
Abstract
This study aimed to elucidate the natural history of senile myoclonic epilepsy, a type of myoclonic epilepsy associated with Alzheimer’s disease in adult Down syndrome patients. Twelve Down syndrome patients over the age of 40 years with myoclonic epilepsy and Alzheimer’s disease underwent clinical, neuropsychological, neurophysiological, and neuroradiological study. The kariotypes, APOE polymorphisms, all exons in the PSEN1 and PSEN2 genes, and exons 16 and 17 in the APP gene were determined for all patients. CSF Aβ42, p-tau181, and t-tauAg were determined for two patients. Three main stages appeared during the course of the syndrome. The first stage was characterized by dementia onset (mean age: 51 ± 6.6 years), diffuse EEG abnormalities during sleep, and cerebral atrophy determined using neuroimaging. During the second stage, myoclonic epilepsy manifested (mean age: 51.4 ± 7.2 years) with myoclonic jerks time-locked to diffuse epileptiform abnormalities upon awakening, which was controlled with antiepileptic drugs. During the third stage (mean age: 54.8 ± 7.6 years), myoclonic seizures were replaced with nonepileptic myoclonus, and cerebellar signs, severe dementia, and photosensitivity developed. All patients showed complete trisomy 21. Mutations were ruled out on the APP, PSEN1, and PSEN2 genes, and APOE analysis revealed ε3/ε3 homozygosity. CSF biomarkers showed a decrease in Aβ42 and an increase in p-tau181. The natural history of senile myoclonic epilepsy is consistent with progressive myoclonus epilepsy. Chromosome 21 is implicated in its pathophysiology; however, other genetic and/or environmental risk factors cannot be excluded. The absence of the APOE type 4 allele could predict its progression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Down syndrome (DS) patients over the age of 40 years typically show clinical, neuroimaging, and neuropathological evidence of Alzheimer’s disease. A study of 53 DS patients showed Alzheimer’s disease in 8 % of patients aged 35–49 years, in 55 % of patients aged 50–59 years, and in 75 % of patients over 60 years [1]. The increased prevalence of Alzheimer’s disease is caused by an overexpression of the amyloid precursor protein (APP) gene due to the triplication of chromosome 21 [2], which leads to an increased accumulation of β-amyloid. Epilepsy may also be present in adult DS patients and late-onset epilepsy during the fifth and sixth decades of life is often associated with dementia [3]. Prasher and Corbett [4] and Lai and Williams [1] noted an epilepsy prevalence greater than 80 % in DS patients with dementia, whereas Mc Vicker et al. [5] reported a 46 % prevalence in DS patients over the age of 50 years. Therefore, the development of late-onset epilepsy is strongly suggestive of comorbid dementia, and an underlying common pathogenetic basis has been suggested [6]. Indeed, the increased accumulation of β-amyloid peptides triggers synaptic degeneration with abnormal synchronization of neuronal networks [7]. The electro-clinical features of epilepsy in DS patients with dementia are heterogeneous [1, 8–11]: generalized tonic–clonic, myoclonic, and partial complex seizures, which are easily controlled by antiepileptic drugs (AEDs), have been reported to be associated with variable EEG patterns. Nevertheless, case reports and small studies [9, 12–20] have stressed a homogeneous syndrome that is identified in the literature as “late onset myoclonic epilepsy in Down’s syndrome (LOMEDS)” or “senile myoclonic epilepsy” (Table 1). This syndrome is characterized by dementia, myoclonic jerks upon awakening, generalized tonic–clonic seizures, with evolution towards erratic myoclonus, full dependency and death within a few years, thereby apparently resembling a progressive myoclonus epilepsy (PME) [13, 15, 16, 20]. However, the disease spectrum is not well understood, and the current lack of clinical, neurophysiological, and molecular studies prevents the inclusion of this syndrome within the PME category.
The purpose of this study was to investigate the clinical and longitudinal outcomes and neurophysiological and molecular features of this disease to delineate the natural history of senile myoclonic epilepsy.
To our knowledge, this is the first study monitoring the course and characterizing the natural history of senile myoclonic epilepsy.
Methods
Twelve DS patients (5 males, 7 females) over the age of 40 years who were diagnosed with myoclonic epilepsy and Alzheimer’s disease in DS patients were prospectively monitored between January 2010 and December 2013 at the Epilepsy Center of the University of Foggia.
The patients are part of a prospective, ongoing clinical and neurophysiological study of DS patients aged >40 years.
This syndrome had manifested in seven patients prior to study entry, and five patients were identified during prospective observations. All patients were followed up twice per year and received full clinical, neurophysiological, and neuropsychological evaluations.
The mean age of the population at the start of the study was 54.1 ± 7.8 years (median 53.5, range 43–69).
The local ethics committee on human experimentation approved the study, and a written informed consent was obtained from relatives or a legal guardian of the patients.
Clinical and neurophysiological evaluation
Clinical data included demographic information (age, gender, family history, personal antecedents, systemic disorders) and details of the epilepsy diagnosis (age at seizure onset, seizure type, and frequency obtained from epilepsy diaries completed by caregivers, follow-up durations, and responses to the therapy).
The neurophysiological examination consisted of waking and sleep electroencephalography (EEG), long-term video-EEG/polygraphic monitoring, and assessment of multimodal evoked potentials (brainstem, somatosensory). The parameters of the video-EEG/polygraphic recordings included video-EEG (electrodes were placed based on the 10–20 International System with bipolar montage); an electromyogram (EMG) of both deltoid muscles, the right and left flexor and extensor muscles of the hand, and both tibialis anterior muscles; an EKG; and thoracic respiration (monitored using a strain gauge). Polygraphic EMG signals were recorded using pairs of surface electrodes with standard belly-tendon placement. Signals were acquired digitally (sampling frequency: 512 Hz; band-pass filters: 1.6–210 Hz; MicroMed System, Mogliano Veneto, Italy). The relationship between EEG and EMG bursts was analyzed by applying jerk-locked back-averaging. Myoclonus severity was scored used a simplified myoclonus rating scale [21]. Brainstem auditory evoked potentials (BAEPs) and somatosensory evoked potentials (SEPs) were recorded using standard laboratory procedures.
Neuropsychological assessment
Previous intellectual disability was assessed by reviewing of reported intelligence tests, whereas the diagnosis of dementia was generated using the modified ICD-10 criteria for adults with intellectual disabilities [22] based on DSM-IV-TR [23]. Moreover, patients were assessed using the Dementia Screening Questionnaire for Individuals with Intellectual Disabilities (DSQIID) [24], which is an observer-rated questionnaire used to screen for dementia in adults with DS. The DSQIID is comprised of 53 items that evaluate behavior and symptoms that are typically associated with dementia in adults with DS. A cut-off score of 20 was used to indicate for a positive diagnosis of dementia. The informant was either a relative or a carer who had known the patient with DS.
Imaging study
Three DS patients underwent brain magnetic resonance imaging (MRI; 1.5-Tesla System), and 9 underwent brain CT scanning.
Biomarkers
Genetic analyses
The diagnosis of DS was confirmed by karyotyping all DS patients. Apolipoprotein E (APOE) genotypes were determined based on polymerase chain reaction (PCR) amplifications followed by restriction enzymatic digestions. Moreover, all exons in the presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes, as well as exons 16 and 17 of the amyloid precursor protein (APP) gene, were analyzed by PCR followed by direct DNA sequencing.
Cerebrospinal fluid (CSF) analysis
CSF Aβ42, p-tau181 and t-tauAg were quantified using the Innotest ELISA in samples from patients 4 and 7, who provided consent for CSF analyses. ROC curves were used to determine of the cut-offs for single values, and a logarithm which correlates the IATI index β-amyloid 1-42/(240 + 1.18 × t-tau) with the value of p-tau181 was calculated.
Results
Clinical features
Table 2 summarizes the clinical features of the patients.
Syndrome onset
Dementia appeared earlier than myoclonic epilepsy, and the average elapsed time between dementia and seizure onset was 6.9 months (range 0.0–36). In patients 3, 9, 10, and 12, cognitive deterioration and seizures occurred temporally very close together, whereas in the remaining patients dementia preceded seizure onset by 10.3 months (range 5–36).
Dementia
The mean age at dementia onset was 51 ± 6.6 years (median 49.5, range 43–65). In patients 1–8, the earliest and primary sign of cognitive deterioration was the loss of the ability to conduct activities of daily living and the ability to perform tasks necessary for independent coping. Thereafter, a gradually progressive decline, including mental slowing and spatiotemporal disorientation, was reported in all patients (cut-off DSQIID score >20). The dementia was not recognized accurately or early; specifically, half of the patients (1, 3, 7, 8, 9, and 10) had been misdiagnosed with psychiatric disturbances, three (1, 3, 7) of whom had been treated with anti-psychotic drugs. Behavioral disturbances were reported rarely and were often misdiagnosed; specifically, in patients 3 and 8 behavioral signs were initially attributed to exacerbations of pre-existing psychiatric disturbances, whereas in patient 7 behavioral signs were attributed to the effects of levetiracetam.
Epilepsy
The mean age at onset of epilepsy was 51.4 ± 7.2 years (median 49.5, range 43–68). The first sign manifested as apparently generalized tonic–clonic seizures either on awakening from nocturnal sleep (patients 10 and 11) or within short time of awakening (patients 1–9 and 12). Nevertheless, careful clinical history revealed that in almost all patients the apparently generalized tonic–clonic seizures were preceded by myoclonic jerks. Subsequently, after a period ranging from a few weeks to 6 months, all patients presented monthly with myoclonic jerks, typically upon at awakening, that were more marked in the upper body (including arms, hands, and sometimes spreading to the head and trunk), symmetrical or asymmetrical, and rarely massive (patients 1 and 2); however, if massive, they were associated with falls. In patient 7, sudden auditory stimuli-evoked bilateral massive myoclonic jerks were associated with falls. Over months and years of progression, rare tonic–clonic and myoclonic seizures were replaced by nonepileptic myoclonus. Ten of patients were treated (reduction in seizure frequency ≥80 %) using AED monotherapy (seven with levetiracetam, one with valproate, one with lamotrigine, and one with oxcarbazepine), whereas patients 9 and 11 (reduction in seizure frequency ≥80 %) were treated with levetiracetam and carbamazepine, and levetiracetam and phenobarbital, respectively.
Other neurological features
Following progression of dementia and epilepsy, all patients developed cerebellar signs, which were mild in patients 4, 7, 9, 10, and 12 and moderate-severe in the other patients. In particular, an ataxic syndrome with incoordination and tremor, that was associated with nonepileptic myoclonus, developed at a later stage of illness at a mean age of 54.8 ± 7.6 years (median 53, range 45–69), with a mean period of 2.5 ± 1.1 years (median 3, range 1–4) from myoclonic epilepsy onset.
At the time of the last observation, myoclonus had occurred in all patients with the exception of patients 10 and 12, who showed a more recent onset of disease. Myoclonus was multi-focal, subcontinuous, rarely massive, and precipitated by movements, and it did not occur exclusively upon awakening. The mean myoclonus severity score was 4.1 (range 2–5).
Follow-up
The mean duration of follow-up after dementia onset was 3.5 ± 2.1 years (median 3.5, range 1–7). The disease course was inevitably progressive. At the time of the last follow-up, patients 1, 3, and 8 (died of pneumonia, the mean duration of symptoms was 6 years, range 4–7) and patients 2, 5, 6, and 11 (the mean duration of symptoms was 4.7 years, range 4–6) showed severe dementia with complete dependence on others for activities of daily living and were mute and bedridden. Myoclonic seizures were rare and responsive to AEDs. Incoordination, tremor, and nonepileptic myoclonus emerged in all cases.
Patients 4, 7, 9, 10, and 12 (the mean duration of symptoms was 2 years) showed an intermediate dementia with an inability to perform activities of daily living without help. Myoclonic seizures were rare and responsive to AEDs; cerebellar signs were mild or moderate, and myoclonus was absent (patients 10 and 12) or was low in severity score (patients 4, 7, and 9). For patient 7, probable sudden unexpected death in epilepsy (SUDEP) was reported after a 15-month duration of symptoms.
Neurophysiological features
Table 3 summarizes the video-EEG/polygraphic findings.
At the time of dementia onset, EEG background activity showed mild generalized slowing. The intermittent photic stimulation (IPS) at 1–30 Hz was normal. Physiological sleep patterns were always detected. In four patients (1, 2, 3, and 9) the sleep patterns were associated with sporadic diffuse spike-and- wave (SW) or polyspike-and-wave (PSW) discharges ( Fig. 1).

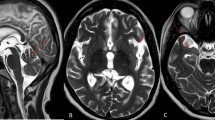
Stage 1: dementia onset. In the upper panel, EEG recordings in patients 1, 2, 3, and 9 are shown. Sleep revealed stereotypical EEG abnormalities in different patients. In the lower left panel, polygraphic recording are shown from patient 3 that are characterized by epileptiform abnormalities during sleep and that were not associated with myoclonic jerks (R. and L. Flex right and left flexor muscles of the hand, R. and L. Ext right and left extensor muscles of the hand, R. and L. Tib. A right and left muscles of the tibialis anterior). In the lower right panel, CT images of the brains of patients 3 and 11 reveal diffuse cerebral atrophy
At the time of epilepsy onset, EEG background activity was slow and associated with diffuse SW or PSW bursts typically upon awakening. Brief sequences of myoclonic jerks (corresponding to brief EMG bursts) were associated with EEG paroxysms in all patients. Nevertheless, in patients 1 and 2, unilateral myoclonia occurred without evident EEG correlates. In these cases jerk-locked back-averaging EEG analysis triggered from myoclonic jerks revealed a clear cortical spike at the centroparietal electrodes. IPS responses at 1–30 Hz were normal. Physiological sleep patterns were absent, whereas SW or PSW discharges persisted without activation (Fig. 2).

Stage 2: myoclonic epilepsy onset. In the upper panel, polygraphic recordings from patient 1 are shown. A cluster of myoclonic jerks correlating with epileptiform abnormalities are shown following tonic–clonic, apparently generalized, seizure upon awakening (R. and L. Flex right and left flexor muscles of the hand, R. and L. Ext right and left extensor muscles of the hand, Thorax thoracic breath). In the lower panel, video-polygraphic recordings from patients 2 and 3 are shown. Myoclonic jerks were prevalent in the right extensor muscle of the hand in patient 2 (left panel) and in the left flexor and extensor muscles of the hand in patient 3 (right panel) (R. and L. Ext right and left extensor muscles of the hand, R. and L. Tib. A right and left tibialis anterior muscles, Thorax thoracic breath)
As the disease progressed, the EEG background activity slowed further, with intermixed paroxysmal activity during awake periods and disorganized sleep. In patients with a longer clinical history, IPS triggered bursts of generalized SW or PSW bursts that were associated with myoclonic jerks, typically in the upper limbs, at a 1:1 ratio at frequencies of up to 12–15 Hz. In patient 1, IPS revealed a focal photic reflex myoclonus, and a back-average of the EEG, triggered from the onset of myoclonus by the right extensor muscle of the hand, showed a contralateral positive–negative transient in the fronto-central region that preceded the myoclonus EMG discharge by 20 ms. In patient 6, IPS at a frequency of 18 Hz evoked myoclonic jerks followed by a secondary tonic–clonic seizure. Polygraphic recordings revealed action myoclonus during posture maintenance and, in particular, erratic myoclonic jerks at rest that were associated with contralateral central spikes (Fig. 3).

Stage 3: progressive neurological deterioration. In the upper panel, an EMG recording from patient 1 is shown, showing erratic, parcellar myoclonic jerks at rest (R. and L. Flex right and left flexor muscles of the hand, R. and L. Ext right and left extensor muscles of the hand, R. and L. Tib. right and left tibialis anterior muscles). The lower panel shows the results of ILS in patients 1 (right panel focal photic reflex myoclonus, prevalent in the right extensor muscle of the hand) and 3 (left panel bilateral myoclonic jerks) (R and L. Delt right and left deltoid muscles, R. and L. Flex right and left flexor muscles of the hand, R. and L. Ext right and left extensor muscles of the hand, R. and L. Tib. A right and left tibialis anterior muscles, Thorax thoracic breath)
BAEPs and SEPs were evaluated in patients 3, 4, 7, and 12. They were generally unremarkable with the exception that the amplitude of cortical components in SEPs was moderately enlarged in patients 4 and 7.
Imaging findings
Table 3 summarizes the imaging features of patients.
Brain MRIs/CTs revealed diffuse cerebral atrophy in all patients (Fig. 1), that was associated with noncommunicating hydrocephalus in patient 8.
Biomarker results
Figure 4 summarizes the genetic and CSF features of the patients.

Biomarkers results. In the upper panel, the main results of genetic and CSF analyses are shown (APP amyloid precursor protein gene, PSEN1 and 2 presenilin 1 and 2 genes, APO E apolipoprotein E, NP not present). In the lower panel, the karyotypes of patients 4 and 5 are shown, as well as T-tau, Aβ1-42, and P-tau(181-P) in patients 4 and 7. The IATI (INNOTEST Amyloid Tau Index) is defined as Ab1-42/(240 + 1.18 × T-tau). The red line in the upper chart indicates IATI = 1
All patients showed a complete trisomy 21.
Mutations were ruled out by sequencing the APP, PSEN1, and PSEN2 genes in all patients.
No subject was homozygosus for the type 4 APOE allele (ε4/ε4). Homozygosity APOE ε3/ε3 was observed in all patients.
In patients 4 and 7, the CSF Aβ42 was 565 and 235.8 pg/ml (normal range 499–1,088), p-tau181 was 94.10, and 70.90 pg/ml (normal value <33), t-tauAg was 954 and 1,409.30 ng/ml (normal value <300), and Aβ42/p-tau181 ratio was 6 and 3.3 (normal value >7), respectively. Using ROC curve analyses, the CSF Aβ42 and p-tau181 values in these patients met the requirements for clinical use in discriminating Alzheimer’s disease from normal aging and other specific neurological disorders [25].
Discussion
The findings of this study indicate that
-
1.
The natural history of senile myoclonic epilepsy is compatible with PMEs;
-
2.
Chromosome 21 is implicated in its pathophysiology, although the existence of other genetic and/or environmental risk factors that have yet to be identified cannot be excluded.
A progressive myoclonus epilepsy
PMEs are a group of inherited disorders defined by the association of myoclonus and epilepsy with progressive neurological deterioration [26]. Five disease entities, Unverricht-Lundborg disease, Lafora’s disease, neuronal ceroid lipofuscinoses, mitochondrial disorders, and sialidoses, constitute the majority of PMEs cases. However, other forms of PMEs have been reported, and Alzheimer’s disease beginning in the third or fourth decade is a rare cause of the PME phenotype. Melanson et al. [27] presented two patients with PME that manifested at approximately 30 years of age in whom the clinical features, MRI findings, and brain neuropathological findings were typical of Alzheimer’s disease. In 1990 and 1994, the first reports of myoclonic epilepsy associated with Alzheimer’s disease in adult DS patients were published in abstract form [12, 13]. Pedersen [12] described 14 patients with adult-onset (third and fourth decade) generalized tonic/clonic and myoclonic seizures with symptoms occurring most often in the morning and that were associated with Alzheimer’s disease. Genton and Paglia [13] evaluated three adult DS patients (aged 41, 46, and 61 years) with myoclonic seizures that occurred initially upon awakening and were associated with Alzheimer’s disease. Thereafter, other small studies [9, 14–20] noted myoclonic epilepsy associated with dementia in adult DS patients and hypothesized that senile myoclonic epilepsy was a PME [13, 15, 16, 20]. Nevertheless, the lack of studies investigating the natural history of senile myoclonic epilepsy patients prevented this syndrome from being considered a PME. Indeed, although myoclonic epilepsy with Alzheimer’s-type dementia emerged in the 31 patients previously reported in the literature (Table 1), the clinical and neurophysiological progression of this syndrome were not characterized and a biomarker study were not performed. In this study, three progressive disease stages have been outlined, the characteristics of which are consistent with a PME phenotype in DS patients with Alzheimer’s disease and support the peculiarity of the association between PME and Alzheimer’s disease in DS patients. The first stage is characterized by dementia onset, with a loss of cognitive abilities and withdrawal from social interactions, followed by progressive decline. Dementia is not easily recognized, particularly in its early stages, and several patients had originally been misdiagnosed with psychiatric disturbances. The second stage is characterized by the appearance of a myoclonic epilepsy close to or a few months after from dementia onset. In addition to the early stages of PME, the onset of myoclonic epilepsy may mimic juvenile myoclonic epilepsy with early-morning myoclonic jerks time-locked to EEG diffuse paroxysmal discharges. As with dementia, a diagnosis of myoclonic epilepsy may be missed if the history is not solicited, and the first generalized tonic–clonic seizure often helps generate the diagnosis. In the later stage of illness (at a mean age of 54.8 ± 7.6 years, after a mean of 2.5 ± 1.1 years from myoclonic epilepsy onset), myoclonic seizures were replaced by nonepileptic myoclonus and by progressive neurological deterioration with progressive dementia, cerebellar signs, and photosensitivity, pointing to a diagnosis of PME. The intensity and rate of progression of the dementia, myoclonus, and cerebellar signs varied between individuals, although the duration of disease typically influenced the speed at which the disease progressed. After a mean duration of symptoms of 4-7 years, 63 % of patients showed severe dementia, with complete dependence on caregivers for activities of daily living, incoordination, and nonepileptic myoclonus. The remaining 37 % of patients (mean symptom duration of 2 years) showed intermediate dementia characterized by withdrawal from social interactions and were unable to perform activities of daily living without help, whereas cerebellar signs were mild or moderate and myoclonus was absent or showed a low severity score. Epileptic seizures are not typically influenced by the duration of the disease. Myoclonic seizures, which are more frequent during the early stages of the disease, often decrease in frequency during the following 2–4 years and may cease entirely following treatment with appropriate AEDs. Levetiracetam and valproate appear to be the most effective and may be considered as the first-line agents for this syndrome. At this late stage of the disease, it is important to clearly define myoclonic seizures and nonepileptic myoclonus. The majority of the myoclonic movements were not time-locked to EEG discharges, and a total of six back-averages performed on five patients excluded time-locked premyoclonic potentials suggesting a subcortical origin of these myoclonic phenomena. The possibility of either a cortical or subcortical origin of myoclonus in PME has been demonstrated previously [28], and the myoclonus features in our patients closely resemble other forms of PME. Finally, during this late phase, photoparoxysmal response (PPR) and photic reflex myoclonus may arise in patients, particularly in those with long clinical histories. In fact, PPR was usually absent at first observation and occurred later at follow-up, and the duration of epilepsy was not significantly different between patients with PPR and those without PPR.
Chromosome 21
The gene encoding APP is located on the proximal/mid-part of the long arm of chromosome 21 (21q21.3), and its overexpression is considered a factor critical for the early onset of brain amyloidosis noted in DS subjects. Our DS patients showed complete trisomy 21, and quantification, by ELISA, of the CSF abundance of Aβ42, p-tau181, and htauAg in two affected cases revealed a pattern typical of Alzheimer’s disease. Although brain neuropathological data are missing, the pathology is most likely an amyloidosis like Alzheimer’s disease due to APP accumulation.
Our biomarker data support a role of chromosome 21 and the APP locus in epileptogenesis in senile myoclonic epilepsy, presumably similar to that observed in Alzheimer’s disease patients. Indeed, high brain levels of β-amyloid may interfere with normal neuronal and synaptic activity. Recent experimental data [7, 29, 30] demonstrate that high levels of β-amyloid in the brain can cause epileptiform activity and cognitive deficits in transgenic mouse models of Alzheimer’s disease, with β-amyloid-induced neuronal hyperexcitability triggering progressive epilepsy. Nevertheless, the specific biology involved in the progression of senile myoclonic epilepsy remains unclear. The APOE type 4 allele is a well-known risk factor of Alzheimer’s disease, but contradictory data on the influence of the APOE type 4 allele on the disease progression exist in the literature [31]. Analysis of the APOE allele distribution in our study showed interesting findings because no patient was homozygous for ApoE ε4; however, surprisingly, we observed that all patients were homozygosus for APOE ε3. The lack of patients carrying the epsilon 4 allele could be due to chance, as its occurrence in populations from southern Europe is very low. Nevertheless, our data could also confirm previous findings that claim that the absence of the type 4 allele may predict rapid progression and short survival in some Alzheimer’s disease patients [32, 33]. However, the hypothesis of APOE as a predictor of progression should be confirmed in a much larger cohort of patients. Finally, no mutations were detected through the sequencing of all exons in the PSEN1 and PSEN2 genes and exons 16 and 17 in the APP gene.
Mutations in cystatin B (CSTB), a gene located on chromosome 21, are associated with Unverricht-Lundborg disease, a PME that shares features with senile myoclonic epilepsy [16]. An increase in CSTB does not induce any spontaneous epileptic activity and neither increase or decrease the propensity of trisomy mice to myoclonic seizures [34]. Nevertheless, over expression of CSTB akin to its deficiency may produce epileptic phenotype via the perturbation of some molecular pathway balance [34].
Therefore, the possible contribution of other genes located on chromosome 21 and the existence of other genetic and/or environmental risk factors that have yet to be identified cannot be excluded; thus, senile myoclonic epilepsy may be a complex genetic and environmental disease.
In conclusion, senile myoclonic epilepsy should now be added as a cause of PME. It is important for neurologists, especially epileptology experts, to be aware of this disease entity to avoid misdiagnoses and administration of inappropriate therapeutics. Future studies in larger series are needed to confirm the role about the APOE alleles on the disease progression and the prevalence of this progressive condition. In fact, it is evident that senile myoclonic epilepsy is not a rarity and could be one of the most common forms of PME in the light of the markedly increased life expectancy of DS patients.
References
Lai F, Williams RS (1989) A prospective study of Alzheimer’s disease in Down syndrome. Arch Neurol 46:849–853
Lott IT, Head E (2001) Down syndrome and Alzheimer’s disease: a link between development and aging. Ment Retard Dev Disabil Res Rev 7:172–178
Puri BK, Ho KW, Singh I (2001) Age of seizure onset in adults with Down’s syndrome. Int J Clin Pract 55:442–444
Prasher VP, Corbett JA (1993) Onset of seizures as a poor indicator of longevity in people with Down’s syndrome. Int J Geriatr Psychiatry 8:923–927
Mc Vicker RW, Shanks OEP, McClelland RJ (1994) Prevalence and associated features of epilepsy in adults with Down’s syndrome. Br J Psychiatry 164:528–532
Menendez M (2005) Down syndrome, Alzheimer’s disease and seizures. Brain Dev 27:246–252
Noebels J (2011) A perfect storm: converging paths of epilepsy and Alzheimer’s dementia intersect in hippocampal formation. Epilepsia 52:39–46
Evenhuis HM (1990) The natural history of dementia in Down’s syndrome. Arch Neurol 47:263–267
Vignoli A, Zambrelli E, Chiesa V et al (2011) Epilepsy in adult patients with Down syndrome: a clinical video EEG study. Epileptic Disord 13:125–132
Lefter S, Costello DJ, McNamara B, Sweeney B (2011) Clinical and EEG features of seizures in adults with Down syndrome. J Clin Neurophysiol 28:469–473
Lott IT, Doran E, Nguyen VQ et al (2012) Down syndrome and dementia: seizures and cognitive decline. J Alzheimers Dis 29:177–185
Pedersen (1990) Epilepsy of late onset in Down’s syndrome: a new epileptic syndrome. Epilepsia 31:613
Genton P, Paglia G (1994) Senile myoclonic epilepsy: late onset of myoclonic seizures associated with dementia in three Down syndrome patients. Epilepsia 35:13
Li LM, O’Donoghue MF, Sander JW (1995) Myoclonic epilepsy of late onset in trisomy 21. Arq Neuropsiquiatr 53(4):792–794
Vignatelli L, Meletti S, Ambrosetto G (1999) “Epilessia mioclonica progressiva” in paziente affetta da sindrome di Down con malattia di Alzheimer. Boll Lega It Epil 106(107):215–216
Moller JC, Hamer HM, Oertel WH, Rosenow F (2001) Late onset myoclonic epilepsy in Down’s syndrome (LOMEDS). Seizure 10:303–305
De Simone R, Daquin G, Genton P (2006) Senile myoclonic epilepsy in Down syndrome: a video and EEG presentation of two cases. Epileptic Disord 8(3):223–227
Crespel A, Gonzalez V, Coubes P, Gelisse P (2007) Senile myoclonic epilepsy of Genton: two cases in Down syndrome with dementia and late onset epilepsy. Epilepsy Res 77:165–168
Sangani M, Shahid A, Amina Shahram, Koubeissi M (2010) Improvement of myoclonic epilepsy in Down syndrome treated with Levetiracetam. Epileptic Disord 12(2):151–154
De Simone R, Puig XS, Gelisse P et al (2010) Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer’s disease in Down syndrome. Seizure 19:383–389
Magaudda A, Gelisse P, Genton P (2004) Antimyoclonic effect of levetiracetam in 13 patients with Unverricht-Lundborg disease: clinical obervations. Epilepsia 45:678–681
Aylward EH, Burt DB, Thorpe LU et al (1997) Diagnosis of dementia in individuals with intellectual disability. J Intellect Disabil Res 41:152–164
American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders (IV-TR). 4th edn-text revisited, Washington, DC
Deb S, Hare M, Prior L, Bhaumik S (2007) Dementia screening questionnaire for individuals with intellectual disabilities. Br J Psichiatry 190:440–444
Hulstaert F, Blennow K, Ivanoiu A et al (1999) Improved discrimination of AD patients using β-amyloid (1-42) and tau levels in CSF. Neurology 52:1555–1562
Marseille consensus group (1990) Classification of progressive myoclonus epilepsies and related disorders. Ann Neurol 28:113–116
Melanson M, Nalbantoglu J, Berkovic S et al (1997) Progressive myoclonus epilepsy in young adults with neuropathologic features of Alzheimer’s disease. Neurology 49:1732–1733
Tassinari CA, Rubboli G, Shibasaki H (1998) Neurophysiology of positive and negative myoclonus. Electroencephalogr Clin Neurophysiol 107:181–195
Palop J, Mucke L (2009) Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol 66:435–440
Minkeviciene R, Rheims S, Dobszay M et al (2009) Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29:3453–3462
Schmidt C, Wolff M, Weitz M et al (2011) Rapidly progressive Alzheimer’s disease. Arch Neurol 68:1124–1130
Schmidt C, Redyk K, Meissner B et al (2010) Clinical features of rapidly progressive Alzheimer’s disease. Dement Geriatr Cogn Disord 29:371–378
Giannattasio C, Poleggi A, Puopolo M et al (2008) Survival in Alzheimer’s disease is shorter in women carrying heterozygosity at codon 129 of the PRNP gene and no APOE epsilon 4 allele. Dement Geriatr Cogn Disord 25:354–358
Brault V, Martin B, Costet N, Bizot JC, Hérault Y (2011) Characterization of PTZ-induced seizure susceptibility in a down syndrome mouse model that overexpresses CSTB. PLoS ONE 6(11):e27845
Conflicts of interest
None of the authors has any conflict of interest to disclose.
Ethical standard
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
A full list of Apulian Study Group on Senile Myoclonic Epilepsy investigators is given in the Appendix.
Appendix
Appendix
Apulian Study Group on Senile Myoclonic Epilepsy:
Chairs: Giuseppe d’Orsi, Luigi M. Specchio (Epilepsy Center, Clinic of Nervous System Diseases, University of Foggia, Ospedali Riuniti, Foggia).
Collaborators: Elena Carapelle, Maria Teresa Di Claudio, Angela Lopopolo, Francesca Pacillo, Maria Grazia Pascarella, Marina Trivisano (Epilepsy Centre, Clinic of Nervous System Diseases, University of Foggia, Ospedali Riuniti, Foggia); Michele Falcone (1st Laboratory analysis, Ospedali Riuniti, Foggia); Gianpaolo Grilli (Radiological Unit, Ospedali Riuniti, Foggia, Italy); Potito Salatto (Department of Anesthesia and Intensive Care, University of Foggia, Foggia); Gabriella De Stefano (Neurology Ward, Opera Don Uva, Foggia); Flavia Meola (Rehabilitation Centre, Manfredonia); Davide Seripa (Geriatric Unit and Gerontology-Geriatric Research Laboratory, Department of Medical Sciences, “Casa Sollievo della Sofferenza”, San Giovanni Rotondo, Foggia); Vincenzo Demaio, Mauro Minervini, Salvatore Ottaviano (Neurology Ward, Opera Don Uva, Bisceglie); Teresa Francavilla, Angela La Neve, Concetta Luisi (Epilepsy Centre, Neurology Hospital “Amaducci”, University of Bari, Bari, Italy).
Rights and permissions
About this article

Cite this article
d’Orsi, G., Specchio, L.M. & On behalf of the Apulian Study Group on Senile Myoclonic Epilepsy. Progressive myoclonus epilepsy in Down syndrome patients with dementia. J Neurol 261, 1584–1597 (2014). https://doi.org/10.1007/s00415-014-7376-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7376-x