
Abstract
Elevated cerebrospinal fluid (CSF), Neurofilament Light (NF-L) and phosphorylated Heavy (pNF-H) chain levels have been found in Amyotrophic Lateral Sclerosis (ALS), with studies reporting a correlation of both neurofilaments (NFs) with the disease progression. Here, we measured NF-L and pNF-H concentrations in the CSF of ALS patients from a single tertiary Center and investigated their relationship with disease-related variables. A total of 190 ALS patients (Bulbar, 29.9%; Spinal, 70.1%; M/F = 1.53) and 130 controls with mixed neurological diseases were recruited. Demographic and clinical variables were recorded, and ΔFS was used to rate the disease progression. Controls were divided into two cohorts: (1) patients with non-inflammatory neurological diseases (CTL-1); (2) patients with acute/subacute inflammatory diseases and tumors, expected to lead to significant axonal and tissue damage (CTL-2). For each patient and control, CSF was taken at the time of the diagnostic work-up and stored following the published guidelines. CSF NF-L and pNF-H were assayed with commercially available ELISA-based methods. Standard curves (from independent ELISA kits) were highly reproducible for both NFs, with a coefficient of variation < 20%. We found that CSF NF-L and pNF-H levels in ALS were significantly increased when compared to CTL-1 (NF-L: ALS, 4.7 ng/ml vs CTL-1, 0.61 ng/ml, p < 0.001; pNF-H: ALS, 1.7 ng/ml vs CTL-1, 0.03 ng/ml, p < 0.0001), but not to CTL-2. Analysis of different clinical and prognostic variables disclosed meaningful correlations with both NF-L and pNF-H levels. Our results, from a relatively large ALS cohort, confirm that CSF NF-L and pNF-H represent valuable diagnostic and prognostic biomarkers in ALS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurofilaments light chain (NF-L) and heavy chain (NF-H) are important, neuron-specific, cytoskeletal proteins present in the cell bodies and axons, which ensure structural stability and axonal polarization of these cells [1]. They are encoded by two independent genes, located on chromosome 8p21 and 22q12.2, respectively [1, 2].
Growing evidence indicates that NF-L and phosphorylated NF-H (pNF-H) are non-specific markers of axonal damage, which are reported to be increased in cerebrospinal fluid (CSF) and blood of several neurodegenerative disorders [1, 3,4,5,6,7,8,9,10,11,12,13,14,15,16]. In particular, increased CSF and blood NF-L levels have been proposed as diagnostic markers for atypical parkinsonian disorders [15, 16], Alzheimer disease [14], frontotemporal dementia [6, 8, 12, 13], HIV-related neurodegeneration [17].
Among the neurodegenerative diseases, amyotrophic lateral sclerosis (ALS) is characterized to be a crippling, severely disabling and rapidly progressive disorder, due to the degeneration of the upper and lower motor neurons. Survival is relatively short, being some 3 years from symptom onset [18].
A biological marker for ALS is, therefore, highly needed, both for diagnosis (about 7% of ALS diagnoses actually turn out to be other diseases [19]) and for monitoring the disease progression. ALS diagnosis and evaluation of disease progression are in fact mainly based on a clinical ground [20, 21]. Furthermore, a biomarker for ALS might represent a useful quantitative end-point for clinical trials.
Given the evidence that cortical (upper) motor neurons (UMN) and bulbar/spinal (lower) motor neurons (LMN) have relatively long axons, much effort has been devoted to assay the levels of NF-L and/or pNF-H in the patients’ biological fluids (i.e., CSF and blood), as markers of axonal degeneration in ALS [2, 11, 22].
Several reports have found that both neurofilaments are increased in the CSF and blood [3, 23,24,25,26,27,28,29,30,31,32,33,34,35], leading to the suggestion that these intermediate filaments might represent useful biomarkers to differentiate ALS from either ALS-mimics [23, 27, 31] or other neurological and neurodegenerative diseases [3, 11, 24, 26, 32]. Furthermore, it has been shown that NF-L might have prognostic relevance, as the increased blood and CSF levels seem to correlate with a shorter survival [29, 33,34,35]. These data raised the key question as to whether blood and/or CSF neurofilament light chain and phosphorylated heavy-chain proteins are now ready to enter into the clinic as: (1) biomarkers for ALS diagnosis, and (2) biochemical outcome measures in clinical trials [11, 36].
In this retrospective study, we assessed the diagnostic relevance of CSF NF-L and pNF-H levels by comparing a relatively large cohort of ALS patients, from a single tertiary center, with controls affected by other neurological diseases.
Controls were further divided into two cohorts, i.e., (1) patients with different non-acute and/or progressive diseases affecting the Peripheral (PNS) or the Central Nervous System (CNS); and (2) patients with acute/subacute inflammatory diseases of the nervous system and brain tumors/metastases, which are expected to lead to significant axonal and tissue damage.
Patients and methods
All patients involved in this study (ALS and disease controls) underwent a lumbar puncture during the diagnostic work-up. An informed written consent was signed according to the current guidelines of our University Hospital “P Giaccone”, Palermo. The informed consent contains a statement that “the biological material may also be used for research purposes”. The study protocol was approved by our institutional Ethics Committee. All the clinical and biological assessments were carried out in accordance with the World Medical Association Declaration of Helsinki.
ALS patients and disease controls
This study included 190 sporadic ALS patients (of which 10 were associated with frontotemporal dementia, ALS-FTD) and 130 controls affected by other neurological disorders. Enrollment was performed in two cohorts during the diagnostic work-up at the ALS Clinical Research Center, Department of Experimental Biomedicine and Clinical Neurosciences (BioNeC), University of Palermo, Italy, between 2006 and 2016.
All ALS patients underwent genetic testing for the major ALS-related genes, i.e., FUS, TARDBP, SOD1, Angiogenin, C9orf72. Patients with familial ALS and/or with mutation/expansions of the above genes were not included in this study.
Clinical follow-up of the control subjects was performed for at least 6 months. ALS patients were regularly followed up at the ALS Clinical Research Center with periodic visits at 3–6 months interval.
Patients with ALS were diagnosed according to El-Escorial revised criteria [20], complemented by the Awaji neurophysiological criteria [37]. In patients with associated ALS-FTD, the dementing disease was diagnosed according to the current consensus criteria [38, 39].
Severity of symptoms was scored with the revised version of the ALS Functional Rating Scale (ALSFRS-R) [21], where the higher score (48 points) indicates the absence of functional deficits and the lower score (0 points) defines a locked-in patient with permanent mechanical ventilation. Disease progression was evaluated with the ΔFS, identified by the following formula: (ALSFRS-R at onset − ALSFRS-R at time of diagnosis)/diagnostic delay (months) [40]. According to the ΔFS, three rates of progression could be calculated, i.e., slow (ΔFS < 0.5), intermediate (ΔFS ≥ 0.5–< 1), and rapid (ΔFS ≥ 1). Seated Forced Vital Capacity (FVC %) was used to assess respiratory function.
ALS patients were submitted to lumbar puncture and cerebrospinal fluid analysis as routine procedure of the diagnostic work-up. All functional evaluations were made at the time of the diagnostic work-up, which in our ALS population occurs some 1 year after the clinical onset [41].
Controls were patients with heterogeneous neurological disorders, who were also submitted to lumbar puncture and cerebrospinal fluid analysis during their diagnostic work-up. We did not include in this study patients with Alzheimer disease and other dementias, Parkinson disease and atypical parkinsonism, and adult-onset neurogenetic disorders (e.g., spinocerebellar ataxias, Huntington disease, hereditary spastic paraplegia, etc.).
The control cohort was divided into two groups, according to pathogenesis: (1) the control group 1 (CTL-1, n = 82) includes different non-inflammatory, non-acute onset neurological disorders (see: S-Table 1 for a full list). Note that CTL-1 includes patients with ALS-mimic diseases (in Italic in the list); (2) the control cohort 2 (CTL-2, n = 48) is formed by patients with acute/subacute inflammatory disorders and tumors/metastases of the nervous system (see: S-Table 2 for a full list). These disorders usually give significant neuronal/axonal death or degeneration and, thus, an increased level of cytoskeletal proteins is expected.
Demographic and clinical characteristics of the ALS patients and the two control cohorts are presented in Table 1.
For subgroup biomarker analysis, patients with ALS were divided into three phenotypes, according to the site of onset (spinal vs bulbar; the term spinal-onset includes also patients with respiratory-onset and generalized-onset) and to the presence of an associated frontotemporal dementia. The demographic and clinical characteristics of the spinal-onset ALS (ALS-S), bulbar-onset ALS (ALS-B), and patients with associated ALS-FTD are shown in Table 2. Note that in the ALS-FTD group, which included eight ALS-S and two ALS-B, the dementia always preceded the onset of motor symptoms.
A cognitive and/or behavioral impairment was, however, present in some 40% of our non-demented ALS population (Spataro et al., unpublished results), a result consistent with the current literature [42,43,44,45].
CSF collection and analysis
CSF was collected between 8:00 a.m. and 10:00 a.m. by lumbar puncture during the diagnostic work-up from both fasted ALS patients and disease controls. Each sample was processed within 1 hr from collection by centrifugation, and then aliquoted in small polypropylene tubes following standard procedures [46]. All aliquots were coded to ensure anonymity, and stored at – 80 °C until further analysis. The collected CSFs from ALS and control patients underwent routine analysis, which included cell (leukocyte) count (cells/mm3), total protein (mg/dl) and glucose (mg/dl) quantification, the CSF/serum albumin concentration ratio (Qalb), and evaluation of oligoclonal bands.
The CSF parameters of the ALS patients and the two control cohorts are shown in Table 3. While the glucose level was comparable between groups, the total protein, the Qalb and the number of cells (lymphocytes) were significantly higher in the CTL-2 group.
Neurofilament assays
Single-batch ELISA kits from two different commercial sources were used for the NF-L assays (i.e., MyBioSource, San Diego, USA and UmanDiagnostics AB, Umeå, Sweden).
For pNF-H determinations, we used a single-batch ELISA kit from BioVendor Research and Diagnostic Product, Czech Republic.
Analyses were performed according to each manufacturer’s instructions.
All samples from ALS patients and disease controls (CTL-1 and CTL-2) were coded, so that the analyst was unaware of any patient specific clinical and demographic data.
Determination of NF-L
Samples were diluted 1:2 and run in duplicate, together with freshly prepared standards. For both ELISA kits, absorbance measurements were carried out at 450 nm with a reference wavelength set at 630 nm, using a EuroClone plate reader.
Determination of pNF-H
Samples were diluted 1:3 and 1:10 with dilution buffer, and run in duplicate, together with freshly prepared standards. Absorbance values were determined by reading the plates at 450 nm, with the reference wavelength set to 630 nm, using a BioTek plate reader.
Calculation of the neurofilament levels and quality control tests
CSF NF-L and pNF-H levels for all groups were calculated by referring to the corresponding standard curves. Reproducibility of the results was assessed by calculating the average coefficient of variation (CV) within plates and between plates.
The mean intra-assay CV was < 10%, whereas the inter-assay CV was < 15% for both NF-L and pNF-H.
Statistical analysis
Analysis of data was carried out with SIGMAPLOT 12.0 software package (Systat Software Inc., San Jose, CA, USA), GRAPHPAD PRISM 5.01 software (GraphPad Inc., La Jolla, CA, USA), and XLSTAT 2017 software (Addinsoft Inc., New York, NY, USA).
Receiver Operating Characteristic (ROC) curves were used to calculate the sensitivity and specificity of the NF-L and pNF-H data in the CSF of ALS patients vs different control subgroups (i.e., CTL-1, CTL-2, ALS-mimics). The optimal cutoff was calculated with the Youden Index. For each cutoff, sensitivity, specificity, the area under curve (AUC) with 95% confidence interval (CI), the likelihood ratio (LR) and the predictive values were analyzed with XLSTAT 2017 software.
Demographic and biochemical variables and neurofilament levels in ALS and control groups were expressed as median with interquartile ranges (IQR).
Nonparametric data comparisons were performed using Mann–Whitney Rank Sum Test or, where appropriate, with the Kruskal–Wallis One-Way Analysis of Variance on Ranks.
Differences between groups were evaluated using the Chi square test. Survival data were expressed as median with IQR. Survival analysis was restricted to cases with survival time ≤ 120 months, to better represent the ALS population, and performed with the Kaplan–Meier method. Survival curves were compared with the Log-Rank test.
All correlations were analyzed with Spearman’s Rank Correlation Coefficient.
p values < 0.05 were considered significant.
Results
The concentrations of NF-L and pNF-H were assessed in the CSF of ALS patients and of a relatively large cohort of control patients, with heterogeneous neurological diseases. The control cohort was further divided into two main subgroups, according to the disease etiopathogenesis (see “Patients and methods” section).
While all assessments of pNF-H were carried with the same ELISA kit (i.e., Biovendor), CSF NF-L levels were initially assayed using a commercial ELISA Kit from MyBioSource. Preliminary evaluation of this kit showed a very good reproducibility for both the standard curve and the sample analysis (S-Table 3 and S-Table 4). However, we were surprised to detect no differences in CSF NF-L concentrations between ALS and the two control groups (Table 4).
CSF pNF-H levels were instead increased in ALS with respect to both CTL-1 and CTL-2 controls; however, only the difference in the pNF-H levels between ALS and CTL-1 reached significance [ALS: 1.7 ng/ml (IQR = 0.76–3.17) vs CTL-1: 0.03 ng/ml (IQR = 0.00–0.31); p < 0.05, Kruskal–Wallis One-Way ANOVA on Ranks with post hoc Dunn’s analysis, Table 4].
The evidence that CSF NF-L levels in ALS, assayed with the MyBioSource ELISA kit, were not significantly different from the two control groups was in striking contrast with previously published works, carried out with different ELISA kits, both home-made or from commercial sources [3, 10, 11, 22,23,24, 26, 30, 31, 33,34,35]. In particular, the commercial UmanDiagnostics ELISA kit (producer: UmanDiagnostics AB, Umeå, Sweden; distributor: IBL, Hamburg, Germany) was the most frequently chosen to quantify NF-L [16, 24, 30,31,32, 34, 35].
We, therefore, adopted the ELISA kit from UmanDiagnostics AB to replicate the NF-L assessments in the CSF of our ALS patients and in the two control disease groups. The results were completely different because, using this tool, the median NF-L levels were significantly higher in the ALS cohort when compared to the CTL-1 group [ALS: 4.7 ng/ml (IQR = 1.18–7.98) vs CTL-1, 0.61 ng/ml (IQR = 0.31–2.67); p < 0.05, Kruskal–Wallis One-Way ANOVA on Ranks with post hoc Dunn’s analysis, Table 4]. Conversely, no significant differences were found in the NF-L levels between ALS and CTL-2.
These data suggest that neurofilaments are not reliable biomarkers when ALS is compared to diseases in which an acute/subacute neuronal/axonal damage or death is a main feature.
A further remarkable conclusion from these experiments is that NF-L levels in the CSF may greatly vary in relationship to the ELISA kit used to make the assay. We postulate that the discrepancy we found between kits may be ascribed to the different affinity of the kit-specific antibodies for NF-L protein.
Given that the UmanDiagnostics ELISA kit for the CSF NF-L assessment gave results consistent with the published literature [16, 24, 34, 35], all subsequent experiments were, therefore, carried out with this commercial tool.
To supplement the analysis of CSF neurofilaments in ALS, we measured NF-L and pNF-H levels in patients divided into two subgroups according to the site of onset (i.e., ALS-S and ALS-B). ALS-FTD was analyzed as a separate cohort. Table 5 shows that CSF neurofilaments do not discriminate ALS patients according to the site of onset, or to the presence of an associated dementia. Nevertheless, both neurofilament levels appeared lower in the ALS-FTD group, though the difference did not reach significance.
As the diagnostic delay (DD) in ALS-FTD is significantly longer than in ALS-S or ALS-B (Table 2), we hypothesized that the lower levels of neurofilaments in this ALS subgroup might be related to this variable. The Spearman correlation analysis actually confirmed the existence of a small, however, significant, inverse correlation between the diagnostic delay and CSF neurofilaments (DD vs NF-L: r = − 0.20, p = 0.006; DD vs pNF-H: r = − 0.25, p = 0.0006).
CSF NF-L and pNF-H levels were then studied in ALS according to different demographic and clinical variables. As shown in Table 6, both neurofilament levels were unrelated to age or sex. However, we observed a clear-cut relationship between rate of disease progression, as measured by ΔFS [40], and both NF-L and pNF-H levels. ALS patients were categorized into three subgroups, according to ΔFS, i.e., slow (< 0.5), intermediate (≥ 0.5–< 1), and rapid (≥ 1) progression, which was found to inversely correlate with survival in our cohort (S-Fig. 1). For both neurofilaments, CSF levels were significantly different in the three progression cohorts, with higher levels found in the rapid progressing patients (Table 6). The Spearman correlation analysis further confirmed the positive relationship between ΔFS and CSF NF-L and pNF-H levels (NF-L, r = 0.312, p = 0.00001; pNF-H, r = 0.315, p = 0.00001, Fig. 1).

Correlation between CSF neurofilament levels and disease progression (∆FS). Disease progression positively correlated with both CSF NF-L (a) and pNF-H (b) levels. NF-L and pNF-H data are expressed in ng/ml and ∆FS is determined with the following formula: (ALSFRS-R at onset − ALSFRS-R at time of diagnosis)/diagnostic delay (months). Correlation is performed using the Spearman Rank Correlation Coefficient and linear regression analysis
Taken together, the above results strongly suggest that both CSF neurofilaments may have a prognostic value, the higher levels at diagnosis being predictive of a relatively short survival.
To further confirm this hypothesis, we performed survival curves of ALS patients dichotomized according to the median CSF values of NF-L or pNF-H. Kaplan–Meier curves showed that survival is significantly shorter for patients with higher CSF NF-L or pNF-H levels at diagnosis (Fig. 2). Note that the CSF NF-L and pNF-H levels in ALS showed a positive correlation (NF-L vs pNF-H, n = 190, r = 0.70, p = 0.0000002, S-Fig. 2), indicating that a patient with a high level of one neurofilament is likely to have a high level of the other neurofilament protein too.

CSF NF-L and pNF-H levels at diagnosis have a prognostic value in ALS. Kaplan–Meier survival curves of ALS patients stratified according to the median CSF values of NF-L (4.7 ng/ml) or pNF-H (1.7 ng/ml) at diagnosis. Statistical analysis shows a significantly shorter survival of patients with higher CSF NF-L or pNF-H levels (p < 0.001; Log-Rank test)
We conclude that both CSF NF-L and pNF-H levels at diagnosis have a prognostic value in ALS, and they might be adopted as important surrogate biomarkers in clinical trials.
The CTL-1 group included also some ALS-mimics, that is, diseases which may enter into the differential diagnosis with ALS (i.e., cervical spondylotic myelopathy, motor neuropathy/plexopathy, multifocal motor neuropathy, S-Table 1). CSF neurofilament levels of patients with ALS were, therefore, compared with the CTL-1 subgroup of ALS mimics.
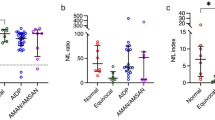
As shown in Fig. 3, the CSF levels for both neurofilaments were once again higher in ALS than in the ALS-mimics (Fig. 3a, NF-L: ALS, 4.7 ng/ml [IQR = 1.18 − 7.98] vs ALS-mimics, 1.11 ng/ml [IQR = 0.46 − 4.60], p < 0.001, Mann–Whitney Rank Sum Test; Fig. 3b, pNF-H: ALS, 1.70 ng/ml [IQR = 0.76 − 3.17] vs ALS-mimics, 0.30 ng/ml [IQR = 0.00 − 1.23], p < 0.001, Mann–Whitney Rank Sum Test).

CSF NF-L and pNF-H levels represent useful biomarkers to differentiate ALS from ALS-mimic patients. ALS-mimics are included in the CTL-1 control group and show diseases that may enter into the differential diagnosis of ALS (i.e., cervical spondylotic myelopathy, motor neuropathy, multifocal motor neuropathy, see S-Table 1 for more details). CSF NF-L (a) and pNF-H (b) levels are significantly higher in ALS patients when compared to the ALS-mimics group. Data are expressed in ng/ml [NF-L: ALS: 4.7 ng/ml (IQR = 1.18–7.98), ALS-mimics: 1.11 ng/ml (IQR = 0.46–4.60) p < 0.001; pNF-H: ALS: 1.70 ng/ml (IQR = 0.76–3.17), ALS-mimics: 0.30 ng/ml (IQR = 0.00–1.23), p < 0.001, Mann–Whitney Rank Sum Test]
To study the sensitivity and specificity of the observed differences in CSF neurofilament levels between ALS and controls, we performed several ROC analyses. First, we asked which cutoff value of both CSF neurofilaments would better discriminate between ALS and non-inflammatory/non-tumor neurological diseases (CTL-1).
The ROC curves shown in Fig. 4 indicate that the best cutoff value for neurofilament levels to distinguish the two groups is as follows: (1) NF-L, 1.838 ng/ml with an AUC of 0.775 CI (0.713–0.837), sensitivity 76.3% and specificity 72.8% and a Youden index of 0.49 (Fig. 4a); (2) pNF-H, 0.460 ng/ml with an AUC of 0.869 CI (0.814–0.924), sensitivity 84% and specificity 82.9% and a Youden index of 0.669 (Fig. 4b).

ROC curves of CSF NF-L and pNF-H to discriminate between ALS and CTL-1 patients. ROC curve analyses have been performed to determine levels of CSF neurofilaments that best differentiate ALS versus non-inflammatory/non-tumor neurological diseases (CTL-1). Data are expressed in ng/ml; AUC area under the curve, CI 95% confidence interval, NPV negative predictive value, PPV positive predictive value
The ROC values for the two neurofilament biomarkers to discriminate ALS vs all different control subgroups (i.e., CTL-1, CTL-2, and ALS-mimics) are instead summarized in Table 7. Both NF-L and pNF-H cutoff values yielded a grading of AUC, sensitivity and specificity, in which differentiation of ALS from controls was as follows: CTL-1 > ALS-mimics > CTL-2.
We conclude that both neurofilaments show a relatively good performance as diagnostic biomarkers for ALS to discriminate this motor neuron disorder from non-inflammatory/non-degenerative/non-tumor disease controls (CTL-1) and, with less extent, from ALS-mimics. pNF-H appears slightly more efficient than NF-L.
Discussion
Our study aimed to characterize the diagnostic and prognostic performance of the CSF levels of two neurofilaments in ALS, i.e., NF-L and pNF-H.
We demonstrated that both cytoskeletal proteins are significantly increased in the CSF of ALS patients with respect to controls affected by different neurological diseases. In particular, NF-L and pNF-H were able to efficiently discriminate ALS vs controls affected by other neurological disorders, not specifically linked to progressive neuronal cell death/axonal damage or acute inflammation (i.e., the CTL-1 cohort). pNF-H showed better sensitivity and specificity than NF-L, with a higher AUC (0.869 pNF-H vs 0.775 NF-L).
These results add to the growing literature in the field [3, 10, 11, 22,23,24, 26, 30, 31, 33,34,35], and they strongly support a role for these two cytoskeletal proteins as very promising biomarkers in ALS.
When ALS patients were compared to disease controls with significant neuronal cell death/axonal damage due to different causes (i.e., tumors, metastases, inflammation; the CTL-2 cohort), the ability of the CSF neurofilaments to discriminate between the two groups was greatly reduced. The median CSF levels of NF-L and pNF-H were in fact not different between ALS and CTL-2, with the ROC analysis showing reduced specificity and AUC values for both neurofilaments.
This clearly demonstrates that neurofilaments are non-specific markers of those diseases characterized by neuronal cell death/axonal degeneration, especially when evolving at a relatively fast pace. ALS is a disease whose natural history is generally shorter than other neurodegenerative disorders, like Alzheimer disease or Parkinson disease. Only few other neurodegenerative diseases, e.g., multisystem atrophy and prion encephalopathies, show a disease progression comparable to, or even more aggressive than, ALS. In these disorders, growing evidence show that CSF neurofilaments and other cytoskeletal proteins, like the Tau protein, are strongly increased [16, 47, 48].
Neurofilament light-chain and phosphorylated heavy-chain proteins can, therefore, be considered biomarkers of neuronal degeneration and death, especially when the underlying pathological process evolves rapidly. When the neurodegenerative process causing neuronal/axonal damage is relatively slow, the impact of the neurofilaments as biomarkers is likely to be minor. In this context, the CSF neurofilament levels are not strictly related to the number of neurons involved in the pathological process.
CSF NF-L and pNF-H levels in the ALS cohort were also higher when confronted to ALS-mimics, with a sensitivity, specificity and AUC roughly comparable to the disease controls. Other reports found similar outcomes [31, 34, 35]. pNF-H showed, however, a better performance than NF-L in discriminating ALS from ALS-mimics.
ALS-mimics are neurological diseases that are frequently involved in the differential diagnosis of ALS [19]. Among them, cervical spondylosis and cervical spondylotic myelopathy, motor mono–poly-neuropathies, plexopathies and multifocal motor neuropathy seem the most challenging [19, 49]. In this work, we did not include other motor neuron diseases, like adult-onset lower motor neuron disease, bulbo-spinal muscular atrophy, hereditary spastic paraparesis, primary lateral sclerosis, etc., which would require a dedicated study.
CSF NF-L and pNF-H are, therefore, promising ALS biomarkers that can support the diagnosis, provided that the shared guidelines for ALS diagnosis are followed [20, 37, 50]. It remains, however, an open question whether CSF and/or blood neurofilaments are biological markers predictive of an incoming disease onset in asymptomatic carriers [30].
In our ALS cohort, both CSF neurofilaments were not able to discriminate between ALS phenotypes (i.e., spinal-onset vs bulbar-onset vs ALS-FTD). Furthermore, age at onset or sex did not show significant relationship with both CSF NF-L and pNF-H levels. Similar observations have been made in other studies [24, 29, 31, 35]. These analyses were made at diagnosis, and all patients were in a mild–moderate stage of disability (the median ALSFRS-R of our ALS cohort was 38/48; range 30–42). This further confirms that neurofilaments are non-specific markers for the intrinsic mechanism of neuronal degeneration, which is likely to be unaffected by demographic variables.
The severity of the disease, in terms of rate of progression, is instead strictly related to both neurofilament levels, in that the more rapid the disease evolution, as measured by ΔFS [40], the higher the NF-L and pNF-H levels in the CSF. As an indirect support to this result, we showed that high levels of both CSF neurofilaments at diagnosis were associated with a reduced survival. This strongly suggest that both NFs have a potential as prognostic biomarkers of the disease [11, 24, 29,30,31, 35].
It is now a matter of fact that ALS is highly heterogeneous, with different phenotypes [51] and, more important, with different rates of progression [40, 52]. Both variables are in part implicated in the failure of the most recent clinical trials in the disease [53].
There is suggestion that future clinical trials should be performed by selecting more homogeneous groups of patients, with the aim to move towards a more personalized therapy. The early randomized clinical trial with riluzole found a better survival effect in bulbar–onset patients [54], and a recent pilot clinical trial with tauroursodeoxycholic acid enrolled only spinal-onset ALS patients to reduce phenotypic variability [55]. Along with ΔFS, which allows the grouping of patients according to their rate of progression [40], CSF neurofilaments appear at present the best prognostic biomarkers in ALS. A recent commentary [36] and a meta-analysis [11] have in fact advocated the potential usefulness of CSF neurofilament levels as potential outcome measure in clinical trials.
A question raises as to whether both neurofilament light-chain and phosphorylated heavy-chain proteins should stably enter in the ALS clinic [11, 36]. The results from our work represent a further support to it.
Our study unveiled, however, a relevant issue that must be considered when using commercial ELISA kits for a biomarker assays. We found a big variability between commercial kits.
For neurofilament light-chain assay, we initially adopted an ELISA kit from MyBioSource (San Diego, USA), which gave excellent standard curve and high reproducible data. However, we did not found differences between ALS and disease controls, with a range of CSF NF-L levels nonetheless similar to those reported in other works [24, 29,30,31, 33]. These results appeared skewed with respect to the wide published literature on the field, and raised the suspicion that the antibody used for this ELISA kit was either at low affinity for his ligand or it was cross-reacting with other molecules in the CSF.
A rapid survey of the literature showed that several research groups were using home-made kits [3, 23] or, more often, a commercial kit by UmanDiagnostics AB (Umeå, Sweden) purchased either directly from the producer [24, 29, 33,34,35] or from the distributor IBL (Hamburg, Germany) [30,31,32]. Some works reported using an electrochemioluminescence detection to increase the sensitivity of the assay, especially in blood samples [26, 29, 33]. The S-TAB 5 summarizes the different ELISA systems used by the different research groups with the corresponding ROC curves (ALS vs controls) for both neurofilaments.
The replication ELISA assay with the UmanDiagnostics AB kit on the same CSF samples from our ALS cohort and disease controls showed a completely different scenario, with a significant difference in NF-L levels between groups. Thus, the level of the NF-L protein in a biological fluid may strongly depend on the commercial kit and from the primary antibody used to detect the specific antigen. Note, however, that the two different commercial kits (i.e., MyBioSource and UmanDiagnostics) were both able to give highly reproducible standard curves and data.
There are many questions to be answered before neurofilaments can be routinely utilized as biological markers in the ALS clinic. In particular, multicenter studies should be performed with the aim to: (1) achieve a validation of these biomarkers; a recent work attempted a first validation for both neurofilaments light-chain and phosphorylated heavy-chain proteins [32], but the number of CSF samples provided by each participating center was small, and some assay variability was observed between the two analyzing centers; (2) make a comprehensive comparison between different commercial ELISA kits, to verify which one gives the best results in terms of specificity and sensitivity in order to distinguish ALS from disease controls and ALS-mimics; at present, from the published studies and the present research, the ELISA kit from UmanDiagnostics [24, 29,30,31,32,33,34,35], to assay NF-L, and from BioVendor, to assay pNF-H, appear the most promising candidates; (3) to evaluate the neurofilament biomarkers in subgroups of ALS patients, made homogeneous by phenotype and rate of disease progression. Given that CSF neurofilaments are non-specific biomarkers of cell death/axonal degeneration, it is important to clearly identify those ALS patients for whom the biomarker might have relevance both in diagnostic and prognostic terms.
In conclusion, we have shown that neurofilaments light-chain and phosphorylated heavy-chain proteins are increased in the CSF of ALS patients, the higher levels being associated with a more rapidly evolving disease and a shorter survival. Our results add to the recent growing literature supporting the two cytoskeletal proteins as very promising diagnostic and prognostic biomarkers for ALS. More multicenter validation studies are, however, needed before neurofilaments can definitely enter into the ALS clinic.
References
Lee MK, Cleveland DW (1996) Neuronal intermediate filaments. Annu Rev Neurosci 19:187–217
Petzold A (2005) neurofilament phosphoforms: surrogate markers of axonal injury, degeneration and loss. J Neurol Sci 233:183–198
Rosengren LE, Karlsson J-E, Karlsson J-O et al (1996) Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem 67:2013–2018
Hu YY, He SS, Wang XC et al (2002) Elevated levels of phosphorylated neurofilament proteins in cerebrospinal fluid of Alzheimer disease patients. Neurosci Lett 320:156–160
Pijnenburg YAL, Janssen JC, Schoonenboom NSM et al (2007) CSF neurofilaments in frontotemporal dementia compared to early onset Alzheimer’s disease and controls. Dement Geriatric Cogn Dis 23:225–230
Mattsson N, Rüetschi U, Pijnenburg YAL et al (2008) Novel cerebrospinal fluid biomarkers of axonal degeneration in frontotemporal dementia. Mol Med Rep 1:757–761
Ganesalingam J, An J, Bowser R et al (2013) pNfH is a promising biomarker in ALS. Amyotrophic Lat Scler Frontotemp Degen 14:146–149
Landqvist Waldö M, Santillo AF, Passant U et al (2013) Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurology 13:54e1–54e8
Scherling CS, Hall T, Berisha F et al (2014) Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 75:116–126
Abdelhak A, Junker A, Brettschneider J et al (2015) Brain-specific cytoskeletal markers in cerebrospinal fluid: is there a common pattern between amyotrophic lateral sclerosis and primary progressive multiple sclerosis? Int J Mol Sci 16:17565–17588
Xu Z, Henderson RD, David M, McCombe PA (2016) Neurofilaments as biomarkers for amyotrophic lateral sclerosis: a systematic review and meta-analysis. PLoS ONE 11(e0164625):e1–e18
Meeter LH, Dopper EG, Jiskoot LC et al (2016) Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 3:623–626
Wilke C, Preische O, Deuschle C et al (2016) Neurofilament light chain in FTD is elevated not only in cerebrospinal fluid, but also in serum. J Neurol Neurosurg Psychiatry 87:1270–1272
Mattsson N, Andreasson U, Zettenberg H et al (2017) association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 74:557–566
Hu X, Yang Y, Gong D (2017) Cerebrospinal fluid levels of neurofilament light chain in multiple system atrophy relative to Parkinson’s disease: a meta-analysis. Neurol Sci 38:407–414
Hansson O, Janelidze S, Hall S et al (2017) Blood-based NfL. A biomarker for differential diagnosis of parkinsonian disorder. Neurology 88:930–937
Krut JJ, Mellberg T, Price RW et al (2014) Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS ONE 9(e88951):e1–e7
Spataro R, Lo Re M, Piccoli T et al (2010) Causes and place of death in Italian patients with Amyotrophic Lateral Sclerosis. Acta Neurol Scand 122:217–223
Traynor BJ, Codd MB, Corr B et al (2000) Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol 57:109–113
Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 1:293–299
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 169:13–21
Menke RAL, Gray E, Lu C-H et al (2015) CSF neurofilament light chain reflects corticospinal tract degeneration in ALS. Ann Clin Transl Neurol 2:748–755
Reijn TS, Abdo WF, Schelhaas HJ, Verbeek MM (2009) CSF neurofilament protein analysis in the differential diagnosis of ALS. J Neurol 256:615–619
Tortelli R, Ruggieri M, Cortese R et al (2012) Elevated cerebrospinal fluid neurofilament light levels in patients with amyotrophic lateral sclerosis: a possible marker of disease severity and progression. Eur J Neurol 19:1561–1567
Boylan KB, Glass JD, Crook JE et al (2013) Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as potential prognostic biomarker in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 84:467–472
Gaiottino J, Norgren N, Dobson R et al (2013) Increased neurofilament light chain blood levels in neurodegenerative neurological disease. PLoS ONE 8(e75091):e1–e9
Gonçalves M, Tillack L, de Carvalho M et al (2014) Phosphoneurofilament heravy chain and N-glycomics from the cerebrospinal fluid in amyotrophic lateral sclerosis. Clin Chim Acta 438:342–349
Lu C-H, Petzold A, Topping J et al (2015) plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry 86:565–573
Lu C-H, Macdonald-Wallis C, Gray E et al (2015) Neurofilament light chain. A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84:2247–2257
Weydt P, Oeckl P, Huss A et al (2016) Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol 79:152–158
Steinacker P, Feneberg E, Weishaupt J et al (2016) Neurofilaments in the diagnosis of motoneuron diseases: a prospective study of 455 patients. J Neurol Neurosurg Psychiatry 87:12–20
Oeckl P, Jardel C, Salachas F et al (2016) Multicenter validation of CSF neurofilaments as diagnostic biomarkers for ALS. Amyotr Lat Scler Frontotemp Degen 17:404–413
Steinacker P, Huss A, Mayer B et al (2017) diagnostic and prognostic significance of neurofilament light chain NF-L, but not progranulin and S100B, in the course of amyotrophic lateral sclerosis: data from the German MND-net. Amyotr Lat Scler Frontotemp Degen 18:112–119
Gaiani A, Martinelli I, Bello L et al (2017) Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis. Neurofilament light chain levels in definite subtypes of disease. JAMA Neurol 74:525–532
Poesen K, De Schaepdryver M, Stubendorff B et al (2017) neurofilaments markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 88:2302–2309
Turner MR, Gray E (2015) Are neurofilaments heading for the ALS clinic? J Neurol Neurosurg Psychiatry 87:3–4
De Carvaho M, Dengler R, Eisen A et al (2008) Electrodiagnosis criteria for diagnosis of ALS. Clin Neurophysiol 119:497–503
Neary D, Snowden JS, Gustafson L et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Bang J, Spina S, Miller BL (2015) Frontotemporal dementia. Lancet 386:1672–1682
Kimura F, Fujimura C, Ishida S et al (2006) Progression rate of ALSFRS-R at the time of diagnosis predicts survival time in ALS. Neurology 66:265–267
Cellura E, Spataro R, Taiello AC, La Bella V (2012) Factors affecting the diagnostic delay in amyotrophic lateral sclerosis. Clin Neurol Neurosurg 114:550–554
Robinson KM, Lacey SC, Grugan P, Glosser G, Grossman M, McCluskey LF (2006) Cognitive functioning in sporadic amyotrophic lateral sclerosis: a six month longitudinal study. J Neurol Neurosurg Psychiatry 77:668–670
Phukan J, Pender NP, Hardiman O (2007) Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 6:994–1003
Kasper E, Zydatiss K, Schuster C, Machts J, Bittner D, Kaufmann J et al (2016) No change in executive performance in ALS patients: a longitudinal neuropsychological study. Neurodegener Dis 16:184–191
Burkhardt C, Neuwirth C, Weber M (2017) Longitudinal assessment of the Edinburgh Cognitive and Behavioural Amyotrophic Lateral Sclerosis Screen (ECAS): lack of practice effect in ALS patients. Amyotroph Lateral Sclerosis Frontotemp Degener 18:202–209
Teunissen CE, Petzold A, Bennett JL et al (2009) A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 73:1914–1922
Abdoa FW, Bloema BR, Van Geel WJ et al (2007) CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson’s disease. Neurobiol Aging 28:742–747
Steinacker P, Blennow K, Halbgebauer S et al (2016) Neurofilaments in blood and CSF for diagnosis and prediction of onset in Creutzfeldt-Jakob disease. Sci Rep 6:38737 e1–38737 e6
Kang DX, Fan DS (1995) The electrophysiological study of differential diagnosis between amyotrophic lateral sclerosis and cervical spondylotic myelopathy. Electromyogr Clin Neurophysiol 35:231–238
Andersen PM, Abrahams S, Borasio GD et al (2012) EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS) – revised report of an EFNS task force. Eur J Neurol 19:360–375
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nature Rev Neurol 10:661–670
Appel V, Stewart SS, Smith RG, Appel SH (1987) A rating scale for amyotrophic lateral sclerosis: description and preliminary experience. Ann Neurol 22:328–333
Mitsumoto H, Brooks BR, Silani V (2014) Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 13:1127–1138
Bensimon G, Lacomblez L, Meininger V, for the ALS/Riluzole study group (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. New Engl J Med 330:585–591
Elia AE, Lalli S, Monsurrò MR et al (2015) Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol 23:45–52
Acknowledgements
This work was supported by funds from the Italian Ministry of Health for the Ricerca Corrente to IRCCS Istituti Clinici Scientifici Maugeri SpA SB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest related to the Ms JOON-D-17-01506.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rossi, D., Volanti, P., Brambilla, L. et al. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J Neurol 265, 510–521 (2018). https://doi.org/10.1007/s00415-017-8730-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-017-8730-6