
Abstract
Myelin oligodendrocyte glycoprotein antibodies (MOG-Ab) have been reported in acute demyelinating encephalomyelitis (ADEM), optic neuritis (ON), and neuromyelitis optica spectrum disorders (NMOSD) in adults and pediatrics. We aimed to delineate the common features of MOG-Ab-related disorders in children and adults, and report uncommon presentations. Twenty-seven consecutive pediatric and adult patients testing positive for MOG-Ab, with a minimum follow-up of 6 months, were included. Comprehensive epidemiological, clinical, radiological, and laboratory data were retrospectively analyzed. Additionally, we compared radiological features between ADEM MOG-Ab-positive patients, and a group of ADEM MOG-Ab-negative ones, recruited during the same period. Among the whole cohort, 13 (48.1%) were pediatric, and 14 (51.9%) were female. MOG-Ab-related disorders comprised eight ADEM, eight ON, five isolated myelitis, four with NMOSD and two patients with multiple sclerosis, at last follow-up. After a median follow-up of 17.8 months, 11 (40.7%) patients presented a relapse. The most frequent clinical phenotype at onset was encephalopathy in pediatrics (53.9%) and myelitis in adults (50%) (p = 0.013). There were no other differences between both groups. When comparing ADEM MOG-Ab positive and negative patients, bilateral thalamic lesions were more often found in the positive group (p = 0.010). Unusual presentations were identified in three patients: patchy spinal cord gadolinium-enhancing lesions, an associated teratoma, and one presented with status epilepticus. MOG-Ab-related disorders shared common clinical and prognostic features, but encompass a spectrum wider than recently reported.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelin oligodendrocyte glycoprotein-auto-antibodies (MOG-Ab) have been mainly described in pediatric patients with acute demyelinating encephalomyelitis (ADEM) or optic neuritis (ON) [1,2,3,4,5], as well as pediatric and adults diagnosed with neuromyelitis optica spectrum disorders (NMOSD) without aquaporin-4 antibodies (AQP4-Ab) [6,7,8].
Patients presenting with demyelinating diseases and MOG-Ab in serum were initially characterized by a younger age at onset, lower risk to relapse and a more benign course than AQP4-Ab-positive ones [6,7,8]. However, recent studies with longer follow-up plead against these first described features [9, 10]. Clinical manifestations at onset are reported different between pediatric and adults patients: a higher predisposition to encephalopathy in children [11, 12], in comparison to ON or transverse myelitis in adults [13, 14]. However, most of the studies describing characteristics of MOG-Ab-positive patients usually mix features from adults and pediatrics, without having performed a direct comparison between both groups.
Some patients develop a different evolution with less common clinical or radiological features, or a fulminant course [9, 13, 15, 16]. Since MOG-Ab-related disorders are an emerging entity, there is a need for a deeper comprehensive description of atypical cases as the whole spectrum has likely not been fully described.
In the present study, we aimed to delineate the common features of MOG-Ab-related disorders between children and adults, and we report new uncommon presentations.
Methods
Patients
Between January 2014 and January 2016, 177 serum samples from adult and pediatric patients presenting with a suspected acquired demyelinating syndrome (ADS) admitted to Hôpital Neurologique Pierre Wertheimer and Hôpital Pédiatrique Mère Enfant of Lyon (France) were routinely analyzed for AQP4 and MOG-Ab by cell-based assay (CBA). All patients had a minimum of 6 months follow-up. We defined ADS as an acute clinical episode of the central nervous system (CNS) lasting for more than 24 h involving the optic nerve, brain, brainstem or spinal cord associated with T2 lesions on magnetic resonance imaging (MRI).
We registered epidemiological features such as age, gender, ethnicity, concomitant autoimmune diseases, infections or vaccinations 1 month prior to disease onset. We noted the topography of the first episode, neurological symptoms as well as the number and location of relapses. Relapse was defined as a new episode at any CNS structure at least 1 month after the first episode, and sustained at least 24 h in the absence of fever or infection.
Clinical disability at nadir of symptoms and last visit was measured by the Expanded Disability Status Scale (EDSS). An EDSS < 3.0 at last visit was considered as a good outcome. For ON, visual acuity (VA) was evaluated by the visual functional system, and severe visual disability was defined as VA ≤ 0.2 both at nadir and last visit.
Treatments at the acute phase and type of immunosuppressant were also registered. Analysis of brain and spinal cord MRI was performed by a specialized neuroradiologist blinded to the patient sero-status. MRI studies included axial and sagittal images of the brain and spinal cord obtained by T1-weighted (W), T2-W, FLAIR and T1-W post-contrast sequences. Brain MRI at first episode was classified based on Paty’s criteria and topography of lesions was noted: juxtacortical, periventricular, corpus callosum, putamen, thalamus, cerebellar peduncles and brainstem tegmentum.
Longitudinally extensive transverse myelitis (LETM) was defined as ≥3 contiguous vertebral segments in the sagittal plane. Available brain and spinal cord MRI follow-up studies were compared to the initial MRI, and were categorized in four subgroups: complete resolution, improvement, new lesions and no change.
Cerebrospinal fluid (CSF) was analyzed for cell count (pleocytosis >5 cells/mm3), protein content, IgG index and oligoclonal bands (OCB).
At the end of the follow-up, patients were classified as ADEM [17], NMOSD [18], MS [19] or other limited NMO-like phenotypes (i.e.; isolated monophasic or relapsing myelitis or ON).
In addition, aiming to characterize features associated with ADEM MOG-Ab positive, we compared this group to monophasic ADEM tested seronegative for AQP4 and MOG-Ab during the same period of recruitment. All data were prospectively entered in the database adapted from the EDMUS system and software (Eugène Devic European Network, EDEN), and retrospectively analyzed [20].
Autoantibody detection
Aquaporin-4 Ab was detected using a cell-based assay [21]. MOG-Ab was detected by cell-based assay using an in-house method [22]. Briefly, HEK293 cells were transfected with pEGFP-N1-hMOG plasmid (kind gift from Markus Reindl, Innsbruck, Austria) for 48 h. Transfected cells were incubated with patients serum diluted at 1:640. This cut-off was selected to avoid false-positive signal detected with healthy control in previous works [23]. Bound IgG was detected with a fluorescent secondary antibody, APC-Goat anti human IgG-Fcγ fragment-specific (1:100 dilution, Jackson ImmunoResearch). Evaluation of signal intensity was performed by flow cytometry. Titration of positive samples was performed using serial dilution until loss of positive signal (from 1:160 to 1: 20.000).
Statistical analysis
At first episode, patients were classified into two groups: pediatric [admitted at neuropediatrics department (<16 years old)] and adult groups. Categorical and continuous variables were compared with nonparametric test (Fisher exact and Mann–Whitney U test, respectively). Statistical significance was set at two-tailed p value <0.05. All statistical analyses were performed using STATA (64-bits) software.
Results
General cohort features
We found a positive result for MOG-Ab in the serum of 27 patients. None of them were positive for AQP4-Ab. Flow chart for patient selection is depicted in Fig. 1.

Flowchart showing the clinical phenotype at onset and last follow-up in MOG seronegative patients. ADS acquired demyelinating syndrome, ADEM acute demyelinating encephalomyelitis, NMOSD neuromyelitis optica spectrum disorders, LETM longitudinal extensive transverse myelitis
After a median follow-up of 17.8 [interquartile range (IQR) 11.5–68.3] months, eight patients were finally diagnosed with ADEM, eight with isolated ON (four monophasic and four relapsing), five with isolated monophasic myelitis (four LETM), four with NMOSD and two patients with MS (Table 1).
Among the 27 MOG-Ab-positive patients, the median age of presentation was 16.8 (IQR 6.8–33.7) years. Patients were mainly Caucasians 25 (92.6%) with a female:male ratio of 1:0.9 (Table 1).
The most frequent clinical phenotypes at onset were myelitis, encephalopathy and unilateral ON (33.3 vs 29.6 vs 25.9%, respectively). The median EDSS at nadir was four (IQR 2.5–5.0); three patients presented with a severe motor attack at onset (two paraplegic, and one tetraplegic needing intubation). Eight out of 10 (80%) patients presenting with an initial ON had severe visual impairment (VA ≤ 0.2). During the follow-up, 11 (40.7%) patients relapsed either in form of ON (37%) or brainstem symptoms (3.7%). All but three patients relapsed within 8 months (median 3.8; IQR 2.0–22.5 months) from disease onset. No other CNS topographies were affected at relapse (Table 1).
Median time from onset of symptoms to MOG sampling was 1.4 months (IQR 0.4–21.9) (Table 1). When analyzing only those samples taken within 3 months from onset of symptoms, encephalopathy was related to higher MOG-Ab titers in comparison to the other phenotypes at onset [median 6280 (IQR 2560–20.000) vs 640 (640–2560), p = 0.043]. Similarly, ADEM showed to have higher MOG-Ab titers than the remaining phenotypes at last follow-up [median 10.000 (IQR 2560–20.000) vs 640 (640–2560), p = 0.014 (Supplementary Fig. 1).
Brain MRI at onset was abnormal in 16 (59%) patients. Among them, 7 (43.8%) and 5 (31.2%) patients showed bilateral thalamic or brainstem tegmental lesions, respectively. Eleven (73.3%) out of 15 patients with an available brain MRI during follow-up showed either complete resolution or improvement of the lesions. Spinal cord MRI performed at the acute phase disclosed LETM in 10 patients, and one patient showed T2-W patchy non-extensive lesions with gadolinium enhancement. Complete resolution or improvement of the spinal cord lesions was observed in 6 (60%) of these patients (Table 2).
All but one patient were under intravenous (i.v.) methylprednisolone (MTP) at the acute phase of disease, and steroids were further gradually tapered over next months. Immunosuppressive or immunomodulatory therapies were started in 12 patients (44.4%): mycophenolate mofetil in seven, azathioprine in two and rituximab in one patient. One MS patient was under teriflunomide, and the other received glatiramer acetate being further switched to natalizumab due to lack of efficacy (clinical and radiological features of MS patients are depicted in Table 3 and Fig. 2; case description in the supplementary material).

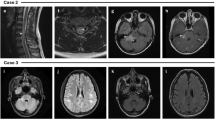
Radiological features of MOG antibodies-positive patients with MS and uncommon features. Patient MS 1 a, b showed typical MS-like lesions; patient MS 2, c showing mesencephalic and d gadolinium-enhancing MS-like lesions; patient ID 1 e patchy T2-W spinal cord hyperintensities and f gadolinium-enhancing lesions; patient ID 2 with underlying teratoma, g diffuse occipital white matter brain FLAIR hyperintensities and h extensive T2-W spinal cord lesion; patient ID 3 i, symmetric bilateral thalamic lesions and j tegmental FLAIR hyperintensities
We were able to test MOG-Ab during the follow-up in eight patients [median 7.1 months (IQR 2.2–11.3)]. Seven patients tested positive (three patients with relapsing ON, two NMOSD, one monophasic LETM and one ADEM) and one was negative (monophasic non-LETM).
At last visit, 23 (85.2%) had a good clinical outcome with a median EDSS of 1 (IQR 0–2.0), and only one patient with an initial clinical phenotype of unilateral ON persisted with severe visual sequelae, after 45 months of follow-up.
Comparison between MOG-Ab-positive pediatric and adult groups
No differences regarding gender and previous infections or vaccinations were observed between pediatric and adults. One adult had a previous myasthenia gravis.
Clinical phenotype at onset was more frequently characterized by encephalopathy in pediatrics than in adults [7/13 (53.9%) vs 1/14 (7.1%), p = 0.013]. On the contrary, myelitis was the initial phenotype in only three pediatrics (23.1%) and seven (50%) adults (Table 1).
When performing MOG-Ab titers, titers were significantly higher in the pediatric group [2560 (IQR 2560–20.000)] compared to adults [640 (IQR 640–2560)], p = 0.020 (Table 1; supplementary Fig. 1).
There were no differences with respect to the proportion of patients with an abnormal brain MRI at onset between both groups. However, six out of nine (66.7%) children disclosed bilateral thalamic lesions compared to only one (14.3%) adult, among patients with an initial abnormal brain MRI (p = 0.060) (Table 2).
In terms of severity at last follow-up, there were no differences, and both groups evolved with a general good outcome.
Comparison between ADEM MOG-Ab-positive and ADEM MOG-Ab negatives
We noted that among the ADEM MOG-Ab-positive patients (n = 8), 7 (87.5%) disclosed bilateral thalamic lesions in the first brain MRI in comparison with only one patient in the ADEM MOG-Ab-negative group (p = 0.010). We did not find other epidemiological or clinical differences between these two groups (Table 4).
MOG-Ab-positive patients with uncommon presentations
Unusual presentations were observed in three MOG-Ab-positive patients: (1) one presented with patchy gadolinium-enhancing lesions along the whole spinal cord; (2) one had an underlying ovarian teratoma associated with ADEM; (3) one presented with status epilepticus at onset of symptoms (Table 5; Fig. 2; supplementary data for case description).
Discussion
The present study encompassed a cohort of 27 MOG-Ab-positive patients presenting with an initial CNS demyelinating episode. Patients developed general features of MOG-Ab spectrum diseases characterized by young age at onset, ON and a general good outcome.
Although the MOG-Ab clinical spectrum seems to be highly restricted to ADEM or limited NMO-like phenotypes, two patients were diagnosed with MS: one adult developed a relapsing severe ON with typical MS-like brain lesions, and one pediatric patient presented with a relapsing brainstem syndrome. Despite that the latter relapsed under glatiramer acetate, both showed a good response to natalizumab and teriflunomide. The diagnostic classification in the pediatric MS may be a matter of discussion since the patient would have been diagnosed by others as multiphasic ADEM [4]. One recent study has found that up to 13% of MOG-Ab-positive patients may have OCB in CSF and up to 33% fulfill 2010 McDonald criteria [9]. Whether MOG-Ab-positive patients fulfilling the current McDonald criteria should be classified as MS is still under debate. Moreover, it has been suggested that MS patients with MOG-Ab share distinctive clinical features, characterized by severe or recurrent ON, brainstem syndrome and myelitis [24]. Patients also presented with a high relapse rate and did not properly response to disease-modifying therapies. Overall, MOG-Ab-related disorders are newly described, and further studies are needed to clarify classification, prevalence, phenotypical features and therapeutic options in MS patients with MOG-Ab.
MOG-Ab-related disorders were previously known for having fewer relapses in comparison to AQP4-Ab-positive patients [6,7,8]. We underline that up to 40% of patients relapsed in our study, despite the relatively short follow-up duration. Not unexpected, relapses involved the optic nerve in all but one patient. Moreover, we also found that most of the relapses occurred within 8 months after disease onset. In line with our observation, a recent study supports that most of MOG-Ab-positive patients will develop a relapse disease course, having the second attack only few months after the disease onset [9]. A dependency on steroids has been previously noted in MOG-Ab-related disorders [25, 26], and steroids cessation may have been related to the short time to relapse in the present study. Although controlled prospective studies evaluating the optimal therapeutic options are not available in MOG-Ab-related disorders, we believe that the prompt introduction of an immunosuppressant therapy together with an individualized steroid tapering could have a beneficial effect in reducing the number of further episodes.
In the present study, we noted that most of patients presenting with ON developed a severe initial visual impairment, and three adults presented with a fulminant motor attack. MOG-Ab-positive patients with a high degree of disability at onset have been described, leading in some cases to neurologic sequelae or even death [9, 15, 16, 25, 27]. In the present cohort, 15% of patients remained disabled at last visit, reinforcing the need to identify prognostic factors at onset in order to select those individuals at high risk of disability where more aggressive acute treatments, such as plasma exchange, could be beneficial.
The encephalopathic onset of symptoms seems to be directly related to the bilateral thalamic affection observed in all but one ADEM MOG-Ab-positive. Bilateral thalamic lesions are described up to 63% of ADEM patients, occurring more frequently in pediatrics than adults [28, 29]. These patients are characterized by showing a complete resolution of the brain lesions and experiencing a good prognosis. In the only study focused on the radiological features of ADEM MOG-Ab-positive patients, up to 89.5% of patients presented with lesions either in thalamus or basal ganglia [3]. Other interesting radiological findings were the presence of brainstem lesions involving the pontine tegmentum in one-third of patients, as others have remarked [13, 25]. Whether there is a predilection for selected anatomical brain areas in MOG-Ab-related disorders is a matter of controversy. Age-related radiological differences could be the result of different myelination stages in the course of development [30].
Interestingly, MOG-Ab titers were found higher in pediatrics compared to adults, as a result of an ADEM overrepresentation within this former group. This finding suggests a more intense underlying immunity against MOG protein and reflects the more extensive affection, both clinical and radiological, observed in younger patients [3, 11].
The presence of uncommon clinical or radiological features in some MOG-Ab-positive patients must be pointed out, since MOG-Ab-related disorders is a recent entity, and the clinical spectrum is likely not to be entirely described. One patient presented with an atypical spinal image characterized by patchy gadolinium-enhancing lesions along the whole spinal cord. Thus, although LETM and less frequently partial myelitis have been related to MOG-Ab-related disorders [6, 8, 9, 13, 22], clinicians should also be aware of other spinal cord inflammatory lesions in these patients. Interestingly, an underlying teratoma was found in one ADEM MOG-Ab-positive patient presenting with severe clinical symptoms other than those typical for N-Methyl-d-aspartate receptor (NMDA-R) encephalitis. Contrary to the often existing overlapping observed between demyelinating syndromes and NMDA-R encephalitis [31], neither serum nor CSF NMDA-R antibodies were found. There is only a previous case reported of ovarian teratoma in patients with MOG-Ab [9]. Whether mechanisms of breakdown of immunologic tolerance or mimicry towards MOG protein expressed in the teratoma are involved in the present case are unknown, so far. Finally, we should keep in mind that in spite of their general good prognosis observed, MOG-Ab-related disorders may initiate with a live threatening picture in form of status epilepticus, requiring intubation and careful monitoring.
Altogether, we observe that MOG-Ab related disorders share common clinical and prognostic features that may differ between adults and pediatrics at onset. Since this is a recently described entity, MOG-Ab encompasses a clinical and radiological spectrum much wider than recently reported.
References
Pröbstel AK, Dornmair K, Bittner R et al (2011) Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 77:580–588
Rostasy K, Mader S, Schanda K et al (2012) Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 69:752–756
Baumann M, Sahin K, Lechner C et al (2014) Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry 86:265–272
Baumann M, Hennes E-M, Schanda K et al (2016) Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Scler 22:1821–1829
Huppke P, Rostasy K, Karenfort M et al (2013) Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler 19:941–946
Sato DK, Callegaro D, Lana-Peixoto MA et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82:474–481
Kitley J, Waters P, Woodhall M et al (2014) Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71:276–283
Höftberger R, Sepulveda M, Armangue T et al (2014) Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. 21:866–874
Jarius S, Ruprecht K, Kleiter I et al (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation 13:280
Havla J, Kümpfel T, Schinner R et al (2016) Myelin-oligodendrocyte-glycoprotein (MOG) autoantibodies as potential markers of severe optic neuritis and subclinical retinal axonal degeneration. J Neurol 264:139–151
Fernandez-Carbonell C, Vargas-Lowy D, Musallam A et al (2016) Clinical and MRI phenotype of children with MOG antibodies. Mult Scler 22:174–184
Ketelslegers IA, Van Pelt DE, Bryde S et al (2015) Anti-MOG antibodies plead against MS diagnosis in an acquired demyelinating syndromes cohort. Mult Scler 21:1513–1520
Sepúlveda M, Armangue T, Martinez-Hernandez E et al (2016) Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol 263:1349–1360
van Pelt ED, Wong YYM, Ketelslegers IA et al (2015) Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol 23:580–587
Di Pauli F, Höftberger R, Reindl M et al (2015) Fulminant demyelinating encephalomyelitis: insights from antibody studies and neuropathology. Neurol Neuroimmunol Neuroinflam 2:e175
Spadaro M, Gerdes LA, Mayer MC et al (2015) Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol 2:295–301
Krupp LB, Tardieu M, Amato MP et al (2013) International pediatric multiple sclerosis study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 19:1261–1267
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189
Polman CH, Reingold SC, Banwell B et al (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302
Confavreux C, Compston DA, Hommes OR et al (1992) EDMUS, a European database for multiple sclerosis. J Neurol Neurosurg Psychiatry 55:671–676
Marignier R, Bernard-Valnet R, Giraudon P et al (2013) Aquaporin-4 antibody-negative neuromyelitis optica: distinct assay sensitivity-dependent entity. Neurology 80:2194–2200
Cobo-Calvo Á, Sepúlveda M, Bernard-Valnet R et al (2015) Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: clinical and prognostic implications. Mult Scler. 22:312–319
Mader S, Gredler V, Schanda K et al (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflam 8:184
Spadaro M, Gerdes LA, Krumbholz M et al (2016) Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflam 3:e257
Kim S-M, Woodhall MR, Kim J-S et al (2015) Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflam 2:e163
Ramanathan S, Reddel SW, Henderson A et al (2014) Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflam 1:e40
Piccolo L, Woodhall M, Tackley G et al (2016) Isolated new onset “atypical” optic neuritis in the NMO clinic: serum antibodies, prognoses and diagnoses at follow-up. J Neurol 263:370–379
Tenembaum S, Chamoles N, Fejerman N (2002) Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology 59:1224–1231
Ketelslegers IA, Visser IER, Neuteboom RF et al (2011) Disease course and outcome of acute disseminated encephalomyelitis is more severe in adults than in children. Mult Scler 17:441–448
Hasegawa M, Houdou S, Mito T et al (1992) Development of myelination in the human fetal and infant cerebrum: a myelin basic protein immunohistochemical study. Brain Dev 14:1–6
Titulaer MJ, Höftberger R, Iizuka T et al (2014) Overlapping demyelinating syndromes and anti-N-methyl-d-aspartate receptor encephalitis. Ann Neurol 75:411–428
Acknowledgements
A. Cobo-Calvo is supported by a grant from the Fundación Alfonso Martín Escudero. The authors thank the group of NeuroBioTec from Hôpital Civils de Lyon for supporting this study. This work has been done by using data from the Observatoire Français de la Sclérose en Plaques (OFSEP) which is supported by a grant provided by the French State and handled by the “Agence Nationale de la Recherche”, within the framework of the “Investments for the Future” programme, under the reference ANR-10-COHO-002 Observatoire Français de la Sclérose en plaques (OFSEP), and by the Eugene Devic Foundation against Multiple Sclerosis (EDMUS Foundation).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Cobo-Calvo, Ruiz, d’Indy, Poulat, Carneiro, Nicolas and Desportes report no disclosures. Durand-Dubief serves on scientific advisory board for Merck Serono and has received funding for travel and honoraria from Biogen Idec, Merck Serono, Novartis, Sanofi-Genzyme, Roche and Teva. Deiva received travel funding from Biogen Idec, Merck Serono, and Genzyme. Vukusic has received consulting and lecturing fees, travel grants and research support from Biogen, Geneuro, Genzyme, Novartis, Merck Serono, Roche, Sanofi Aventis and Teva Pharma. Marignier serves on scientific advisory board for MedImmune and has received funding for travel and honoraria from Biogen Idec, Merck Serono, Novartis, Sanofi-Genzyme and Teva.
Ethical standards
The ethics committee of Lyon University Hospital approved this study. All samples were stored at −80 °C at Neurobiotec (Hospices Civils de Lyon, France, no. 0033-00046, AC-2013-1867, NFS96-900).
Informed consent
Informed consent for storage and use of these samples for research was obtained from all patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cobo-Calvo, Á., Ruiz, A., D’Indy, H. et al. MOG antibody-related disorders: common features and uncommon presentations. J Neurol 264, 1945–1955 (2017). https://doi.org/10.1007/s00415-017-8583-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-017-8583-z