
Abstract
Longitudinally extensive transverse myelitis (LETM) is a characteristic feature of Neuromyelitis Optica (NMO), but it can also occur in several other inflammatory diseases of the central nervous system (CNS). An IgG autoantibody that binds to aquaporin-4 (AQP4), the predominant water channel of the CNS, is a reliable biomarker of the NMO spectrum disorders, and if detected predicts the recurrence of the myelitis. In this study, we compared the clinical and neuroimaging characteristics of AQP4-IgG+ and AQP4-IgG− LETM patients. Thirty-seven first-ever LETM patients were retrospectively evaluated and divided into two groups according to the presence of AQP4 autoantibodies. AQP4-IgG was detected in the serum and in the cerebrospinal fluid of sixteen patients. The female to male ratio was higher in AQP4-IgG+ patients. Intractable nausea and vomiting and paroxysmal tonic spasms often accompanied the LETM in AQP4-IgG+ patients. T2-weighted spinal cord MRI revealed that inflammatory lesions extending into the brainstem and involving the central grey matter occurred more frequently in AQP4-IgG+ LETM patients. Hypointense lesions on T1-weighted spinal cord MRI were detected more frequently in the seropositive group, and their presence correlated with attack severity. In conclusion, this study provides clinical and spinal cord neuroimaging clues that can help distinguishing AQP4-IgG+ LETM patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Longitudinally extensive transverse myelitis (LETM) is a characteristic feature of Neuromyelitis Optica (NMO) [1, 2], but it can also occur in several other inflammatory diseases of the central nervous system (CNS), such as acute disseminated encephalomyelitis, sarcoidosis and HTLV-1-associated myelopathy [3].
NMO is an autoimmune astrocytopathy that preferentially affects the optic nerve and spinal cord [1]. An IgG biomarker that binds to aquaporin-4 (AQP4), the predominant water channel of the CNS, distinguishes the spectrum of NMO from multiple sclerosis (MS) and other inflammatory diseases of the CNS [2]. This autoantibody binds to the AQP4 extracellular domain and is considered to have pathogenic potential [4, 5]. The detection of the AQP4-IgG autoantibody predicts the recurrence of the myelitis and the development of optic neuritis [6–8]. However, the clinical and radiological characteristics of isolated LETM associated with AQP4 autoantibodies remain to be fully elucidated. In this study, we evaluated the neuroimaging and the clinical features of LETM in relation to the AQP4-IgG serostatus.
Patients and methods
Study subjects
In this retrospective study with longitudinal follow-up, we performed a chart and neuroimaging review of patients with a first episode of isolated LETM admitted at the Multiple Sclerosis Center of the Catholic University between 1998 and 2012. The following inclusion criteria were adopted:
(a) inflammatory transverse myelitis as defined by the transverse myelitis working group [9]; (b) inflammatory lesions of the spinal cord extending over at least three contiguous vertebral segments; (c) available spinal cord MRI images performed at the time of the LETM onset (within 2 weeks).
Informed consent was given by all the patients to get their records and MRI images reviewed. All patients included in the study underwent neurological examination and MRI before starting the immunosuppressive therapy. NMO was diagnosed according to the 2006 diagnostic criteria [10]. Cerebrospinal fluid (CSF) analysis was performed during the acute phase of the myelitis in all patients. Serological and CSF evaluation excluded viral and bacterial infections in all patients at the time of LETM. Patients’ neurological disability at the attack nadir was assessed using the Kurtzke Expanded Disability Status Scale (EDSS).
AQP4-IgG testing
Sera and CSF obtained from all patients were tested for AQP4-IgG by a cell-based assay (CBA) (Euroimmun, Luebeck, Germany) [11].
Sera and CSF that yielded a negative result were also tested by an in-house Flow Cytometry assay employing HEK293 cells transiently transfected with cDNA encoding the M23 isoform of AQP4, as previously described [12].
Neuroimaging
Brain and spinal cord MRI were acquired using a 1.5-Tesla scanner. Duration of the disease at MRI examinations was not different between AQP4-IgG+ and AQP4-IgG− patients (days from attack onset to MRI [mean ± SD]: 2.43 ± 1.09 vs. 2.25 ± 0.96). Brain coronal, sagittal and axial Fluid Attenuated Inversion Recovery and T2-weighted images, as well as T1-weighted images before and after gadolinium administration have been evaluated. Spinal cord sagittal and axial T2-weighted images as well as T1-weighted images before and after gadolinium administration have been evaluated.
Statistical analysis
Statistical power (1-β) was assessed as post-hoc analysis by means of G*Power [13]. Fisher’s exact test was performed to compare demographic, clinical data and MRI findings between AQP4-IgG+ and APQ4-IgG− patients with LETM. The nonparametric Mann–Whitney U test was employed for between-group comparisons. The Spearman’s rank correlation was used to assess the correlation between T1-hypointense lesions of the spinal cord and disability.
Results
Patients
Clinical record review identified 37 patients (22 females, 67 %) with a first-ever episode of isolated LETM. The demographic and clinical data of the patients included in the study are summarized in Table 1. Patients' mean age was 46.6 years old (range 8–78 years). The follow-up mean was 44 months (range 7–146 months). CSF samples from all patients were tested for AQP4-IgG. AQP4-IgG was detected in both the serum and the CSF of 16 patients (43 %). Sera and CSF that resulted negative by the CBA were tested by the Flow Cytometry assay. However, none of the samples tested negative by the CBA resulted positive by the Flow Cytometry assay.
The female to male ratio was higher in AQP4-IgG+ patients (p = 0.0006). There was no difference in the mean age and months of follow-up between the seropositive and seronegative patients. Oligoclonal bands were detected in 3/16 (19 %) AQP4-IgG+ patients and in 5/21 (24 %) AQP4-IgG− patients.
At the last follow-up, 15 patients were diagnosed as having NMO (three patients received a diagnosis of seronegative NMO), fulfilling the Wingerchuck et al. [1] criteria, and four patients received a diagnosis of NMO Spectrum Disorder (NMOSD) (relapsing LETM and AQP4-IgG seropositivity). Four AQP4-IgG+ patients had a previous history of optic neuritis and converted to clinically defined NMO when the LETM occurred.
All seronegative NMO patients had a monophasic course. AQP4-IgG− patients were treated with intravenous methylprednisolone (1 g/day for 5 days) followed by a tapering dose of prednisone per os for 6 months.
The diagnoses of the remaining AQP4-IgG− patients at the last follow-up were: monophasic LETM in 13 patients (idiopathic, 10; sarcoidosis, 1; paraneoplastic [Yo-IgG+], 1; Sjogren’s syndrome, 1) and relapsing LETM in five patients.
Clinical characteristics
There was no difference between the AQP4-IgG+ and AQP4-IgG− LETM patients in the frequency of clinical symptoms such as weakness, sensory disturbances or sphincteric dysfunction. However intractable nausea and vomiting (56 % of AQP4-IgG+ vs. 0 % of AQP4-IgG− patients, p = <0.0001) and paroxysmal tonic spasms (38 % of AQP4-IgG+ vs. 5 % of AQP4-IgG negative patients, p = 0.0287) occurred more frequently in the seropositive group. We did not find any significant difference between the two groups in the disability assessed by EDSS, at attack nadir (e.g. the highest EDSS documented). A higher percentage of seropositive patients experienced a relapse during the follow-up compared to seronegative patients (p = 0.03). In particular, the occurrence of optic neuritis (p = 0.003) and the recurrence of LETM (p = 0.023) were more frequently observed in the AQP4-IgG+ group during the follow-up. AQP4-IgG+ patients experienced a higher number of attacks during the follow-up (p = 0.0056).
MRI findings
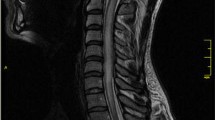
The MRI abnormalities of AQP4-IgG+ and AQP4-IgG− patients are summarized in Table 2. There was no difference in the frequency of brain lesions, in the length of spinal cord lesions and in the frequency of the cervical or the thoracic cord involvement between AQP4-IgG+ and AQP4-IgG− patients. None of the patients included in the study had more than one spinal cord lesion. A contiguous inflammatory lesion extending into the medulla and involving the area postrema region was observed in 10/16 (66 %) of the AQP4-IgG+ patients (Fig. 1) and in none of AQP4-IgG− LETM patients (p = <0.0001). Nine AQP4-IgG+ patients with the medullospinal inflammatory lesion experienced intractable nausea and vomiting during the attack. The spinal cord central grey matter was more frequently involved in seropositive patients (15/16, 94 % vs. 12/21, 57 %; p = 0.0230). Post-contrast T1-weighted images revealed no difference in the frequency of gadolinium-enhancing lesions between seropositive and seronegative patients. Hypointense lesions on T1-weighted images were detected more frequently in AQP4-IgG+ patients (12/16, 75 % vs. 7/21, 33 %; p = 0.0201). All LETM patients (p = 0.0087) and AQP4-IgG+ LETM patients (p = 0.0077) with T1-hypointense lesions presented a higher degree of disability at the attack nadir. The EDSS score at the attack nadir significantly correlated with T1-hypointensity in all LETM patients (p = 0.0090; r = 0.42) and in AQP4-IgG+ (p = 0.0099; r = 0.65) patients but not in AQP4-IgG− patients (p = 0.069; r = 0.4) (Fig. 2).

Representative spinal cord MRI images of two different AQP4-IgG+ (A, B, C, F) and one AQP4-IgG− (D, E) LETM patients. Sagittal T2-weighted spinal cord MRI images of AQP4-IgG+ patients show longitudinally extensive inflammatory lesions involving the cervical cord and extending into the medulla oblongata (A, C). Axial T2 weighted images (insert in A and D) show the preferential involvement of the central grey matter in a AQP4-IgG+ patient (insert in A) and the involvement of the posterior columns in a AQP4-IgG− patient (insert in D). Hypointense lesions are evident on sagittal T1-weighted MRI images of the AQP4-IgG+ patients (B, F) but not in the AQP4-IgG− patient (E)

Expanded disability status scale (EDSS) score of patients with longitudinally extensive transverse myelitis (LETM) at attack nadir. A significant statistical difference in the EDSS score was observed in LETM patients (A) and AQP4-IgG+ patients (B) with hypointense lesions on spinal cord T1-weighted MRI images compared to patients with T1-isointense lesions. No significant difference in EDSS score was observed between AQP4-IgG− patients with and without hypointense lesions on T1-weighted MRI images (C). A significant correlation between the presence of T1-hypointense lesions and EDSS was observed in all LETM (D) and in AQP4-IgG+ patients (E), but not in AQP4-IgG− patients (F)
Discussion
In this study, we observed distinctive clinical and neuroimaging characteristics in patients with first-ever LETM harboring AQP4 autoantibodies.
Intractable nausea and vomiting and paroxysmal tonic spasms often accompanied the LETM in AQP4-IgG+ patients. These findings are consistent with previous studies demonstrating that both intractable nausea and vomiting and paroxysmal tonic spasms are distinctive clinical manifestations of NMO [14–16]. Thus, the presence of these clinical symptoms may be of help in distinguishing the AQP4-IgG+ LETM patients.
The neuroimaging findings revealed a different topological distribution of the lesions in the vertical and horizontal planes of the spinal cord in AQP4-IgG+ LETM patients. In T2-weighted sagittal MRI images, inflammatory lesions extending into the brainstem were more frequent in seropositive patients. This finding can be explained by the preferential involvement of the area postrema in NMOSD, as demonstrated by pathological and clinical studies [14, 15, 17]. AQP4 is densely expressed on astrocytes endfeet at interfaces between CNS parenchyma and fluid compartments, both CSF and blood [2], and in circumventricular organs (e.g. area postrema), regions of the CNS where the blood–brain barrier is lacking. The absence of an intact blood–brain barrier may facilitate the access of circulating IgG and cytokines to these areas [18]. Interestingly, 90 % of the AQP4-IgG+ patients with inflammatory lesions of the area postrema included in the study experienced intractable nausea and vomiting. Our findings are consistent with the area postrema playing a key role in the pathophysiology of NMOSD.
In axial T2-weighted MRI images of the spinal cord, central grey matter-predominant involvement was observed more frequently in AQP4-IgG+ patients as previously reported [19].
Moreover, AQP4-IgG seropositivity was associated with spinal cord lesion T1 hypointensity. This finding is consistent with previous studies describing a higher frequency of T1 hypointensity in NMO spinal cord lesions [20, 21]. Correlative MRI histopathology studies, conducted on MS patients, demonstrated that chronic T1-hypointense lesions represent areas of intrinsic tissue injury, profound axonal loss and matrix disruption, while in acute lesions, T1 hypointensity is thought to reflect the entity of oedema and demyelination [22, 23]. Furthermore, it has been shown that in MS patients, lesion hypointensity on T1-weighted spinal cord MRI images is associated with disability and atrophy [24].
In this study, we found a correlation between T1 hypointensity of the spinal cord lesions and attack severity. Further studies are needed in order to ascertain the correlation of chronic T1 hypointensity and long-term disability and spinal cord atrophy in LETM patients.
In conclusion, this study provides clinical and spinal cord neuroimaging clues that can help distinguishing AQP4-IgG+ LETM patients in the clinical setting. The detection of AQP4 autoantibodies predicts the occurrence of optic neuritis or the recurrence of LETM [6–8]. Hitherto, the early recognition of NMOSDs is important, because it offers the unique opportunity to modify, with the appropriate immunotherapy, the course of a severe disease, preventing the accumulation of disability.
References
Wingerchuck DM, Lennon VA, Lucchinetti CF, Pittock SL, Weinshenker BG (2007) The spectrum of Neuromyelitis Optica. Lancet Neurol 6(9):805–815
Iorio R, Lennon VA (2012) Neural antigen-specific autoimmune disorders. Immunol Rev 248(1):104–121
Kitley JL, Leite MI, George JS, Palace JA (2012) The differential diagnosis of longitudinally extensive transverse myelitis. Mult Scler 18(3):271–285
Iorio R, Fryer JP, Hinson SR, Fallier-Becker P, Wolburg H, Pittock SJ, Lennon VA (2013) Astrocytic autoantibody of neuromyelitis optica (NMO-IgG) binds to aquaporin-4 extracellular loops, monomers, tetramers and high order arrays. J Autoimmun 40:21–27
Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69(24):2221–2231
Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, Lennon VA (2006) Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 59(3):566–569
Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, Weinshenker BG (2008) NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 70(23):2197–2200
Jarius S, Frederikson J, Waters P, Paul F, Akman-Demir G, Marignier R, Franciotta D, Ruprecht K, Kuenz B, Rommer P, Kristoferitsch W, Wildemann B, Vincent A (2010) Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci 298(1–2):158–162
Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG (2011) Therapeutics and Technology Subcommittee of American Academy of Neurology. Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 77(24):2128–2134
Wingerchuck DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66(10):1485–1489
Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, Wandinger KP (2010) Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci 291(1–2):52–56
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, Palace J, Mandrekar JN, Vincent A, Bar-Or A, Pittock SJ (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78(9):665–671
Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*Power 3: a flexible statistical power analysis program for the social, behavioral and biomedical sciences. Behav Res Methods 39:175–191
Apiwattanakul M, Popescu BF, Matiello M, Weinshenker BG, Lucchinetti CF, Lennon VA, McKeon A, Carpenter AF, Miller GM, Pittock SJ (2010) Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 68:757–761
Iorio R, Lucchinetti CF, Lennon VA, Farrugia G, Pasricha PJ, Weinshenker BG, Pittock SJ (2013) Intractable nausea and vomiting from antibodies targeting brain water channels. Clin Gastroent Hepatol 11:240–245
Usmani N, Bedi G, Lam BL, Sheremata WA (2012) Association between paroxysmal tonic spasms and neuromyelitis optica. Arch Neurol 69(1):121–124
Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, Newell K, Mandler RN, Pittock SJ, Weinshenker BG, Lucchinetti CF (2011) Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76:1229–1237
Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, Rommer P, Kleiter I, Stich O, Reuss R, Rauer S, Zettl UK, Wandinger KP, Melms A, Aktas O, Kristoferitsch W, Wildemann B (2011) Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 306(1–2):82–90
Chang KH, Lyu RK, Chen CM, Wu YR, Chang HS, Huang CC, Kuo HC, Chu CC, Hsu WC, Ro LS. Distinct features between longitudinally extensive transverse myelitis presenting with and without anti-Aquaporin 4 antibodies. Mult Scl Journ Epub ahead of print doi: 10.1177/1352458512451659
Filippi M, Rocca MA (2004) MR imaging of Devic’s neuromyelitis optica. Neurol Sci 25(Suppl 4):S371–S373
Nakamura M, Miyazawa I, Fujihara K, Nakashima I, Misu T, Watanabe S, Takahashi T, Itoyama Y (2008) Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol 255(2):163–170
van Walderveen MA, Kamphorst W, Scheltens P, van Waesberghe JH, Ravid R, Valk J, Polman CH, Barkhof F (1998) Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis. Neurology 50:1282–1288
Filippi M, Rocca MA, Barkhof F, Bruck W, Chen JT, Comi G, De Luca G, De Stefano N, Erickson BJ, Evangelou N, Fazeks F, Geurts JJ, Lucchinetti C, Miller DH, Pelletier D, Popescu BF, Lassmann H (2012) Attendees of the Correlation between Pathological MRI findings in MS workshop. Lancet Neurol 11(4):349–360
Losseff NA, Wang L, Miller DH, Thompson AJ (2001) T1 hypointensity of the spinal cord in multiple sclerosis. J Neurol 248(6):517–521
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standard
All human studies must state that they have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iorio, R., Damato, V., Mirabella, M. et al. Distinctive clinical and neuroimaging characteristics of longitudinally extensive transverse myelitis associated with aquaporin-4 autoantibodies. J Neurol 260, 2396–2402 (2013). https://doi.org/10.1007/s00415-013-6997-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-013-6997-9