
Abstract
Establishing a molecular diagnosis of autosomal recessive cerebellar ataxias (ARCA) is challenging due to phenotype and genotype heterogeneity. We report the validation of a previously published clinical practice-based algorithm to diagnose ARCA. Two assessors performed a blind analysis to determine the most probable mutated gene based on comprehensive clinical and paraclinical data, without knowing the molecular diagnosis of 23 patients diagnosed by targeted capture of 57 ataxia genes and high-throughput sequencing coming from a 145 patients series. The correct gene was predicted in 61 and 78 % of the cases by the two assessors, respectively. There was a high inter-rater agreement [K = 0.85 (0.55–0.98) p < 0.001] confirming the algorithm’s reproducibility. Phenotyping patients with proper clinical examination, imaging, biochemical investigations and nerve conduction studies remain crucial for the guidance of molecular analysis and to interpret next generation sequencing results. The proposed algorithm should be helpful for diagnosing ARCA in clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
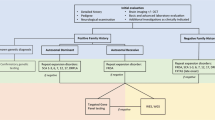
Autosomal recessive cerebellar ataxias (ARCAs) are heterogeneous and complex inherited neurodegenerative disorders that affect the cerebellum, the spinal cord and the peripheral nerves. Molecular diagnosis of the ARCAs is challenging due to both phenotypic and genetic heterogeneity [1]. It was formerly a step by step approach with serial sequencing of several genes by the Sanger technique. The advent of affordable next generation sequencing (NGS) technologies allows now sequencing of exomes or large gene panels for diagnosis of rare neurological diseases such as ARCAs [2]. However, massive amount of data with multiple rare variants in several genes in a single patient increases the complexity of the analysis. A collaborative cross-talk between molecular geneticists and clinicians is even more necessary than before for NGS diagnosis validation in clinical practice. Age of onset, ataxia progression rate and careful clinical examination combined with laboratory, morphological and neurophysiological investigations [including vitamin E, alpha-fetoprotein (AFP), albumin, cholestanol, brain MRI, nerve conduction studies (NCS)] are useful for reaching a diagnostic conclusion. A new classification of ARCAs also has been established comprising 3 groups: ARCA with pure sensory neuropathy, ARCA with sensorimotor axonal neuropathy and ARCA without neuropathy. An algorithm for the diagnosis of ARCAs based on these items has been proposed by our group according to personal experience and literature data [3] (Fig. 1). We conducted a blind study to validate this clinical practice-based algorithm on a series of patients with molecular diagnosis of ARCA. These patients were part of a cohort of patients consecutively investigated with targeted capture sequencing of a panel of 57 ataxia genes.

Algorithm for the diagnosis of autosomal recessive cerebellar ataxias (simplified and adapted [3]). The combination of natural history, associated clinical signs and paraclinical data (such as brain MRI, nerve conduction studies and several biomarkers) leads to one or a few diagnosis. yr years, EMG electroneuromyography
Patients and methods
Between 2010 and 2012, 145 unrelated index patients were recruited in 12 tertiary centers for movement disorders: 130 in France and 15 in Algeria. Inclusion criteria for the NGS analysis were the combination of: (1) progressive cerebellar ataxia; (2) age at onset before 60 years; (3) molecular analysis negative for Friedreich ataxia and other investigations depending on clinical assessment; (4) recessive inheritance or sporadic cases. Written informed consent was obtained from all participants and local ethics committee approved the study. Hundred and forty-five consecutive patients were analyzed by a targeted exon-capture strategy coupled with multiplexing and high-throughput sequencing of 57 genes causing ataxia when mutated (listed in supplementary file-A). Library preparation, targeted capture and sequencing were realized as previously reported [4]. NGS analysis is detailed in supplementary file-B [5].
Hundred and thirty-four patients were presenting ataxia starting before 40 years and 11 patients had late onset ataxia, starting after 40 years. Our cohort was mainly comprised of sporadic cases: 85 patients (59 %) had neither familial history of ataxia nor consanguinity. Fifty-four patients (37 %) had a recessive pedigree (2 or more affected in the kindred or isolated case with parental consanguinity). A molecular diagnosis was made in 27/145 patients (19 %) with mutations in ARCA genes. Among the 27 patients with ARCA molecular diagnosis, 4 were excluded due to the lack of available data. Molecular data of these 4 patients and clinical data of 118 patients without diagnosis are summarized in supplementary file C [6] and D, respectively.
We selected the remaining 23 ARCA patients with an established molecular diagnosis to assess the validity of the clinical practice-based algorithm. Two movement disorders specialists (MA, CT) performed independently a blind analysis based on the clinical (age at onset, current age, current disability based on scale of assessment and rating of ataxia (SARA) [7] score and/or spinocerebellar degeneration functional score (SDFS) [1], exhaustive clinical examination abnormalities including ocular motor signs, movements disorders, pyramidal signs, mental retardation) and paraclinical (biomarkers—especially vitamin E, AFP, albumin, cholestanol-, brain MRI, NCS findings) but molecular data: each patient had to be categorized in one of the three ARCA groups as described above and ranking of the two most probable disease causing genes was achieved. Inter-rater agreement was assessed by computing a weighted Kappa coefficient (K) with ‘“squared”‘ weights. All mutated genes affecting the 23 patients were ordered according to the data of the literature as followed on a ordinal scale to take account of the degree of disagreement [group without neuropathy: (1) SYNE1 (2) ANO10, (3) ADCK3; group with pure sensory neuronopathy: (1) POLG, (2) TTPA; group with axonal sensorimotor neuropathy (1) APTX, (2) SETX, (3) CYP27A1, (4) SACS]. Genes that showed larger number of associated phenotypic differences were separated by greater distance on the ordinal scale. Confidence interval was calculated using the adjusted bootstrap percentile (BCa) method based on 10,000 replicates. A “z” test was performed to assess if the classification which produced the Kappa statistic is significantly better than a random result (K = 0). A p value <0.05 was considered statistically significant. Analyses were performed using R software version 3.1.0 (R Project for Statistical Computing) with the “irr” package.
Results
Twenty-three patients were investigated with the clinical practice-based algorithm. The age of onset ranged from 1 to 47 years (median 16). Genetic analysis identified two pathogenic mutations in ANO10 (6 patients), in SETX (4), in SYNE1 and ADCK3 (3 each), in SACS and APTX (2 each) and in TTPA, CYP27A1, POLG (1 each) (Table 1). The correct ARCA group was found in all patients by the two assessors. The gene ranked first by the first assessor (MA) was correct in 14/23 cases (61 %) and 18/23 cases (78 %) for the second assessor (CT). The most frequent error (11 errors/14) was misdiagnosis within the ARCA group without neuropathy probably due to the closely overlapping phenotypes of pure cerebellar ataxias (especially ARCA1, ARCA3 and to some extent ARCA2), with a wide range of age at onset. Five errors were shared by both experts. Considering the two most probable genes according to the assessors, the correct diagnosis was identified in 18/23 (78 %) and 21/23 (91 %), respectively. There was a high inter-rater agreement [K = 0.85 (0.55–0.98) p < 0.001] on the first gene ranked by the assessors confirming the algorithm’s reproducibility.
Discussion
We report on the validation of a clinical practice-based algorithm in a series of patients, based on a blind analysis of clinical and paraclinical data. Given the high percent of correct diagnosis in our study (the assessors were able to find the good molecular diagnosis in 2/3 and 3/4 of cases, respectively), it is expected that this algorithm will be useful in clinical practice for neurologists and geneticists. Moreover, the algorithm allowed us to identify nine distinct entities, including entities that belong to the three different groups of the new classification of ARCAs [3]. One hundred forty-five patients suspected with ARCA were included in the study which is a high number since Friedreich ataxia was previously excluded. Therefore, the evaluation of the 23 patients with a molecularly confirmed diagnosis is relevant.
Confirmation of diagnosis by NGS can be straightforward in presence of clear-cut mutations, even with few clinical data. However, NGS data analysis frequently reveals several variants of unknown significance especially missense mutations in one or more ARCA-causing genes. In these difficult cases, proper phenotyping of ARCA patients, including precise clinical examination, biochemical investigations, brain imaging and NCS, is still necessary for guidance of genetic analysis and interpretation of the NGS data: these data have to be in agreement with an already described phenotype in order to confirm the variant’s pathogenicity. Similarly, appropriate knowledge of the several entities and of their description in the literature is recommended for the best management of such scarce diseases.
Despite the relevance of the algorithm, it is not infallible since few errors were made. ANO10 and SYNE1 mutations are responsible for close phenotypes [8, 9] with pure cerebellar ataxia and slow progression and may be difficult to distinguish without genetic analysis especially because there is no biomarker. Some errors were therefore done during the blinded assessment regarding these entities. Such algorithm is limited by the presence of atypical phenotypes associated with mutations in known genes. For instance, most patients with autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS) experience their first symptoms before 5 years of age, but in very few patients the first signs may occur after 20 or 30 years. Atypical phenotypes have also been reported regarding peripheral neuropathy which was lacking in 1/11 patient in a recent series of ARSACS [10]. Lack of neuropathy is also a rare feature in ataxia with oculomotor apraxia type 2 (AOA2), representing only 2.5 % of cases in a series of 90 patients [11]. Only one patient with ADCK3 confirmed mutations presented a mild axonal neuropathy (out of 34 patients described in the litterature [12–18]). In the same way, few patients with AOA2 or ataxia telangiectasia presented with the very unusual lack of elevated AFP serum level [11, 19]. However, the overall clinical and paraclinical assessment mostly lead to only a few possible diagnoses, confirming that the phenotype in one subtype of ARCA is relatively homogeneous. Few clinical findings (such as oculomotor apraxia, vertical supranuclear gaze palsy, spastic paraplegia, telangiectasia) as well as reliable biomarkers (such as vitamin E, AFP and albumin serum level) may also be suggestive of one or few diseases. The good results of our blinded assessment support this statement.
Herein, the percentage of positive diagnosis (19 %) is similar to previous studies on NGS in cerebellar ataxias [2] but remains low for several reasons including selection of patients with onset before 60 years of age (whereas ARCAs mostly occur before 30), exclusion of patients with Friedreich ataxia, absence of important ataxia genes in the gene panel, such as WFS1 and SPG7, and the fact that many genes have not been identified yet. It is also possible that a few sporadic cases have in fact polyglutamine SCA due to marked anticipation, particularly for SCA2 and SCA7. Polyglutamine expansions should therefore also be tested, along with Friedreich ataxia expansions.
Web resources
UCSC Genome Browser: http://genome.ucsc.edu/index.html
Ensembl Genome Browser: http://www.ensembl.org/index.html
Exome Variant Server (EVS), NHLBI GO Exome Sequencing Project (ESP), Seattle, WA: http://evs.gs.washington.edu/EVS (June, 2013)
References
Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, Ginglinger E, Boulay C, Courtois S, Drouot N, Fritsch M, Delaunoy JP, Stoppa-Lyonnet D, Tranchant C, Koenig M (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace. Eastern France: implications for clinical management Neurogenetics 11:1–12
Németh AH, Kwasniewska AC, Lise S, Parolin Schnekenberg R, Becker EB, Bera KD, Shanks ME, Gregory L, Buck D, Zameel Cader M, Talbot K, de Silva R, Fletcher N, Hastings R, Jayawant S, Morrison PJ, Worth P, Taylor M, Tolmie J, O’Regan M; UK Ataxia Consortium, Valentine R, Packham E, Evans J, Seller A, Ragoussis J (2013) Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 136:3106–3118
Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366:636–646
Redin C, Le Gras S, Mhamdi O, Geoffroy V, Stoetzel C, Vincent MC, Chiurazzi P, Lacombe D, Ouertani I, Petit F, Till M, Verloes A, Jost B, Chaabouni HB, Dollfus H, Mandel JL, Muller J (2012) Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alström syndromes. J Med Genet 49:502–512
Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, Stoetzel C, Blavier A, Laporte J, Muller J (2015) VaRank: a simple and powerful tool for ranking genetic variants. PeerJ 3:e796. doi:10.7717/peerj.796
Hamza W, Ali Pacha L, Hamadouche T, Muller J, Drouot N, Ferrat F, Makri S, Chaouch M, Tazir M, Koenig M, Benhassine T (2015) Molecular and clinical study of a cohort of 110 Algerian patients with autosomal recessive ataxia. BMC Med Genet 16:36
Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, Giunti P, Globas C, Infante J, Kang JS, Kremer B, Mariotti C, Melegh B, Pandolfo M, Rakowicz M, Ribai P, Rola R, Schöls L, Szymanski S, van de Warrenburg BP, Dürr A, Klockgether T, Fancellu R (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66:1717–1720
Renaud M, Anheim M, Kamsteeg EJ, Mallaret M, Mochel F, Vermeer S, Drouot N, Pouget J, Redin C, Salort-Campana E, Kremer HP, Verschuuren-Bemelmans CC, Muller J, Scheffer H, Durr A, Tranchant C, Koenig M (2014) Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol 71:1305–1310
Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA (2007) Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 39:80–85
Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schüle R, Haack TB, Schöning M, Biskup S, Rudnik-Schöneborn S, Senderek J, Hoffmann KT, MacLeod P, Schwarz J, Bender B, Krüger S, Kreuz F, Bauer P, Schöls L (2013) Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 8:41
Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schöls L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M’Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M (2009) Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 132:2688–2698
Lagier-Tourenne C, Tazir M, López LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, Anheim M, Lynch DR, Thibault C, Plewniak F, Bianchetti L, Tranchant C, Poch O, DiMauro S, Mandel JL, Barros MH, Hirano M, Koenig M (2008) ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet 82:661–672
Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, Munnich A, Rötig A (2008) CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet 82:623–630
Gerards M, van den Bosch B, Calis C, Schoonderwoerd K, van Engelen K, Tijssen M, de Coo R, van der Kooi A, Smeets H (2010) Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 10:510–515
Horvath R, Czermin B, Gulati S, Demuth S, Houge G, Pyle A, Dineiger C, Blakely EL, Hassani A, Foley C, Brodhun M, Storm K, Kirschner J, Gorman GS, Lochmüller H, Holinski-Feder E, Taylor RW, Chinnery PF (2012) Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry 83:174–178
Blumkin L, Leshinsky-Silver E, Zerem A, Yosovich K, Lerman-Sagie T, Lev D (2014) Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep 12:103–107
Mignot C, Apartis E, Marques Durr A, Lourenço C, Charles P, Devos D, Moreau C, de Lonlay P, Drouot N, Burglen L, Kempf N, Nourisson E, Chantot-Bastaraud S, Lebre AS, Rio M, Chaix Y, Bieth E, Roze E, Bonnet I, Canaple S, Rastel C, Brice A, Rötig A, Desguerre I, Tranchant C, Koenig M, Anheim M (2013) Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis 8:173
Liu YT, Hersheson J, Plagnol V, Fawcett K, Duberley KE, Preza E, Hargreaves IP, Chalasani A, Laurá M, Wood NW, Reilly MM, Houlden H (2014) Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation. J Neurol Neurosurg Psychiatry 85:493–498
Méneret A, Ahmar-Beaugendre Y, Rieunier G, Mahlaoui N, Gaymard B, Apartis E, Tranchant C, Rivaud-Péchoux S, Degos B, Benyahia B, Suarez F, Maisonobe T, Koenig M, Durr A, Stern MH, Dubois d’Enghien C, Fischer A, Vidailhet M, Stoppa-Lyonnet D, Grabli D, Anheim M (2014) The pleiotropic movement disorders phenotype of adult ataxia-telangiectasia. Neurology 83:1087–1095
H’mida-Ben Brahim D, M’zahem A, Assoum M, Bouhlal Y, Fattori F, Anheim M, Ali-Pacha L, Ferrat F, Chaouch M, Lagier-Tourenne C, Drouot N, Thibaut C, Benhassine T, Sifi Y, Stoppa-Lyonnet D, N’Guyen K, Poujet J, Hamri A, Hentati F, Amouri R, Santorelli FM, Tazir M, Koenig M (2011) Molecular diagnosis of known recessive ataxias by homozygosity mapping with SNP arrays. J Neurol 258:56–67
Lionnet C, Carra C, Ayrignac X, Levade T, Gayraud D, Castelnovo G, Besson G, Androdias G, Vukusic S, Confavreux C, Zaenker C, De Seze J, Collongues N, Blanc F, Tranchant C, Wallon D, Hannequin D, Gerdelat-Mas A, Brassat D, Clanet M, Zephir H, Outteryck O, Vermersch P, Labauge P (2014) Cerebrotendinous xanthomatosis: a multicentric retrospective study of 15 adults, clinical and paraclinical typical and atypical aspects. Rev Neurol (Paris) 170:445–453
Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J, Schulze M, Magri S, Sarto E, Mustafa M, Deconinck T, Haack T, Züchner S, Gonzalez M, Timmann D, Stendel C, Klopstock T, Durr A, Tranchant C, Sturm M, Hamza W, Nanetti L, Mariotti C, Koenig M, Schöls L, Schüle R, de Jonghe P, Anheim M, Taroni F, Bauer P (2016) SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large scale multi-centre study. Brain 139:1378–1393
Acknowledgments
Sequencing was performed by the IGBMC Microarray and Sequencing platform a member of the “France Génomique” consortium (ANR-10-INBS-0009). This study was supported by funds from the Agence Nationale pour la Recherche-Maladies Rares and Maladies Neurologiques et Psychiatriques (ANR-09-MNPS-001-01 to M.K. and A.D.) the ANR/E-rare JTC 2011 “Euro-SCAR” (2011-RARE-004-01 to M.K.) and the Agence de la Biomédecine (to J.-L.M.). M.R. was supported by a fellowship from the “Journées de Neurologie de Langue Française”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Financial disclosure
The authors declare no financial disclosure related to the research covered by this article. Martial Mallaret received travel grants from Ipsen and Merz. Mathieu Anheim received honoraria and travel grants from Actelion.
Conflicts of interest
None.
Additional information
M. Anheim and M. Koenig equally contributed to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
415_2016_8112_MOESM1_ESM.docx
Supplementary file: A-Panel of 57 ataxia mutated genes (in alphabetical order). B- Variants Analysis of NGS data. C-Clinical and molecular data of the patients without evaluation by the clinical practice-based algorithm. D- Comparative clinical data from patients with a molecular diagnosis (positive patients) and without (negative patients) in our series (DOCX 26 kb)
Rights and permissions
About this article

Cite this article
Mallaret, M., Renaud, M., Redin, C. et al. Validation of a clinical practice-based algorithm for the diagnosis of autosomal recessive cerebellar ataxias based on NGS identified cases. J Neurol 263, 1314–1322 (2016). https://doi.org/10.1007/s00415-016-8112-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8112-5