
Abstract
The hexanucleotide repeat expansion GGGGCC in the C9ORF72 gene larger than 30 repeats has been identified as a major genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Recent papers investigated the possible pathogenic role and associated clinical phenotypes of intermediate C9ORF72 repeat expansion ranging between 20 and 30 repeats. Some studies suggested its pathogenicity for typical Parkinson’s disease (PD), atypical parkinsonian syndromes, FTD with/without parkinsonism, and ALS with/without parkinsonism or with/without dementia. In our study, we aimed to screen patients affected by atypical parkinsonian syndromes or PD complicated by psychosis or dementia for the presence of C9ORF72 repeat expansions, and in unrelated age- and sex-matched healthy controls. Consecutive unrelated patients with atypical parkinsonian syndromes and patients with PD complicated by psychosis or dementia were included in this study. Atypical parkinsonian syndromes were further divided into two groups: one with patients who met the criteria for the classic forms of atypical parkinsonism [multiple system atrophy (MSA), Lewy body disease (LBD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD)] ;and patients who did not meet the above criteria, named non-classical atypical parkinsonism with or without dementia. Ninety-two unrelated patients (48 men, 44 women) were enrolled. None of the patients was found to be carriers of C9ORF72 repeat expansions with more than 30 repeats. Intermediate 20–30 repeat expansions were detected in four female patients (4.3 %). Three of them presented clinical features of atypical parkinsonian syndromes, two with non-classical atypical parkinsonism and dementia FTD-like, and one with non-classical atypical parkinsonism without dementia. The other patient presented clinical features of typical PD complicated by psychosis. Among 121 control subjects, none presented long or short expansion for the C9ORF72 gene. Our findings seem to support the hypothesis that the hexanucleotide expansions of C9ORF72 gene with intermediate repetitions between 20 and 29 repetitions could be associated with typical PD with psychosis or dementia and atypical parkinsonisms with dementia (non-classical atypical parkinsonism with dementia FTD-like) or without dementia (non-classical atypical parkinsonism upper MND-like), although the causal relationship is still unclear. In these latter patients, parkinsonism, more or less levodopa responsive, constituted the symptomatological central core at onset.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The large hexanucleotide (GGGGCC) repeat expansion in the first intron of the C9ORF72 (chromosome 9 open reading frame 72) gene has been identified as the most frequent mutation responsible for amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and comorbid FTD-ALS worldwide [1–3]. Subsequent works showed this mutation as causative of these diseases when present with more than 30 repetitions (large expansion) and that subjects carrying this mutation may present signs of atypical parkinsonism in early stages of the disease and increased incidence of parkinsonism with or without features of the FTLD/ALS complex in their relatives [4–10]. A recent work reported that C9orf72 expansions were the most common genetic cause of Huntington disease phenocopies [11].
However, the exact cutoff threshold for pathogenicity of C9ORF72 repeat expansions remains debatable. Furthermore, repeat sizes between 20 and 30 are usually referred to as intermediate alleles with unclear significance [12]. Although several studies have investigated the presence of large C9ORF72 repeat expansions in Parkinson disease (PD) or atypical parkinsonisms, its pathogenic role in these disease is not clear [12–23].
Interestingly, one of these studies identified the presence of an intermediate allele of 24 repeats in a patient with PD [23], and more recently a study found that short expansion (20–30 repeats) in the C9ORF72 gene can represent a risk factor for this disease [24]. Notwithstanding these observations, the same authors observed that long or short C9ORF72 repeat expansions were not associated with stringently selected autopsy-confirmed PD [25]. Recently, no pathologic repeat expansions in the C9ORF72 gene were detected in 100 neuropathologically confirmed cases of multiple system atrophy (MSA) [26]. On this line, also a recent work documented a case of MSA in a family with ALS and C9ORF72 hexanucleotide repeat expansions which met clinical, but not pathological, criteria for MSA [27].
These findings underscored the clinical heterogeneity of PD and supported the hypothesis that the expansions of C9ORF72 repeats in PD patients may be included in this heterogeneity [25]. To investigate whether C9ORF72 gene expansion could have a pathogenetic role for atypical parkinsonian syndromes or PD complicated by psychosis or dementia, we screened for C9ORF72 expansions in our cohort of patients affected by these diseases.
Materials and methods
Subjects
Consecutive unrelated patients with atypical parkinsonian syndromes or PD complicated by psychosis or dementia, attending the Movement Disorders Centre at the University Hospital, Cagliari, Sardinia, Italy, were included in the study. Then, the atypical parkinsonian syndromes were further divided into patients who met the criteria for the classic forms of atypical parkinsonism [MSA, Lewy body disease (LBD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD)] and patients who did not meet the above criteria; the latter were grouped separately.
Neurological examinations, family history, and neuropsychological and neuropsychiatric assessment were performed by four neurologists specialized in movement disorders, motor neuron disease, and dementia (AC, PS, GB, and GF). Diagnosis of PD was established according to the Gelb’s criteria [28], MSA diagnosis according to the Gilman’s criteria [29], PSP diagnosis according to the Litvan’s criteria [30], CBD diagnosis according to the Riley’s criteria [31], and LBD diagnosis according to the McKeith’s criteria [32]. Patients with atypical parkinsonism who did not meet the above criteria were named as non-classical atypical parkinsonism [33] without dementia or with dementia FTD-like. The clinical diagnosis was confirmed after at least 1 year of follow-up. Where necessary, additional elements of differential diagnosis were obtained by neuroimaging, neuropsychological, neurophysiology, and autonomic studies. None of the cases was autopsy confirmed.
Also, unrelated age- and sex-matched healthy Sardinian control individuals were investigated for the presence of C9ORF72 repeat expansions and included in this study.
Screening of hexanucleotide repeat expansion of the C9ORF72 gene
Hexanucleotide repeat number was assessed by repeat-primed PCR (or anchor PCR) using published primers [2]. PCR conditions have been modified from published protocol. Briefly the RP-PCR includes 7 % DMSO, 1 M betaine, 0.18 mM 7-deaza-dGTP and 0.9 mM MgCl2, 0.33 μM FWD, 0.33 μM REV, and 0.033 μM anchor primers. The REV primer includes ~4 GGGGCC repeats which allow it to bind anywhere in the repeat tract, plus a 21 bp ‘clamp’ sequence which binds adjacent to the repeat. The clamp sequence encourages amplification of the entire repeat.
Repeat expansions produce a characteristic sawtooth pattern with a 6-bp periodicity when fragment lengths are analyzed on a capillary-based sequencer.
PCR products were electrophoresed on an ABI 3500xL capillary analyzer and allele scoring was performed using GeneMapper v4.0 software (Applied Biosystems).
The assay allows samples to be categorized into those that carry only wild-type alleles (<20 repeats), intermediate repeats alleles (20–29 repeats), and a pathogenic repeat expansion (>30 repeats).
All subjects in the study, who were shown to be carriers of the intermediate repeats alleles, underwent the traditional ‘sizing PCR’, as follows.
We used the value of 20 repeats as a low threshold for intermediate repeat alleles, as the majority of controls in the literature are reported to have less than 20 repeats [1, 2, 18, 22, 23].
First, to determine the number of GGGGCC units and internal composition of the repeat, ten individuals homozygous for different fragment lengths were sequenced using the primers set, forward TACTCGCTGAGGGTGAACAA and reverse GCCTCCTCACTCACCCACT, designed with Primer3 [v. 0.4.0] [34, 35]. Then the GGGGCC hexanucleotide repeat in C9ORF72 was PCR amplified using the aforementioned forward and reverse primer set, labeling the same forward with a molecule of 5′-6-FAM. The PCR products were finally sized by fragment length analysis on an automated ABI3500xL DNA-analyzer (Applied Biosystems). Allele identification and scoring were performed using GeneMapper v4.0 software (Applied Biosystems).
The frequencies of carriers of the intermediate repeat alleles between the groups of patients with atypical parkinsonian syndromes or PD complicated by psychosis or dementia and control subjects were compared using the Fisher’s exact test. p < 0.05 was established as a statistically significant differential value.
Results
We screened 92 unrelated patients (48 men, 44 women) of Sardinian ancestry for C9ORF72 hexanucleotide repeat expansions using a repeat-primed polymerase chain reaction assay (Table 1). At the time of examination, the patients had a mean age of 73.3 (SD 16.1) years (range 41–90, interquartile range 69–79 years) and disease duration of 6.3 (SD 5.3) years (range 1–19, interquantile range 3–9 years).
Seventy-three cases (79.3 %) had clinical features consistent with atypical parkinsonian syndromes, while 19 (20.7 %) were diagnosed with PD complicated by psychosis or dementia. Among patients with atypical parkinsonian syndromes, 60 had a classical atypical parkinsonism and 13 a non-classical atypical parkinsonism without or with dementia.
None of the patients was found to be carriers of C9ORF72 repeat expansions with more than 30 repeats. Intermediate repeat expansions (20–30 repeats) were found in four female patients (4.3 %).
The frequency and sizes of hexanucleotide repeat expansion GGGGCC in the C9ORF72 gene (regrouped in classes) found in our population of patients are provided as supplemental data (Table S1).
Three of them presented with clinical features of non-classical atypical parkinsonism, (two with dementia FTD-like and one without dementia), while one patient presented clinical features of a typical common form of PD complicated by psychosis.
One hundred and twenty-one control subjects, age- and sex-matched, were studied, but none presented the long or short expansion for the C9ORF72 gene.
Intermediate-length expansions (20–29) were significantly more frequent in cases than in controls (p < 0.034).
The clinical characteristics of patients carrying the C9ORF72 expansion are reported in Table 2.
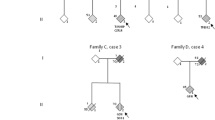
Short illustrative descriptions of these four patients are provided as supplemental data in the case reports section.
Discussion
The aim of this study was to investigate a possible role of C9ORF72 gene in the etiology of atypical parkinsonian syndromes and in PD complicated by psychosis or dementia.
Previous studies suggested that patients affected by ALS and FTD, carrying pathogenic repeat expansions (length of the expansion ≥30) in the C9ORF72 gene, may present features of atypical parkinsonism syndromes, in early stages of their disease, and have increased incidence of parkinsonism with or without features of the FTD/ALS complex in their relatives [4–9].
On the other hand, recent papers documented a wide frequency of psychosis in patients with FTD and C9ORF72 mutation [36, 37] suggesting the search for this mutation in patients with psychiatric disorders [37].
On these bases, other studies investigated a possible role of C9ORF72 mutation in PD susceptibility, giving conflicting results [14, 18, 21]. Some research groups have investigated only the presence of long expansions in PD or atypical parkinsonism without evidence of its role in the pathogenesis of these diseases [12–23]. However, when intermediate expansions have also been considered (20–29 repeats), the presence of a 24 repeats allele was documented in a patient affected by typical PD [22]. Thus, it has been hypothesized that the intermediate expansions of the C9ORF72 mutation might express different pathological conditions from FTD and ALS, playing as a risk factor for typical PD [24]. However, the same authors observed that expanded or intermediate C9ORF72 repeats were not associated with stringently selected autopsy-confirmed PD [25].
In our study, none of the patients was detected with the expanded pathogenic (>30) C9ORF72 repeat expansions, while four patients (4.3 %), carriers of intermediate-sized hexanucleotide C9ORF72 repeats, were identified. These data are in line with previous findings [21–23].
Among these subjects, three, respectively, with 20, 23, and 28 repetitions, suffered from an atypical parkinsonian syndrome, while the fourth, carrying 22 repeats, presented a classical form of PD, complicated by a peculiar transient psychotic disorders.
Interestingly, two patients with atypical parkinsonism syndromes (patient 3 with 23 repeats and patient 4 with 28 repeats) were affected by a severe form of rigid akinetic parkinsonism accompanied since the onset by severe psychiatric disturbances of the dissociative/affective sphere (schizoaffective psychosis, patient 3; and major depression with severe apathy and anhedonia, patient 4) and quickly complicated with cognitive disorders moving toward signs of dementia FTD-like.
Differently, patient 1 (with 20 repeats) presented at onset with features compatible with a typical PD, levodopa responsive, but after 5 years she developed signs of upper motor neuron involvement and bulbar dysfunction, with severe dysarthria and dysphagia, evolving over the years toward a form of upper MND with severe parkinsonism, but without dementia. Finally, patient 2 had a typical PD, optimally responsive to levodopa and with the classical clinical evolution of this disease. In this case, the only data that can be related to the clinical phenotype observed in patients carrying the long expansion of the C9ORF72 were the onset of a psychotic episode [36, 37].
Another interesting observation in our group of four patients with C9ORF72 intermediate repetitions was the presence, in three of them, of a levodopa-responsive parkinsonism: in one case with optimal response (case 2), in another case with clear response for many years, and less noticeable in the very advanced stage (case 1) and in the third case with modest but clear response (case 4).
This means that in the presence of a clear parkinsonism, especially in the early years of illness, the response to levodopa should not be considered as a sufficient condition to make a diagnosis of typical PD.
A limitation of our study is that the cases of our cohort were largely clinically diagnosed without pathologic diagnosis, which is the definitive method for establishing the diagnosis in DLB, MSA, etc. Another limitation is the number of samples, but this is dependent on the frequency of these cases.
In summary, this study seems to support the hypothesis that the hexanucleotide expansions of C9ORF72 gene with intermediate repetitions between 20 and 29 repetitions could be associated with different clinical variable phenotypes ranging from classical PD to atypical parkinsonism; thus, these intermediate repetitions might represent a potential risk factor, although the causal relationship is still unclear. We are of the opinion that these cases of non-classical atypical parkinsonism, in which the parkinsonism is the fundamental core of the symptomatology in the early years of illness, are probably destined, however, to evolve into overlapping forms of FTD (forms with psychosis and increasing cognitive impairment versus dementia) and overlapping forms of upper MND (forms without dementia with severe pyramidal tract signs, in particular bulbar dysfunctions).
Finally, further studies are needed to define the pathological spectrum and the clinical phenotype of C9ORF72 gene when it is expressed with an intermediate number of hexanucleotide repetitions.
References
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(245):256
Renton AE, Majounie E, Waite A et al (2011) ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268
Majounie E, Renton AE, Mok K et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11(4):323–330. doi:10.1016/S1474-4422(12)70043-1
Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C (2013) Current insights into the C9ORF72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci 36(450):459
Takada LT, Pimentel ML, DeJesus-Hernandez M et al (2012) Frontotemporal dementia in a Brazilian kindred with the C9ORF72 mutation. Arch Neurol 69(1149):1153
Savica R, Adeli A, Vemuri P et al (2012) Characterization of a family with c9FTD/ALS associated with the GGGGCC repeat expansion in C9ORF72. Arch Neurol 69(1164):1169
Luigetti M, Quaranta D, Conte A et al (2013) Frontotemporal dementia, parkinsonism and lower motor neuron involvement in a patient with C9ORF72 expansion. Amyotroph Lateral Scler Frontotemporal Degener 14(66):69
Hsiung GY, DeJesus-Hernandez M, Feldman HH et al (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135(709):722
Van Langenhove T, van der Zee J, Gijselinck I et al (2013) Distinct clinical characteristics of C9ORF72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 70(1):9
Lindquist S, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, Svenstrup K, Pinborg L, Vestergaard K, Hjermind L, Stokholm J, Andersen B, Johannsen P, Nielsen J (2012) Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet 83:279–283
Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, Adamson G, Mudanohwo E, McColgan P, Haworth A, Wild EJ, Sweeney MG, Houlden H, Mead S, Tabrizi SJ (2014) C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 82(4):292–299
Schottlaender LV, Polke JM, Ling H et al (2015) The analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonism. Neurobiol Aging 36(2):1221.e1–1221.e6
Theuns J, Verstraeten A, Sleegers K, Wauters E, Gijselinck I, Smolders S et al (2014) Global investigation and meta-analysis of the C9ORF72 (G4C2)n repeat in Parkinson disease. Neurology 83(21):1906–1913
Majounie E, Abramzon Y, Renton AE, Keller MF, Traynor BJ, Singleton AB (2012) Large C9ORF72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol Aging 33(10):2527.e1–2527.e2
Ticozzi N, Tiloca C, Calini D, Gagliardi S, Altieri A, Colombrita C, Cereda C, Ratti A, Pezzoli G, Borroni B, Goldwurm S, Padovani A, Silani V (2014) C9ORF72 repeat expansions are restricted to the ALS-FTD spectrum. Neurobiol Aging 35(4):936.e13–936.e17
Harms MB, Neumann D, Benitez BA et al (2013) Parkinson disease is not associated with C9ORF72 repeat expansions. Neurobiol Aging 34:1519.e1–1519.e2
DeJesus-Hernandez M, Rayaprolu S, Soto-Ortolaza AI et al (2013) Analysis of the C9ORF72 repeat in Parkinson’s disease, essential tremor and restless legs syndrome. Parkinsonism Relat Disord 19(198):201
Xi Z, Zinman L, Grinberg Y et al (2012) Investigation of C9ORF72 in 4 neurodegenerative disorders. Arch Neurol 69(1583):1590
Jiao B, Guo JF, Wang YQ et al (2013) C9ORF72 mutation is rare in Alzheimer s disease, Parkinson’s disease, and essential tremor in China. Front Cell Neurosci 7:164
Akimoto C, Forsgren L, Linder J et al (2013) No GGGGCC hexanucleotide repeat expansion in C9ORF72 in parkinsonism patients in Sweden. Amyotroph Lateral Scler Frontotemporal Degener 14(26):29
Lesage S, Le Ber I, Condroyer C et al (2013) C9ORF72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136(385):391
Yeh TH, Lai SC, Weng YH et al (2013) Screening for C9ORF72 repeat expansions in parkinsonian syndromes. Neurobiol Aging 34(1311):1314
Daoud H, Noreau A, Rochefort D et al (2013) Investigation of C9ORF72 repeat expansions in Parkinson’s disease. Neurobiol Aging 34(1710):1719
Nuytemans K, Bademci G, Kohli MM et al (2013) C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann Hum Genet 77(351):363
Nuytemans K, Inchausti V, Beecham GW, Wang L, Dickson DW, Trojanowski JQ, Lee VM, Mash DC, Frosch MP, Foroud TM, Honig LS, Montine TJ, Dawson TM, Martin ER, Scott WK, Vance JM (2014) Absence of C9ORF72 expanded or intermediate repeats in autopsy-confirmed Parkinson’s disease. Mov Disord 29(6):827–830. doi:10.1002/mds.25838
Scholz SW, Majounie E, Revesz T, Holton JL, Okun MS, Houlden H, Singleton AB (2015) Multiple system atrophy is not caused by C9orf72 hexanucleotide repeat expansions. Neurobiol Aging 36(2):1223
Goldman JS, Kuo SH (2014) Multiple system atrophy and repeat expansions in C9orf72—reply. JAMA Neurol 71(9):1191–1192
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56:33–39
Gilman S, Wenning GK, Low PA et al (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71(670):676
Litvan I, Agid Y, Calne D et al (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47(1):9
Riley DE, Lange AE, Lewis A et al (1990) Cortico-basal ganglionic degeneration. Neurology 40(1203):1212
McKeith IG, Dickson DW, Lowe J et al (2005) Consortium on DLB. Diagnosis and management of dementiawith Lewy bodies: third report of the DLB Consortium. Neurology 2005(65):1863–1872
Stamelou M, Quinn NP, Bhatia KP (2013) “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord 28(9):1184–1199
Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3 new capabilities and interfaces. Nucleic Acids Res 40(15):e115
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23(10):1289–1291
Floris G, Borghero G, Cannas A, Di Stefano F, Costantino E et al (2012) Frontotemporal dementia with psychosis, parkinsonism, visuo-spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: a peculiar phenotype? J Neurol 259(8):1749–1751
Shinagawa S, Nakajima S, Plitman E, Graff-Guerrero A, Mimura M et al (2014) Psychosis in frontotemporal dementia. J Alzheimers Dis 42(2):485–499
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standards statement
This study has been approved by the appropriate institutional authority and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Additional information
A. Cannas, P. Solla, and A. Chio contributed equally to the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cannas, A., Solla, P., Borghero, G. et al. C9ORF72 intermediate repeat expansion in patients affected by atypical parkinsonian syndromes or Parkinson’s disease complicated by psychosis or dementia in a Sardinian population. J Neurol 262, 2498–2503 (2015). https://doi.org/10.1007/s00415-015-7873-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7873-6