of cognitive interference, motor speed, planning and correction found that CAG repeat lengths in the HD disease range (36–109 CAG repeats) were inversely correlated with changes in cognitive scores, particularly when scores were normalized to disease duration [11] or adjusted for age [26, 27, 30, 31].
Recent efforts are establishing updated cognitive testing standards for monitoring disease progression and as outcome measures in clinical trials [30]. HD-CAB (Cognitive Assessment Battery) uses the most sensitive measures of cognitive decline during early disease manifestation consisting of Symbol Digit Modalities Test, Paced Tapping, One Touch Stockings of Cambridge (abbreviated), Emotion Recognition, Trail Making B, and the Hopkins Verbal Learning Test. Further evaluation of correlating cognitive decline with HTT CAG repeats in early HD may potentially reveal sensitive changes in cognitive function to predict individual patient cognitive dysfunction progression, using mathematical adjustments for age, shown in Table 3 as disease burden (DB) scores [30,31,32]. Disease burden (age × [CAG − 35.5]) models a patient’s expected disease state based on CAG repeat number and age at the time of onset or assessment. For example, a 20-year-old patient with a CAG repeat number of 55 would be expected to have similar symptom severity as an 86-year-old with a CAG repeat number of 40.
Overall, findings demonstrate correlation of HTT CAG repeat lengths with declining cognitive function in HD. It is noted that this relationship becomes less variable with increasing CAG length.
Daily living capacity and HTT CAG repeat lengths
Studies relating CAG repeat length with changes in quantitative scales measuring ability to function in daily life utilized the UHDRS Functional Independence Scale (FIS), total functional capacity (TFC), and the HD activities of daily living (ADL) scale [12, 26,27,28], as summarized in Table 4. These measures of daily function showed strong relationships with CAG repeat number [26, 27]. It is noted that Total Functional Capacity was not quite significant (p < 0.1) [26]. Adjusting for age was an important factor in the observed progression in these scores, since age at the time of assessment independently affected rate of decline in daily function [26, 27]. When adjusting for age at onset in a random effects model, Rosenblatt et al. found an increased effect size of 159% in ADL (activities of daily living) [27]. Of interest was one study which found that ten additional repeats were correlated with an 81% change in the UHDRS Functional Independence Scale (FIS) over the 30-month study period (shown in Table 4 as the difference in score per repeat) [26]. The relationship included age at assessment in the regression model [26].
Overall, findings in the field demonstrate association of HTT CAG repeat numbers with the extent of decline in daily living capacity in HD patients, which occurs as the disease progresses.
Weight loss correlates with CAG repeat lengths of HTT
Many studies have utilized weight loss to represent overall deterioration in health. Two studies of 300–500 patients with repeats ranging from 36 to 69 found correlations of repeat length with BMI (body mass index) change and overall weight loss [26, 33]. Ravina et al. found significant differences in weight loss over 30 months for every ten repeats (p < 0.05) [26], and Aziz et al. found that each repeat was associated with − 0.22 change in BMI units per year (p = 0.017) [33]. Similarly, a study of 47 patients with 42–62 repeats reported a significant association between CAG repeat length and age of feeding tube insertion, as well as age of nursing home admission (p < 0.001 for both) [29].
A larger cohort study (n = 5821) from Enroll-HD confirmed a prior study that showed weight loss increased with increasing CAG repeats [34]. Interestingly, the researchers found that a higher BMI at the beginning of HD onset significantly correlated with slower decline of motor, cognitive and TFC that was independent of CAG repeat length (p < 0.001), even if patients were obese or morbidly obese (p < 0.01). Also, results of the van den Burg group [34] suggest that a higher BMI can supersede the impact of a patient’s genetic burden on disease progression.
In summary, these clinical findings illustrate a relationship of the extent of weight loss with HTT CAG repeat numbers.
Higher risk of death correlates with CAG repeat lengths of HTT
HD is ultimately a fatal disease, and three prospective cohort studies have examined the relationship of CAG repeat length with age at death for the subset of patients who died during the study periods [8, 14, 15]. A study of 135 patients, including 41 deceased at ages 18–83, found that each CAG repeat conferred a higher risk of death during the study period, assessed by the hazard ratio (HR) of 1.09 for one triplet repeat increase (p = 0.002) [15]. For the 51 deceased patients in a study of 360 patients, repeat length accounted for 18% of variation in age at death (p ≤ 0.01) [5]. However, another study with 40 deceased patients out of 112 total found that repeat length accounted for 58% of variation in age at death (p = 0.001) [14]. A larger study of 4448 patients confirmed a significant correlation between CAG repeat length and age of death, but the variance of age at onset and the age of death were not explained by CAG repeat length, especially at lower disease-penetrant CAG scores, indicating other factors are involved (p < 0.0001) [35]. A limitation of studies of this type is susceptibility to ascertainment bias, since patients still alive at the end of the study period would naturally be excluded from analysis; thus, length of follow-up and proportion of the cohort deceased may also affect estimated correlations.
Although studies differ in their estimates of the exact contribution of repeat length to age at death, the overall evidence clearly supports a significant relationship linking CAG repeat lengths with risk of mortality.
Brain neurodegeneration and biomarkers correlated with CAG repeat lengths of HTT
Reduced brain volume and neurodegeneration
Post-mortem examination of human HD brains and imaging in live HD patients have revealed massive cell death and neurodegeneration in the striatum, and significant loss of volume occurs in cortical brain regions [1, 4] (Table 5). An average loss of 20% brain volume in terminal HD suggests that cell loss occurs in more brain regions than only striatum and cortex [36, 37]. A variety of studies have examined neuropathology directly, either at autopsy or in living patients by MRI or CT scan [9, 36, 38,39,40]. Studies quantifying striatal atrophy at a single time point (age adjusted) found that CAG repeat length accounted for 66–78% of variance in neuronal cell loss in striatum [38, 39]. Studies measuring rate of atrophy during a 1–2-year time period found significant but more modest relationships with CAG repeat number [9, 40]. For example, an MRI study in living HD patients found 0.3% faster cortical thinning for every CAG repeat in some cortical regions, although not all cortical regions and no subcortical regions had atrophy rates that were significantly associated with repeat number during the 1-year study [40]. The study by Hadzi and colleagues [36] used a cluster analysis approach for neuropathology in HD brains. The “striatal” cluster encompassed regions including striatum, which shows correlation of CAG repeat number with neuropathology. The overall evidence supports the role of mutant HTT CAG repeat length in HD brain neurodegeneration.
Various mathematical correlation models for neuroimaging and CAG or age-adjusted repeat length show significant decreases in brain tissue volume in premanifest HD to advanced HD which include whole brain atrophy, caudate and putamen, midbrain, pons, thalamus, gray and white matter of all cerebrocortical regions, and ventricular expansion. Atrophy was more severe with higher CAG and disease burden (DB) scores [31, 32, 41], and CAP ((age at imaging) × [CAG − 33.6]). CAP is often used for age-adjustment in imaging studies [41, 42] (Table 5). These brain volume studies show that multiple brain regions are degenerated beginning in premanifest HD [20, 31, 32, 43], not only in the striatum, and that brain atrophy occurs in a central to peripheral and posterior to anterior pattern [42]. Loss of white matter integrity also begins in premanifest HD in the corpus callosum [45], subcortical white matter of all cerebrocortical lobes [42], and white matter tracts of the basal ganglia [44]. These results suggest that neuronal connectivity deficit is an early event in HD that is exacerbated by CAG score. Additionally, axonal demyelination occurs more sharply in premanifest HD, and slows in early manifest HD [45].
Cellular and molecular HD brain alterations
Molecular imaging techniques have identified localized changes in receptor density, ionic and glucose balance and increased glial activation that correlate with CAG repeat length in premanifest and early manifest HD (Supplemental Table 1) [32, 46,47,48,49,50]. Decreased blood oxygenation showed significant correlation between CAG repeat and loss of connectivity between the primary motor cortex and motor- and visual-associated areas; connectivity to the posterior cingulate cortex, an area of pain perception and autobiographical information, was significantly increased [41].
Globally in the brain, elevated sodium levels and decreased glucose uptake are correlated with CAG repeats and disease burden [46, 48]. Quantification of brain glucose uptake was also used as a variable in mathematical prediction modeling for HD symptom progression and improved prediction of age at onset of motor symptoms by 37% [46].
Receptor density studies, measured by tracer-tagged receptor agonists, identify changes in neurotransmitter systems in premanifest and early stage HD that are correlated by CAG and age-adjusted repeat scores. Dopamine receptor 2 (D2) density is highly decreased (50%) in the striatum of premanifest to HD grade 2 patients [32]. Prefrontal and premotor cortical cannabinoid receptor 1 (CB1) are significantly decreased in early-to-late-stage HD [50].
Other studies identified neurotransmitter receptor changes in HD patients but did not model a correlation with CAG repeat number (see Pagano et al. for a review [51]). Findings included upregulation of the adenosine receptor A1 in early premanifest HD patients’ thalamic and cerebral cortical regions, but downregulated in later premanifest and manifest HD patients caudate and amygdala [52]. GABA receptors are also decreased in the striatum and thalamus of premanifest HD [41], along with opioid receptors [53]. These data show that neurotransmitter systems are disrupted before a patient experiences HD symptoms. Cell-specific markers also identify significant changes in premanifest HD that are significant with CAG or adjusted scores. Activated microglia are sharply upregulated in the striatum, and also diffusely throughout the cerebral cortex [47, 49, 54].
Overall, brain neuroimaging of brain tissue degeneration and biomarker studies of premanifest through all stages of HD show that HD is a whole brain disease, not just a disease of the striatum. Volumetric studies of neurodegeneration, white matter integrity disruption, and molecular imaging studies of receptor systems, and cell-targeted imaging studies have characterized cellular and molecular HD brain features associated significantly with CAG repeat lengths of the HTT gene.
Discussion of HTT gene expression in human brain
Given the evidence in support of HTT CAG repeat length-dependent clinical features in HD, a logical question to address is what is the distribution of HTT gene expression in human brain regions? Analyses of HTT gene expression can indicate its expression in human striatal and cortical brain regions primarily responsible for the behavioral dysfunctions occurring in HD, compared to other brain regions that also undergo neurodegeneration and molecular alterations (explained in section “Brain neurodegeneration and biomarkers correlated with CAG repeat lengths of HTT”, above).
Gene expression microarray analyses of HTT in normal adult human brain regions have been compiled by the Allen Human Brain Atlas resource (http://human.brain-map.org/). Notably, these data show ubiquitous HTT expression among the 169 human brain regions investigated (Fig. 2 and supplemental Table 2). HTT gene expression was expressed as average log2 expression values among donors. Multiple normalization steps provided log2 gene expression values relative to the normalized mean of all human brain gene expression levels (for 21,245 genes) in human brain (see Technical White Paper “Microarray Data Normalization” of the Allen Brain Atlas resource, http://human.brain-map.org/). HTT expression was present in all regions examined, with some variations in expression levels among the regions.

HTT gene expression in human brain regions. a Human brain regions. HTT gene expression was assessed by microarray analyses in 169 brain regions (from six donor brains). Brain regions are indicated by the second bar (at top of figure) with colors indicating the main brain regions of (top bar): FL frontal lobe, insula, CgG cingulate gyri, HiF hippocampal formation, OL occipital lobe, PL parietal lobe, TL temporal lobe, Amg amygdala, BsFb basal forebrain, Str striatum, Clstr claustrum, Epithal epithalamus; hypothalamus, Thal thalamus, Subthal subthalamus, MES mesencephalon, CbCtx cerebellar cortex, CbN cerebellar nuclei; pons, MY myelencephalon, WM white matter structures, CP choroid plexus of the lateral ventricles. Color keys of these brain regions, and subregions, are shown in Supplemental Table 2. b HTT gene expression in human brain regions. Graphical representation of log2 values of HTT gene expression assessed by microarray analyses (averaged for six donor brains) is shown for 169 brain regions. The colors of the graphed bars correspond to the brain region colors indicated in panel ‘a’. The key for brain regions #1-169 is shown in Supplemental Table 2
Human brain images of HTT gene expression localization overlaid with coronal MRI (magnetic resonance imaging) images and 3D-rendered sagittal images illustrate the expression of HTT in striatum, primary motor cortex (Brodmann area 4), premotor cortex (Brodmann area 6), and globus pallidus (Fig. 3). Examination of these regions in normal human brain is of interest since these areas undergo neurodegeneration in HD. In these normal brain images, HTT is expressed in striatal areas of caudate nucleus, nucleus accumbens, and putamen (Fig. 3) which function in movement and in HD chorea [37, 55]. Expression occurs in the cortical regions of Brodmann areas 4 and 6 that are essential for motor control and mental functions (Fig. 3). HTT expression occurs in hippocampus (Fig. 3) that is important in cognitive functions in normal brain, and in HD cognitive deficits [56, 57]. Additionally, normal human brain regions of cortex (Brodmann areas 1, 2, 3, 17–19, 41), nucleus accumbens, globus pallidus, cerebellar cortex, substantia nigra, superior olive, lateral vestibular nucleus, oculomotor nucleus, and others display abundant HTT expression. In HD, these brain regions undergo neurodegeneration and are involved in behavioral dysfunctions that occur in HD.

Human brain images of HTT gene expression. HTT gene expression localization is illustrated in the human brain regions of a striatum—caudate nucleus, b primary motor cortex Brodmann area 4, c premotor cortex Brodmann area 6, and d hippocampus. Coronal brain MRI images and space filling 3D rendered sagittal brain images show colored overlaid gene expression levels (log2 values) of HTT gene expression. The MNI coronal coordinate is indicated below the coronal slab images (MNI: Montreal Neurological Institute coordinates are spatially organized to standardized stereotaxic space). Images credits: Allen Institute (human.brain-map.org)
These data of HTT expression in normal human brain illustrate that the gene is expressed in HD-affected brain areas (Supplemental Table 3). But HD brains from human subjects have not yet undergone complete HTT expression analyses. It is predicted that HD brains may display HTT expression similar to that in normal brain, since no differences have been found in the HTT gene promoter regions of mutant and normal HTT. Nonetheless, it is critical in future studies to evaluate the distribution of mutant HTT expression in human HD brains.
Overall, in normal human brain, HTT expression occurs in brain regions that are affected in HD by cellular loss and neurodegeneration. However, HTT is also expressed ubiquitously throughout the brain at levels substantially above normalized gene expression levels of all human genes. In HD, because behavioral dysfunctions and neurodegeneration occur in particular brain regions, the selectivity of HTT-mediated dysfunctions will involve the translated mutant huntingtin (Htt) protein and its unique cellular properties compared to normal Htt.
Human brain mutant HTT CAG repeat length mechanisms responsible for clinical HD features
The multiple clinical features displayed by HD patients result from a range of mutant CAG repeat expansions in the HTT gene, inherited in autosomal dominant manner in HD. The important question to address in HD research is what mutant HTT-initiated pathways are responsible for each of the numerous behavioral and brain biomarker deficits in HD? Elucidation of mutant HTT pathways leading to the clinical features will be of high importance in understanding the complex mechanisms responsible for CAG repeat length-dependent outcomes. Tremendous effort has been ongoing in the HD field to elucidate molecular pathways involved in HTT pathogenesis. Cellular pathway systems hypothesized to participate in HD include gene expression, synaptic vesicle trafficking, autophagy and energy metabolism, and are related as illustrated in Fig. 4, based on numerous studies in the field [1,2,3, 58,59,60,61,62,63,64,65,66]. It will be essential to gain understanding of mutant Htt-initiated pathway mechanisms that lead to the multiple clinical features of HD. Such knowledge will facilitate translational research to provide new targeted opportunities for therapeutics development in HD.

Cellular systems linked to mutant Htt (mHtt) in non-human animal models. a Cellular functions regulated by mutant Htt (mHtt) protein interactions. Htt protein interactions involve cellular pathways of transcriptional regulation, synaptic neurotransmitter systems, energy and mitochondria regulation, lysosome and autophagy, protein translation and folding, and are related [3,4,5, 58,59,60,61,62,63,64,65,66]. This illustration is based on numerous mechanistic investigations of mutant Htt-induced protein interactions and molecular pathways in non-human animal model systems. b Clinical HD neurodegeneration and behavioral deficits. Mutant Htt protein interactions with functional cellular pathways results in dysfunctions of motor and cognitive deficits, compromised daily living capacity, weight loss, and brain tissue loss in neurodegeneration. Ongoing HD research is defining the human HD brain mechanisms for mutant Htt induction of human HD clinical deficits
Abbreviations
- HD:
-
Huntington’s disease
- HTT:
-
Huntingtin gene
- Htt:
-
Huntingtin protein
- mHtt:
-
Mutant huntingtin
- polyQ:
-
Polyglutamine
- wt:
-
Wild type
- Q:
-
Glutamine
- r :
-
Pearson correlation coefficient
- QNE:
-
Quantified neurological examination
- UHDRS:
-
Unified Huntington’s Disease Rating Scale
- MMSE:
-
Mini Mental State Examination
- FIS:
-
Functional Independence Scale
- TFC:
-
Total functional capacity
- ADL:
-
Activities of daily living
References
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384
Petersén A, Mani K, Brundin P (1999) Recent advances on the pathogenesis of Huntington’s disease. Exp Neurol 157:1–18. https://doi.org/10.1006/exnr.1998.7006
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. https://doi.org/10.1016/S1474-4422(10)70245-3
Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90:905–981. https://doi.org/10.1152/physrev.00041.2009
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ (2015) Huntington disease. Nat Rev Dis Primers 1:15005. https://doi.org/10.1038/nrdp.2015.5
The Huntington’s Disease Collaborative Research Group (1999) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayán J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, Cha J-H, Irizarry M, Rosas D, Hersch S, Hollingsworth Z, MacDonald M, Young AB, Andresen JM, Housman DE, De Young MM, Bonilla E, Stillings T, Negrette A, Snodgrass SR, Martinez-Jaurrieta MD, Ramos-Arroyo MA, Bickham J, Ramos JS, Marshall F, Shoulson I, Rey GJ, Feigin A, Arnheim N, Acevedo-Cruz A, Acosta L, Alvir J, Fischbeck K, Thompson LM, Young A, Dure L, O’Brien CJ, Paulsen J, Brickman A, Krch D, Peery S, Hogarth P, Higgins DS, Landwehrmeyer B, U.S.-Venezuela Collaborative Research Project (2004) Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci USA 101:3498–3503. https://doi.org/10.1073/pnas.0308679101
Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet 4:398–403. https://doi.org/10.1038/ng0893-398
Aylward EH, Li Q, Stine OC, Ranen N, Sherr M, Barta PE, Bylsma FW, Pearlson GD, Ross CA (1997) Longitudinal change in basal ganglia volume in patients with Huntington’s disease. Neurology 48:394–399
Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Ross CA (1996) Trinucleotide repeat length and clinical progression in Huntington’s disease. Neurology 46:527–531
Illarioshkin SN, Igarashi S, Onodera O, Markova ED, Nikolskaya NN, Tanaka H, Chabrashwili TZ, Insarova NG, Endo K, Ivanova-Smolenskaya IA (1994) Trinucleotide repeat length and rate of progression of Huntington’s disease. Ann Neurol 36:630–635. https://doi.org/10.1002/ana.410360412
Kieburtz K, MacDonald M, Shih C, Feigin A, Steinberg K, Bordwell K, Zimmerman C, Srinidhi J, Sotack J, Gusella J (1994) Trinucleotide repeat length and progression of illness in Huntington’s disease. J Med Genet 31:872–874
Lee J-M, Ramos EM, Lee J-H, Gillis T, Mysore JS, Hayden MR, Warby SC, Morrison P, Nance M, Ross CA, Margolis RL, Squitieri F, Orobello S, Di Donato S, Gomez-Tortosa E, Ayuso C, Suchowersky O, Trent RJA, McCusker E, Novelletto A, Frontali M, Jones R, Ashizawa T, Frank S, Saint-Hilaire MH, Hersch SM, Rosas HD, Lucente D, Harrison MB, Zanko A, Abramson RK, Marder K, Sequeiros J, Paulsen JS, PREDICT-HD study of the Huntington Study Group (HSG), Landwehrmeyer GB, REGISTRY study of the European Huntington’s Disease Network, Myers RH, HD-MAPS Study Group, MacDonald ME, Gusella JF, COHORT study of the HSG (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78:690–695. https://doi.org/10.1212/WNL.0b013e318249f683
Pekmezovic T, Svetel M, Maric J, Dujmovic-Basuroski I, Dragasevic N, Keckarevic M, Romac S, Kostic VS (2007) Survival of Huntington’s disease patients in Serbia: longer survival in female patients. Eur J Epidemiol 22:523–526. https://doi.org/10.1007/s10654-007-9157-7
Rinaldi C, Salvatore E, Giordano I, De Matteis S, Tucci T, Cinzia VR, Rossi F, Castaldo I, Morra VB, Di Maio L, Filla A, De Michele G (2012) Predictors of survival in a Huntington’s disease population from southern Italy. Can J Neurol Sci J Can Sci Neurol 39:48–51
Trembath MK, Horton ZA, Tippett L, Hogg V, Collins VR, Churchyard A, Velakoulis D, Roxburgh R, Delatycki MB (2010) A retrospective study of the impact of lifestyle on age at onset of Huntington disease. Mov Disord Off J Mov Disord Soc 25:1444–1450. https://doi.org/10.1002/mds.23108
Lipe H, Bird T (2009) Late onset Huntington Disease: clinical and genetic characteristics of 34 cases. J Neurol Sci 276:159–162. https://doi.org/10.1016/j.jns.2008.09.029
Andresen JM, Gayán J, Cherny SS, Brocklebank D, Alkorta-Aranburu G, Addis EA, US Venezuela Collaborative Research Group, Cardon LR, Housman DE, Wexler NS (2007) Replication of twelve association studies for Huntington’s disease residual age of onset in large Venezuelan kindreds. J Med Genet 44:44–50. https://doi.org/10.1136/jmg.2006.045153
Andresen JM, Gayán J, Djoussé L, Roberts S, Brocklebank D, Cherny SS, US-Venezuela Collaborative Research Group, HD MAPS Collaborative Research Group, Cardon LR, Gusella JF, MacDonald ME, Myers RH, Housman DE, Wexler NS (2007) The relationship between CAG repeat length and age of onset differs for Huntington’s disease patients with juvenile onset or adult onset. Ann Hum Genet 71:295–301. https://doi.org/10.1111/j.1469-1809.2006.00335.x
Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E, Predict-HD Investigators of the Huntington Study Group (2006) Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol 63:883–890. https://doi.org/10.1001/archneur.63.6.883
Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, Predict-HD Investigators and Coordinators of the Huntington Study Group (2008) Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 79:874–880. https://doi.org/10.1136/jnnp.2007.128728
Alonso ME, Yescas P, Rasmussen A, Ochoa A, Macías R, Ruiz I, Suástegui R (2002) Homozygosity in Huntington’s disease: new ethical dilemma caused by molecular diagnosis. Clin Genet 61:437–442
Dürr A, Hahn-Barma V, Brice A, Pêcheux C, Dodé C, Feingold J (1999) Homozygosity in Huntington’s disease. J Med Genet 36:172–173
Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Lévi A-C, Simpson SA, Delatycki M, Maglione V, Hayden MR, Donato SD (2003) Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain J Neurol 126:946–955
Ha AD, Fung VSC (2012) Huntington’s disease. Curr Opin Neurol 25:491–498. https://doi.org/10.1097/WCO.0b013e3283550c97
Ravina B, Romer M, Constantinescu R, Biglan K, Brocht A, Kieburtz K, Shoulson I, McDermott MP (2008) The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord Off J Mov Disord Soc 23:1223–1227. https://doi.org/10.1002/mds.21988
Rosenblatt A, Kumar BV, Mo A, Welsh CS, Margolis RL, Ross CA (2012) Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov Disord Off J Mov Disord Soc 27:272–276. https://doi.org/10.1002/mds.24024
Rosenblatt A, Liang K-Y, Zhou H, Abbott MH, Gourley LM, Margolis RL, Brandt J, Ross CA (2006) The association of CAG repeat length with clinical progression in Huntington disease. Neurology 66:1016–1020. https://doi.org/10.1212/01.wnl.0000204230.16619.d9
Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K, Orme C, Paulsen J, Penney JB, Siemers E, Shoulson I (2000) Rate of functional decline in Huntington’s disease. Huntingt Study Group Neurol 54:452–458
Stout JC, Queller S, Baker KN, Cowlishaw S, Sampaio C, Fitzer-Attas C, Borowsky B, HD-CAB Investigators (2014) HD-CAB: a cognitive assessment battery for clinical trials inHuntington’s disease 1,2,3. Mov Disord Off J Mov Disord Soc 29:1281–1288. https://doi.org/10.1002/mds.25964
Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, TRACK-HD Investigators (2013) Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 12:637–649. https://doi.org/10.1016/S1474-4422(13)70088-7
van Oostrom JCH, Maguire RP, Verschuuren-Bemelmans CC, Veenma-van der Duin L, Pruim J, Roos RC, Leenders KL (2005) Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology 65:941–943. https://doi.org/10.1212/01.wnl.0000176071.08694.cc
Aziz NA, van der Burg JMM, Landwehrmeyer GB, Brundin P, Stijnen T, EHDI Study Group, Roos R (2008) Weight loss in Huntington disease increases with higher CAG repeatnumber. Neurology 71:1506–1513. https://doi.org/10.1212/01.wnl.0000334276.09729.0e
van der Burg JMM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RAC, Aziz NA (2017) Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol 82:479–483. https://doi.org/10.1002/ana.25007
Keum JW, Shin A, Gillis T, Mysore JS, Abu Elneel K, Lucente D, Hadzi T, Holmans P, Jones L, Orth M, Kwak S, MacDonald ME, Gusella JF, Lee J-M (2016) The HTT CAG-expansion mutation determines age at death but not disease duration in huntington disease. Am J Hum Genet 98:287–298. https://doi.org/10.1016/j.ajhg.2015.12.018
Hadzi TC, Hendricks AE, Latourelle JC, Lunetta KL, Cupples LA, Gillis T, Mysore JS, Gusella JF, MacDonald ME, Myers RH, Vonsattel J-P (2012) Assessment of cortical and striatal involvement in 523 Huntington disease brains. Neurology 79:1708–1715. https://doi.org/10.1212/WNL.0b013e31826e9a5d
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP (1985) Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44:559–577
Culjkovic B, Stojkovic O, Vojvodic N, Svetel M, Rakic L, Romac S, Kostic V (1999) Correlation between triplet repeat expansion and computed tomography measures of caudate nuclei atrophy in Huntington’s disease. J Neurol 246:1090–1093
Penney JB, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH (1997) CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol 41:689–692. https://doi.org/10.1002/ana.410410521
Rosas HD, Reuter M, Doros G, Lee SY, Triggs T, Malarick K, Fischl B, Salat DH, Hersch SM (2011) A tale of two factors: what determines the rate of progression in Huntington’s disease? A longitudinal MRI study. Mov Disord Off J Mov Disord Soc 26:1691–1697. https://doi.org/10.1002/mds.23762
Koenig KA, Lowe MJ, Harrington DL, Lin J, Durgerian S, Mourany L, Paulsen JS, Rao SM, PREDICT-HD Investigators of the Huntington Study Group (2014) Functional connectivity of primary motor cortex is dependent on genetic burden in prodromal Huntington disease. Brain Connect 4:535–546. https://doi.org/10.1089/brain.2014.0271
Wu D, Faria AV, Younes L, Mori S, Brown T, Johnson H, Paulsen JS, Ross CA, Miller MI, PREDICT-HD Investigators and Coordinators of the Huntington Study Group (2017) Mapping the order and pattern of brain structural MRI changes using change-point analysis in premanifest Huntington’s disease. Hum Brain Mapp 38:5035–5050. https://doi.org/10.1002/hbm.23713
Aylward EH, Liu D, Nopoulos PC, Ross CA, Pierson RK, Mills JA, Long JD, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group (2012) Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiatry 71:822–828. https://doi.org/10.1016/j.biopsych.2011.07.030
Orth M, Gregory S, Scahill RI, Mayer IS, Minkova L, Klöppel S, Seunarine KK, Boyd L, Borowsky B, Reilmann R, Bernhard Landwehrmeyer G, Leavitt BR, Roos RA, Durr A, Rees G, Rothwell JC, Langbehn D, Tabrizi SJ, TRACK-On Investigators (2016) Natural variation in sensory-motor white matter organization influences manifestations of Huntington’s disease. Hum Brain Mapp 37:4615–4628. https://doi.org/10.1002/hbm.23332
Phillips O, Sanchez-Castaneda C, Elifani F, Maglione V, Di Pardo A, Caltagirone C, Squitieri F, Sabatini U, Di Paola M (2013) Tractography of the corpus callosum in Huntington’s disease. PloS One 8:e73280. https://doi.org/10.1371/journal.pone.0073280
Ciarmiello A, Giovacchini G, Orobello S, Bruselli L, Elifani F, Squitieri F (2012) 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging 39:1030–1036. https://doi.org/10.1007/s00259-012-2114-z
Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, Tabrizi SJ, Barker RA, Piccini P (2011) Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp 32:258–270. https://doi.org/10.1002/hbm.21008
Reetz K, Romanzetti S, Dogan I, Saß C, Werner CJ, Schiefer J, Schulz JB, Shah NJ (2012) Increased brain tissue sodium concentration in Huntington’s disease—a sodium imaging study at 4 T. NeuroImage 63:517–524. https://doi.org/10.1016/j.neuroimage.2012.07.009
Russell DS, Barret O, Jennings DL, Friedman JH, Tamagnan GD, Thomae D, Alagille D, Morley TJ, Papin C, Papapetropoulos S, Waterhouse RN, Seibyl JP, Marek KL (2014) The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early Huntington disease. JAMA Neurol 71:1520–1528. https://doi.org/10.1001/jamaneurol.2014.1954
Van Laere K, Casteels C, Dhollander I, Goffin K, Grachev I, Bormans G, Vandenberghe W (2010) Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J Nucl Med Off Publ Soc Nucl Med 51:1413–1417. https://doi.org/10.2967/jnumed.110.077156
Pagano G, Niccolini F, Politis M (2016) Current status of PET imaging in Huntington’s disease. Eur J Nucl Med Mol Imaging 43:1171–1182. https://doi.org/10.1007/s00259-016-3324-6
Matusch A, Saft C, Elmenhorst D, Kraus PH, Gold R, Hartung H-P, Bauer A (2014) Cross sectional PET study of cerebral adenosine A1 receptors in premanifest and manifest Huntington’s disease. Eur J Nucl Med Mol Imaging 41:1210–1220. https://doi.org/10.1007/s00259-014-2724-8
Weeks RA, Ceballos-Baumann A, Piccini P, Boecker H, Harding AE, Brooks DJ (1997) Cortical control of movement in Huntington’s disease. A PET activation study. Brain J Neurol 120(Pt 9):1569–1578
Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P (2006) Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 66:1638–1643. https://doi.org/10.1212/01.wnl.0000222734.56412.17
Aylward EH, Harrington DL, Mills JA, Nopoulos PC, Ross CA, Long JD, Liu D, Westervelt HK, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group (2013) Regional atrophy associated with cognitive and motor function in prodromal Huntington disease. J Huntingt Dis 2:477–489. https://doi.org/10.3233/JHD-130076
Barrios FA, Gonzalez L, Favila R, Alonso ME, Salgado PM, Diaz R, Fernandez-Ruiz J (2007) Olfaction and neurodegeneration in HD. Neuroreport 18:73–76. https://doi.org/10.1097/WNR.0b013e3280102302
Braak H, Braak E (1992) Allocortical involvement in Huntington’s disease. Neuropathol Appl Neurobiol 18:539–547
Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC (2017) Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a024240
Jones L, Hughes A (2011) Pathogenic mechanisms in Huntington’s disease. Int Rev Neurobiol 98:373–418. https://doi.org/10.1016/B978-0-12-381328-2.00015-8
Kalathur RKR, Pedro Pinto J, Sahoo B, Chaurasia G, Futschik ME (2017) HDNetDB: a molecular interaction database for network-oriented investigations into Huntington’s disease. Sci Rep 7:5216. https://doi.org/10.1038/s41598-017-05224-0
Saudou F, Humbert S (2016) The biology of Huntingtin. Neuron 89(5):910–926. https://doi.org/10.1016/j.neuron.2016.02.003
Labbadia J, Morimoto RI (2013) Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci 38(8):378–385. https://doi.org/10.1016/j.tibs.2013.05.003
Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, Hughes RE (2007) Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet 3(5):e82
Culver BP, Savas JN, Park SK, Choi JH, Zheng S, Zeitlin SO, Yates JR 3rd, Tanese N (2012) Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem 28:21599–21614. https://doi.org/10.1074/jbc.M112.359307
Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, Ross CA (2012) Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 11:2006–2021. https://doi.org/10.4161/cc.20423
Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, Yang XW (2012) Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 75:41–57. https://doi.org/10.1016/j.neuron.2012.05.024
Novak MJU, Seunarine KK, Gibbard CR, Hobbs NZ, Scahill RI, Clark CA, Tabrizi SJ (2014) White matter integrity in premanifest and early Huntington’s disease is related to caudate loss and disease progression. Cortex J Devoted Study Nerv Syst Behav 52:98–112. https://doi.org/10.1016/j.cortex.2013.11.009
Macdonald V, Halliday G (2002) Pyramidal cell loss in motor cortices in Huntington’s disease. Neurobiol Dis 10:378–386
Thu DCV, Oorschot DE, Tippett LJ, Nana AL, Hogg VM, Synek BJ, Luthi-Carter R, Waldvogel HJ, Faull RLM (2010) Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain J Neurol 133:1094–1110. https://doi.org/10.1093/brain/awq047
Harrington DL, Rubinov M, Durgerian S, Mourany L, Reece C, Koenig K, Bullmore E, Long JD, Paulsen JS, PREDICT-HD investigators of the Huntington Study Group, Rao SM (2015) Network topology and functional connectivity disturbances precede the onset of Huntington’s disease. Brain J Neurol 138:2332–2346. https://doi.org/10.1093/brain/awv145
Seiss E, Praamstra P, Hesse CW, Rickards H (2003) Proprioceptive sensory function in Parkinson’s disease and Huntington’s disease: evidence from proprioception-related EEG potentials. Exp Brain Res 148:308–319. https://doi.org/10.1007/s00221-002-1291-6
Cheng C-H, Soong B-W, Hsu W-Y, Lin Y-Y (2014) Reduced automatic frontal response to auditory deviance in Huntington’s disease as indexed by magnetic mismatch negativity. J Clin Neurosci Off J Neurosurg Soc Australas 21:1773–1778. https://doi.org/10.1016/j.jocn.2014.01.019
Ruocco HH, Bonilha L, Li LM, Lopes-Cendes I, Cendes F (2008) Longitudinal analysis of regional grey matter loss in Huntington disease: effects of the length of the expanded CAG repeat. J Neurol Neurosurg Psychiatry 79:130–135. https://doi.org/10.1136/jnnp.2007.116244
Labuschagne I, Cassidy AM, Scahill RI, Johnson EB, Rees E, O’Regan A, Queller S, Frost C, Leavitt BR, Dürr A, Roos R, Owen G, Borowsky B, Tabrizi SJ, Stout JC, TRACK-HD Investigators (2016) Visuospatial processing deficits linked to posterior brain regions in premanifest and early stage Huntington’s disease. J Int Neuropsychol Soc JINS 22:595–608. https://doi.org/10.1017/S1355617716000321
van den Bogaard SJA, Dumas EM, Ferrarini L, Milles J, van Buchem MA, van der Grond J, Roos R (2011) Shape analysis of subcortical nuclei in Huntington’s disease, global versus local atrophy—results from the TRACK-HD study. J Neurol Sci 307:60–68. https://doi.org/10.1016/j.jns.2011.05.015
Majid DSA, Aron AR, Thompson W, Sheldon S, Hamza S, Stoffers D, Holland D, Goldstein J, Corey-Bloom J, Dale AM (2011) Basal ganglia atrophy in prodromal Huntington’s disease is detectable over one year using automated segmentation. Mov Disord Off J Mov Disord Soc 26:2544–2551. https://doi.org/10.1002/mds.23912
Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel J-PG, Faull RLM (2015) The neuropathology of Huntington’s disease. Curr Top Behav Neurosci 22:33–80. https://doi.org/10.1007/7854_2014_354
de Azevedo PC, Guimarães RP, Piccinin CC, Piovesana LG, Campos LS, Zuiani JR, Tamashiro EM, Pinheiro G, Amato-Filho AC, Cendes F, Lopes-Cendes I, D’Abreu A (2017) cerebellar gray matter alterations in huntington disease: a voxel-based morphometry study. Cerebellum Lond Engl 16:923–928. https://doi.org/10.1007/s12311-017-0865-6
Rüb U, Hentschel M, Stratmann K, Brunt E, Heinsen H, Seidel K, Bouzrou M, Auburger G, Paulson H, Vonsattel J-P, Lange H, Korf H-W, den Dunnen W (2014) Huntington’s disease (HD): degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol Zurich Switz 24:247–260. https://doi.org/10.1111/bpa.12115
Fielding J, Georgiou-Karistianis N, Bradshaw J, Millist L, Churchyard A, Chiu E, White O (2004) Impaired modulation of the vestibulo-ocular reflex in Huntington’s disease. Mov Disord Off J Mov Disord Soc 19:68–75. https://doi.org/10.1002/mds.10658
Acknowledgements
Efforts for this article were supported by grants from the National Institutes of Health (R21 AG038772 to V. Hook) and the Huntington’s Disease Society of America (to S. Podvin, C. Mosier, and V. Hook). H. Reardon was supported by NIH training Grant T32 DA07315 (awarded to V. Hook).
Author information
Authors and Affiliations
Contributions
SP and HTR conducted the literature search and wrote drafts of the manuscript as advised by VH. CM also conducted literature searches. VH organized the topics covered by this review with HTR, SP, and KY. Figures and Tables were compiled by HTR, SP, KY, and VH. The manuscript text was written jointly by SP, HTR, and VH. SP, HTR, KY, and VH edited the manuscript. All authors proofed the manuscript for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Podvin, S., Reardon, H.T., Yin, K. et al. Multiple clinical features of Huntington’s disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration. J Neurol 266, 551–564 (2019). https://doi.org/10.1007/s00415-018-8940-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8940-6