
Abstract
Guillain–Barré syndrome (GBS) is categorized into two major subtypes: acute inflammatory demyelinating polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN). However, a proportion of patients are electrophysiologically unclassified because of electrophysiological findings that do not fulfil AIDP or AMAN criteria, and underlying pathophysiological mechanisms and lesion distributions of unclassified patients are not well defined. The aims of this study are to elucidate disease pathophysiology and lesion distribution in unclassified patients. We retrospectively studied 48 consecutive GBS patients. Patients were classified on the basis of initial electrophysiological findings according to Ho’s criteria. Clinical and serial electrophysiological examinations of unclassified patients were conducted. Twelve (25 %) GBS patients were unclassified. All unclassified patients were able to walk independently at 21 days after onset. No unclassified patients, except one patient with diabetes mellitus, had sensory nerve involvement. Eight patients underwent a follow-up study within 15 days of the initial study. Distal motor latencies (DMLs) of the left median motor nerve were found to be significantly and uniformly decreased compared with initial studies (p = 0.008). DMLs (p < 0.0001) and distal compound action potential (CMAP) durations (p = 0.002) of all nerves were significantly decreased, and distal CMAP amplitudes (p = 0.026) significantly increased compared with initial studies. In unclassified GBS patients, DML values during initial electrophysiological studies would be prolonged compared with expected values in the same patient unaffected by GBS and later improve rapidly with increased distal CMAP amplitudes without the development of excessive temporal dispersions. Lesions are also present in distal nerve segments caused by reversible conduction failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Guillain–Barré syndrome (GBS) is categorized into two major subtypes: acute inflammatory demyelinating polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN). Several studies have increased our understanding of each subtype [1–6].
While the classification is clinically based on electrophysiological findings, 11–41 % of GBS patients are electrophysiologically unclassified because of electrophysiological findings not fulfilling AIDP or AMAN criteria [1, 7–9]. Here, it should be noted that underlying pathophysiological mechanisms and lesion distributions of unclassified patients are not well defined. Regarding pathophysiological mechanisms, it is even unclear whether a single mechanism or a multiple mechanisms underlie the pathophysiology of unclassified GBS. Regarding lesion distributions, the distal nerve terminals and nerve roots are preferentially affected in GBS regardless of AIDP or AMAN [10], possibly due to selective deficiency of the blood–nerve barrier [11]. The same is likely true for unclassified patients; therefore, lesions are expected to be present in distal nerve segments and proximal nerve segments despite conduction abnormalities not fulfilling AIDP or AMAN criteria. However, in unclassified patients, the presence of lesions especially in distal nerve segments is not fully elucidated.
Recent reports have demonstrated the utility of serial electrophysiological studies in determining GBS pathophysiology and related conditions [8, 9, 12]. Therefore, the concept of reversible conduction failure (RCF) has been established as a new subtype of GBS [13]. RCF is characterized by the presence of decreased distal CMAP amplitudes, conduction blocks in intermediate nerve segments, and prolonged DMLs with rapid subsequent normalization of conduction abnormalities [7, 9, 13, 14]. During normalization, the development of increased durations and polyphagia does not occur concurrently with durations instead decreasing, which is not consistent with demyelination. After that, RCF is demonstrated to be a subtype of AMAN rather than AIDP even by pathological findings, which revealed the primary pathology to be axonal [15]. Using serial electrophysiological studies, some, but not all, previous reports also showed that acute motor conduction block neuropathy (AMCBN), which is a rare subtype of GBS and shows reduction of distal CMAP amplitudes and early partial motor conduction block in intermediate nerve segments in single electrophysiological studies, are caused mainly by RCF [14].
Aside from the utility of serial studies, we previously demonstrated the utility of electrophysiologically classifying the AIDP pattern further according to the presence or absence of sensory nerve conduction abnormalities, which we termed motor-sensory AIDP (MS-AIDP) and motor AIDP (M-AIDP) patterns, respectively [16]. The presence of RCF demonstrates limitations of conventional classification systems, where AMAN patients with RCF may be incorrectly classified as having the AIDP pattern because of conduction abnormalities such as prolonged DML, and patients classified as having the AIDP pattern may include patients with two different disease entities: AMAN with RCF and true AIDP. However, our new classification system allowed these two diseases to be distinguished: the M-AIDP and MS-AIDP patterns corresponded to AMAN with RCF and true AIDP, respectively. Therefore, to elucidate underlying pathophysiology and distal nerve segments involvement in GBS patients not fulfilling AIDP or AMAN criteria, we reviewed and compared unclassified GBS patients with other patterns using serial electrophysiological studies and our classification focused on distal nerve segments including distal motor latencies (DMLs).
Materials and methods
Patients
A total of 48 consecutive GBS patients who visited Osaka Medical College Hospital between January 2005 and August 2015 and underwent initial nerve conduction studies within 16 days of onset were retrospectively studied. All patients fulfilled the clinical criteria for GBS [17], except for the criteria regarding areflexia/hyporeflexia as the presence of GBS in patients with normal or exaggerated tendon reflexes has previously been reported [14, 18].
Patient disabilities were evaluated using the Hughes disability grade score as follows: [19] grade 0, healthy; grade 1, minor signs and symptoms, able to run; grade 2, able to walk independently; grade 3, able to walk with a walker or support; grade 4, bed- or chair-bound; grade 5, assisted respiration required for at least part of the day; and grade 6, dead.
This study received institutional review board approval. Informed consent was obtained from all participants according to the Declaration of Helsinki.
Electrophysiological studies
We performed motor and sensory nerve conduction studies using an MEB-9104 Neuropack mu® (Nihon Kouden, Japan) machine according to the methods and reference values of Kimura et al. [20]. Motor nerve conduction studies were conducted for the median, ulnar, peroneal, and tibial nerves. Compound action potential (CMAP) duration was defined as the time period from the onset of the initial negative phase until the return to baseline of the last negative deflection of the CMAP. Antidromic sensory nerve conduction studies were conducted for the median, ulnar, and sural nerves. On the basis of the results of initial first electrophysiological studies, patients were electrophysiologically classified as having the AIDP or AMAN pattern using the electrophysiological criteria of Ho et al. [1]. Although Ho’s criteria set includes unequivocal temporal dispersion for the detection of demyelination, the amount of temporal dispersion of CMAP that should be considered unequivocal is not defined. Therefore, we used a distal CMAP duration of more than 6.6 ms in the median, 6.7 ms in the ulnar, 7.6 ms in the peroneal, and 8.8 ms in the tibial nerves, respectively [21], or a >30 % increased duration of the proximal CMAP compared with the distal CMAP in all nerves [22]. Patients who did not fulfil the electrophysiological criteria of the AIDP or AMAN patterns were electrodiagnostically classified as having unclassified GBS. The absence of F waves was defined as the absence of, or markedly decreased (persistence <20 %), F waves in at least two nerves [23]. Patients were considered to have sensory nerve conduction abnormalities when the sensory nerve action potential (SNAP) amplitude was <50 % of the lower limit of normal in at least two nerves [24]. We further electrophysiologically classified the AIDP pattern according to the presence or absence of sensory nerve conduction abnormalities, which we termed MS-AIDP and M-AIDP patterns, respectively [16]. In the present study, the electrophysiological pattern of AIDP and AMAN were, respectively, expressed as “AIDP pattern (M-AIDP and MS-AIDP pattern)” and “AMAN pattern” as electrophysiological patterns and disease subtypes may not be equivalent. Accordingly, the disease subtype of AIDP and AMAN was, respectively, expressed as AIDP and AMAN (AMAN with axonal degeneration and AMAN with RCF) from now on.
Statistical analyses
Differences in mean values between the two groups were examined using the Mann–Whitney U test. Differences in frequencies between the two groups were examined using Fisher’s exact probability test. Differences within the same group between examination times were examined using the Wilcoxon matched pairs test. Statistical significance was set at p < 0.05. All analyses were performed using the software Graphpad PRISM, version 5.01 (GraphPad Software, San Diego, CA, USA).
Results
Clinical, serological and electrophysiological features
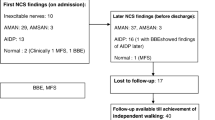
Based on initial electrophysiological findings, 15 (31 %) of the 48 patients were classified as having the “M-AIDP pattern,” 15 (31 %) as having the “MS-AIDP pattern,” 6 (13 %) as having the “AMAN pattern,” and 12 (25 %) were electrophysiologically unclassified. One patient with the “AMAN pattern” exhibited sensory nerve conduction abnormalities and, therefore, corresponded to the “acute motor and sensory axonal neuropathy (AMSAN) pattern” in the strict sense [25]. Table 1 compares the clinical features of unclassified patients and other GBS patients. Patient sex ratio and age did not differ between the two groups. Although the time from onset to the initial electrophysiological studies and Hughes grade scores at initial studies tended to be lower in unclassified patients than in other GBS patients, these differences did not reach statistical significance. The clinical, serological and electrophysiological features of the 12 electrophysiologically unclassified GBS patients are shown in Table 2. Although some unclassified patients were negative for antiganglioside antibodies, the frequency of anti-GM1 antibody in unclassified patients (4 of 10 patients) was significantly higher (p = 0.029) than that in patients with the “MS-AIDP pattern” (0 of 12 patients) and did not differ significantly from those in patients with the “M-AIDP” (6 of 13 patients) and “AMAN” patterns (3 of 5 patients), respectively. Of the 12 unclassified patients, only 2 patients (17 %) had an absence of F waves. Eleven patients (92 %) did not have clinical or electrophysiological sensory nerve involvement. The remaining patient also had diabetes mellitus with involvement attributable to diabetic neuropathy because the “sural sparing pattern,” which is compatible with GBS but not diabetic neuropathy [26, 27], was absent. Eleven of the 12 unclassified patients underwent follow-up studies, all of whom were again classified as having unclassified GBS on all follow-up studies. Moreover, of the 12 unclassified patients, 11 patients (92 %), including those with mild disability, received IVIG treatment, and all were able to walk independently (Hughes grade 2 or less) at 21 days after onset.
Serial electrophysiological studies
Of the 11 unclassified patients undergoing follow-up studies, 8 patients underwent follow-up studies within 15 days of initial studies. The time courses of electrophysiological parameters for the left median nerve are shown in Fig. 1. DMLs were found to be significantly and uniformly decreased compared with initial studies (p = 0.008). In these patients, serial nerve conduction studies were undertaken in a total of 57 motor and 42 sensory nerves. The time courses of electrophysiological parameters of all these nerves are shown in Fig. 2. The values were determined, respectively, not summing the values in each patient, and statistically analyzed. DMLs (p < 0.0001) and distal CMAP durations (p = 0.002) were found to be significantly decreased and distal CMAP amplitudes (p = 0.026) significantly increased compared with initial studies.

Serial findings of DMLs, distal CMAP duration, MCV, distal CMAP amplitudes, and distal SNAP amplitudes in left median nerve. Electrophysiological patterns reported are from initial studies. Follow-up studies were conducted within 15 days of initial studies. Dotted lines indicate the cut-off values of the criteria for demyelination (DMLs and distal CMAP duration), AMAN (distal CMAP amplitude) or sensory nerve conduction abnormalities (distal SNAP amplitude). DMLs distal motor latencies, AMAN acute motor axonal neuropathy, CMAP compound action potential, MCV motor conduction velocity, SNAP sensory nerve action potential

Serial findings of DMLs, distal CMAP duration, MCV, distal CMAP amplitudes, and distal SNAP amplitudes in all nerves tested. Electrophysiological patterns reported are from initial studies. Follow-up studies were conducted within 15 days of initial studies. Values are expressed as mean percentages ± SEM with values at initial recordings normalized to 100 %. DMLs distal motor latencies, CMAP compound action potential, MCV motor conduction velocity, SNAP sensory nerve action potential, SEM standard error of mean
We then compared and analyzed all four electrophysiological patterns of GBS. Because the degree of patient disability significantly differed among the patterns, we studied patients with low disability scores (score 2 or less at the Hughes disability grade score) for each pattern to eliminate the confounding effects of patient disability. Of the 48 GBS patients, 22 patients (46 %) had low disability scores with follow-up studies conducted within 30 days of initial studies in 21 patients: 7 patients (42 nerves) with the “M-AIDP pattern,” 3 patients (20 nerves) with the “MS-AIDP pattern,” 4 patients (23 nerves) with the “AMAN pattern,” and 7 unclassified patients (43 nerves). The time courses of DMLs for all nerves in each pattern are shown in Fig. 3. DMLs were significantly decreased in unclassified patients (p < 0.0001) and patients with the “M-AIDP pattern” (p < 0.0001) compared with initial studies. No difference in DMLs was observed in patients with the “AMAN pattern” between initial and follow-up studies. DMLs were significantly increased in patients with the “MS-AIDP pattern” (p = 0.035) compared with initial studies.

Serial findings of DMLs in all motor nerves in patients with grade 2 or less. Electrophysiological patterns reported are from initial studies. Follow-up studies were conducted within 30 days of initial studies. Values are expressed as mean percentages ± SEM with values of average of healthy subjects normalized to 100 %. Note that the Y axis scale for the unclassified pattern is identical to those of the M-AIDP and AMAN patterns and different from that of the MS-AIDP pattern. M-AIDP motor acute inflammatory demyelinating polyneuropathy, AMAN acute motor axonal neuropathy, MS-AIDP motor-sensory acute inflammatory demyelinating polyneuropathy, DMLs distal motor latencies, SEM standard error of mean
Discussion
The present study showed several features characteristic of electrophysiologically unclassified patients in serial electrophysiological studies. While DMLs at initial studies did not reach the range of demyelination and were occasionally in the normal range, DMLs subsequently decreased rapidly. This finding indicates that in each patient, DMLs at initial studies were prolonged compared with expected values in the same patient unaffected by GBS before subsequently improving rapidly. During the rapid decrease in latencies, amplitudes increased rapidly, while durations did not increase but rather decreased. Taken together, with regard to lesion distributions, these findings indicate distal nerve segments were also affected. With regard to underlying pathophysiology, demyelination is unlikely to explain the features observed in unclassified patients as improvements in prolonged DMLs appeared too rapid to be remyelination. In AIDP, the nadir of conduction abnormalities, including prolonged DMLs, occurred during the 3rd week, with obvious improvement observed during weeks 6–10 in the majority of patients [13, 26, 28]. Further, the development of increased durations was not observed in the recovery stage. Resolution of demyelination usually begins with the development of increased durations and polyphagia, indicative of the presence of remyelinating slow components. This development is one of the most reliable electrophysiological features of the demyelination–remyelination process [29, 30] and occurs regardless of disease severity [31]. We speculate these serial findings in unclassified patients as a result of RCF because the features observed in unclassified patients, as the rapid improvements in prolonged DMLs with increased distal CMAP amplitudes without the development of excessive temporal dispersions, are consistent with those of RCF. To summarize, we demonstrate that serial electrophysiological findings in unclassified patients are caused by RCF with distal nerve segments also affected.
Although RCF is a subtype of AMAN rather than AIDP, AMAN patients with RCF are classified as the “AIDP pattern” using single nerve conduction studies [7, 8, 28], which consequently leads to confusion in both clinical and research fields. To resolve this issue, we previously focused on the fact that AMAN patients with both axonal degeneration and RCF have common clinical features because of a shared disease spectrum [2–5, 28, 32], with the absence of clinical and electrophysiological involvement of sensory nerve, while the involvement of sensory nerve are typically present in AIDP patients. Thereby, we previously demonstrated the utility of classifying GBS into “M-AIDP,” “MS-AIDP,” and “AMAN” patterns for the determination of detailed pathophysiology including AMAN with RCF, as follows: the “M-AIDP pattern” corresponds to AMAN with RCF; the “MS-AIDP pattern” corresponds to true AIDP; and the “AMAN pattern” corresponds to AMAN with axonal degeneration [16]. These findings prompted us to classify GBS into four patterns according to initial electrophysiological findings and compare and analyze serial DML findings between all four patterns. DMLs of both unclassified patients and patients with the “M-AIDP pattern” were found to decrease commonly, possibly reflecting the common underlying pathophysiology, while DMLs were increased in patients with the “MS-AIDP pattern.” These findings validate the utility of our classification and demonstrated electrophysiological findings in unclassified patients, as well as patients with the “M-AIDP pattern,” are caused by RCF.
Aside from the serial electrophysiological findings, the present study identified two features of unclassified patients. Firstly, unclassified patients exhibited the absence of clinical and electrophysiological involvement of sensory nerves, which is consistent with RCF as mentioned above. Secondly, according to previous reports [33, 34], unclassified patients exhibited rapid clinical recovery and were able to walk independently within short time periods. The observed rapid clinical recovery of unclassified patients also confirms the contribution of RCF. RCF is suggested to be a mild subtype of AMAN [5, 14, 35], with rapid clinical recovery known to correlate well with rapid electrophysiological recovery, while AMAN patients with axonal degeneration have prolonged recovery.
In previous studies, the identification of unclassified patients, particularly patients with normal electrophysiological findings, was reported to be associated with early electrophysiological assessment [26, 36–38] and mild disability on assessment [9]. On the other hand, the mechanisms underlying the pathophysiology of unclassified GBS, particularly in cases without absence of F waves, are poorly understood. In the present study, although the duration from onset to electrophysiological study and Hughes grade scores at initial studies tended to be lower in unclassified patients than others, these differences did not reach statistical significance. Therefore, early electrophysiological assessment and mild disability were not considered distinguishing characteristics of unclassified GBS. Further, no unclassified patients changed classification according to the findings of follow-up studies, confirming that early electrophysiological assessment is not a distinguishing factor. On the other hand, the features of unclassified patients were similar to those of patients with RCF. This finding was even shown by serial electrophysiological analysis of patients with similar degrees of mild disability to eliminate the confounding effects of patient disability. Therefore, unclassified GBS was found to be associated with RCF independently of patient disability. As all unclassified patients uniformly demonstrated rapid decreases in DMLs over time, which is one of the most typical electrophysiological features of RCF, it may be at least required for unclassified GBS that lesions are caused by RCF. Therefore, unclassified GBS may almost exclusively comprise patients with a single pathophysiology termed RCF, rather than patients with early electrophysiological assessments or mild disability representing a range of underlying pathophysiologic mechanisms.
Although the pathology and lesion distributions of all unclassified GBS patients are poorly understood, Kuwabara et al. [23]. Performed a detailed study of unclassified GBS patients with the absence of F waves. They certainly observed the presence of lesions in nerve roots based on F-wave abnormalities according to its definition. This study demonstrated that lesions may be caused by axonal dysfunction with both RCF and axonal degeneration, based on two elecrophysiological time courses; F waves rapidly appeared without the prolongation of F-wave latency or F chronodispersion, and the absence of F waves persisted while distal CMAP amplitude decreased leading to revised classifications to the “AMAN pattern.” This finding was confirmed by also demonstrating that patients with IgG antiganglioside antibodies, which are associated with AMAN only [6, 32], had unclassified GBS with the absence of F waves more often than patients without IgG antiganglioside antibodies. Other than AMAN with axonal degeneration and RCF, the authors posited that motor neuron excitability may affect F-wave persistence in a proportion of cases, as previously proposed [39]. We obtained several previously unreported findings in the present study. With regard to subjects, the present study extended the range of subjects to all unclassified GBS patients. With regard to legion distributions, our findings surprisingly demonstrated the presence of lesions in distal nerve segments, some of which could only be detected by serial DMLs findings rather than a single nerve conduction study. With regard to the mechanisms underlying the pathophysiology of unclassified GBS, we demonstrated that associated lesions were caused by a single pathophysiology, namely RCF. This finding is predominantly based on the electrophysiological time course: a rapid decrease in distal motor latencies without excessive temporal dispersions, which is characteristic and specific for RCF and could not be explained by impaired excitability alone.
In the present study, unclassified patients demonstrated a single electrophysiological time course consistent with RCF and did not change classification as a result of any follow-up studies. In contrast, in the study by Kuwabara et al. [23], unclassified patients with the absence of F waves demonstrated two courses consistent with axonal degeneration and RCF. In the course consistent with axonal degeneration, patients accordingly changed classification to the “AMAN pattern.” Two distinguishing features of unclassified patients in the present study may account for these discrepancies. Firstly, the condition of unclassified patients in the present study may be milder than unclassified patients with the absence of F waves in the previous study by Kuwabara et al. In fact, the majority of unclassified patients in the present study did not fulfil the criteria for the absence of F waves. Secondly, the majority of unclassified patients in the present study received IVIG therapy even among those with mild disability. Both factors would be associated with decreased disease progression and development of AMAN with axonal degeneration. In short, the majority of unclassified patients, particularly those receiving IVIG therapy, may have RCF, although a few unclassified patients, particularly those with the absence of F waves on initial studies, appear to develop AMAN with axonal degeneration.
The entity of AMCBN has been proposed as a rare subtype of GBS. While, as mentioned in the Introduction, AMCBN is generally believed to be caused mainly by RCF and to represent an “arrested” AMAN [14], Mangatti et al. suggested in their detailed case report that it is not well clarified whether AMBCN represents an “arrested” AMAN [40]. Therefore, although we demonstrate the pathophysiology of unclassified patients to be RCF, we could not determine whether unclassified patients share a common pathophysiology with AMCBN patients. Moreover, we could not elucidate the similarity and difference in lesion distribution and disease severity between the two. Further study is needed.
This study has several limitations. Our study is small; therefore, for statistical reasons, it is necessary to increase the number of patients in future studies. Also, our study is retrospective and only includes patients from Japan, where AMAN patients are more frequent compared with other western countries. Because the possibility of host susceptibility factors could not be excluded, these observations may apply to only Japanese patients and not patients of other nationalities. Prospective studies in various population groups are required. Moreover, the test for antiganglioside antibodies was performed; however, only antibodies against GM1 and GQ1b were assessed in a limited number of patients. Although our study still showed that IgG anti-GM1 antibody was more frequently found in unclassified patients than in patients with the “MS-AIDP pattern,” which corresponds to true AIDP, more comprehensive tests for antiganglioside antibodies would make more unclassified patients positive for the antibodies and would confirm our conclusion that unclassified patients would be included in AMAN with RCF. Finally, although serial electrophysiological studies were performed, the timing, frequency, and period varied for each patient. In some patients, serial studies were performed only few times over a short period or not performed at all. Further studies, in which more frequent electrophysiological assessment would be undertaken at predetermined time points over a longer period of time, may identify the pathophysiology in greater detail and with greater confirmation.
We previously demonstrated that the pathophysiology of the ‘‘M-AIDP pattern’’ is AMAN with RCF, that the pathophysiology of the ‘‘MS-AIDP pattern’’ is true AIDP, and that the pathophysiology of the ‘‘AMAN pattern’’ is AMAN with axonal degeneration [16]. The findings of the present study demonstrate the pathophysiology of unclassified patients is AMAN with RCF. Taken together, the classification of GBS into the four patterns, which requires only an initial nerve conduction study, allows the determination of detailed pathophysiology, including RCF, in all GBS patients from the early phase of disease. This classification may have utility not only in the clinical setting but also in research fields: the classification makes it possible to make accurate comparisons not only between AMAN and AIDP but also between AMAN with axonal degeneration and AMAN with RCF, leading to the elucidation of the mechanism of disease progression in AMAN.
References
Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, Asbury AK, Blaser MJ, McKhann GM (1995) Guillain–Barre syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain 118(Pt 3):597–605
Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, Zochodne DW (1986) An acute axonal form of Guillain–Barre polyneuropathy. Brain 109(Pt 6):1115–1126
McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, Wu HS, Zhaori G, Liu Y, Jou LP et al (1993) Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol 33:333–342
Yuki N, Hartung HP (2012) Guillain–Barre syndrome. N Engl J Med 366:2294–2304
Kuwabara S, Yuki N (2013) Axonal Guillain–Barre syndrome: concepts and controversies. Lancet Neurol 12:1180–1188
Yuki N, Taki T, Inagaki F, Kasama T, Takahashi M, Saito K, Handa S, Miyatake T (1993) A bacterium lipopolysaccharide that elicits Guillain–Barre syndrome has a GM1 ganglioside-like structure. J Exp Med 178:1771–1775
Kokubun N, Nishibayashi M, Uncini A, Odaka M, Hirata K, Yuki N (2010) Conduction block in acute motor axonal neuropathy. Brain 133:2897–2908
Uncini A, Manzoli C, Notturno F, Capasso M (2010) Pitfalls in electrodiagnosis of Guillain–Barre syndrome subtypes. J Neurol Neurosurg Psychiatry 81:1157–1163
Hiraga A, Kuwabara S, Ogawara K, Misawa S, Kanesaka T, Koga M, Yuki N, Hattori T, Mori M (2005) Patterns and serial changes in electrodiagnostic abnormalities of axonal Guillain–Barre syndrome. Neurology 64:856–860
Brown WF, Snow R (1991) Patterns and severity of conduction abnormalities in Guillain–Barre syndrome. J Neurol Neurosurg Psychiatry 54:768–774
Olsson Y (1990) Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol 5:265–311
Brown WF, Feasby TE (1984) Conduction block and denervation in Guillain–Barre polyneuropathy. Brain 107(Pt 1):219–239
Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, Noda M (1998) IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain–Barre syndrome. Ann Neurol 44:202–208
Capasso M, Caporale CM, Pomilio F, Gandolfi P, Lugaresi A, Uncini A (2003) Acute motor conduction block neuropathy Another Guillain–Barre syndrome variant. Neurology 61:617–622
Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, Hirata K, Baba H, Yuki N (2007) Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci 27:3956–3967
Hosokawa T, Nakajima H, Unoda K, Yamane K, Doi Y, Ishida S, Kimura F, Hanafusa T (2014) An electrophysiological classification associated with Guillain–Barre syndrome outcomes. J Neurol 261:1986–1993
Asbury AK, Cornblath DR (1990) Assessment of current diagnostic criteria for Guillain–Barre syndrome. Ann Neurol 27(Suppl):S21–S24
Yuki N, Kokubun N, Kuwabara S, Sekiguchi Y, Ito M, Odaka M, Hirata K, Notturno F, Uncini A (2012) Guillain–Barre syndrome associated with normal or exaggerated tendon reflexes. J Neurol 259:1181–1190
Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM (1978) Controlled trial prednisolone in acute polyneuropathy. Lancet 2:750–753
Kimura J (1989) electrodiagnosis in diseases of nerve and muscle: principles and practice. F.A. Davis, Philadelphia
Isose S, Kuwabara S, Kokubun N, Sato Y, Mori M, Shibuya K, Sekiguchi Y, Nasu S, Fujimaki Y, Noto Y, Sawai S, Kanai K, Hirata K, Misawa S (2009) Utility of the distal compound muscle action potential duration for diagnosis of demyelinating neuropathies. J Peripher Nerv Syst 14:151–158
Kalita J, Misra UK, Das M (2008) Neurophysiological criteria in the diagnosis of different clinical types of Guillain–Barre syndrome. J Neurol Neurosurg Psychiatry 79:289–293
Kuwabara S, Ogawara K, Mizobuchi K, Koga M, Mori M, Hattori T, Yuki N (2000) Isolated absence of F waves and proximal axonal dysfunction in Guillain–Barre syndrome with antiganglioside antibodies. J Neurol Neurosurg Psychiatry 68:191–195
Feasby TE, Hahn AF, Brown WF, Bolton CF, Gilbert JJ, Koopman WJ (1993) Severe axonal degeneration in acute Guillain–Barre syndrome: evidence of two different mechanisms? J Neurol Sci 116:185–192
Griffin JW, Li CY, Ho TW, Tian M, Gao CY, Xue P, Mishu B, Cornblath DR, Macko C, McKhann GM, Asbury AK (1996) Pathology of the motor-sensory axonal Guillain–Barre syndrome. Ann Neurol 39:17–28
Albers JW, Donofrio PD, McGonagle TK (1985) Sequential electrodiagnostic abnormalities in acute inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve 8:528–539
Kuwabara S, Ogawara K, Misawa S, Mizobuchi K, Sung JY, Kitano Y, Mori M, Hattori T (2004) Sensory nerve conduction in demyelinating and axonal Guillain–Barre syndromes. Eur Neurol 51:196–198
Kuwabara S, Ogawara K, Misawa S, Koga M, Mori M, Hiraga A, Kanesaka T, Hattori T, Yuki N (2004) Does Campylobacter jejuni infection elicit “demyelinating” Guillain–Barre syndrome? Neurology 63:529–533
Clouston PD, Kiers L, Zuniga G, Cros D (1994) Quantitative analysis of the compound muscle action potential in early acute inflammatory demyelinating polyneuropathy. Electroencephalogr Clin Neurophysiol 93:245–254
Baba M, Matsunaga M (1984) Recovery from acute demyelinating conduction block in the presence of prolonged distal conduction delay due to peripheral nerve constriction. Electromyogr Clin Neurophysiol 24:611–617
Kokubun N, Shahrizaila N, Hirata K, Yuki N (2013) Reversible conduction failure is distinct from neurophysiological patterns of recovery in mild demyelinating Guillain–Barre syndrome. J Neurol Sci 326:111–114
Yuki N (2001) Infectious origins of, and molecular mimicry in, Guillain–Barre and Fisher syndromes. Lancet Infect Dis 1:29–37
Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, Swan AV (1998) Electrophysiological classification of Guillain–Barre syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain–Barre Syndrome Trial Group. Ann Neurol 44:780–788
The Italian Guillain-Barre Study Group (1996) The prognosis and main prognostic indicators of Guillain–Barre syndrome a multicentre prospective study of 297 patients. Brain 119(Pt 6):2053–2061
Kuwabara S, Asahina M, Koga M, Mori M, Yuki N, Hattori T (1998) Two patterns of clinical recovery in Guillain–Barre syndrome with IgG anti-GM1 antibody. Neurology 51:1656–1660
Ropper AH, Adelman L (1992) Early Guillain–Barre syndrome without inflammation. Arch Neurol 49:979–981
Gordon PH, Wilbourn AJ (2001) Early electrodiagnostic findings in Guillain–Barre syndrome. Arch Neurol 58:913–917
Vucic S, Cairns KD, Black KR, Chong PS, Cros D (2004) Neurophysiologic findings in early acute inflammatory demyelinating polyradiculoneuropathy. Clin Neurophysiol 115:2329–2335
Yokota T, Inaba A, Yuki N, Ichikawa T, Tanaka H, Saito Y, Kanouchi T (1996) The F wave disappears due to impaired excitability of motor neurons or proximal axons in inflammatory demyelinating neuropathies. J Neurol Neurosurg Psychiatry 60:650–654
Manganelli F, Pisciotta C, Iodice R, Calandro S, Dubbioso R, Ranieri A, Santoro L (2009) Case of acute motor conduction block neuropathy (AMCBN). Muscle Nerve 39:224–226
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standards
This study received institutional review board approval.
Informed consent
Informed consent was obtained from all participants according to the Declaration of Helsinki.
Rights and permissions
About this article

Cite this article
Hosokawa, T., Nakajima, H., Unoda, K. et al. Serial electrophysiological findings in Guillain–Barré syndrome not fulfilling AIDP or AMAN criteria. J Neurol 263, 1709–1718 (2016). https://doi.org/10.1007/s00415-016-8192-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8192-2