
Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a genetically transmitted small vessel disease clinically characterized by migraine, recurrent subcortical strokes, and cognitive and mood disorders. Pathogenic mutations are located on any of the exons of the NOTCH3 gene coding for epidermal-growth factor (EGF)-like repeats of the extracellular domain of the NOTCH3 receptor. Because the gene is large and the mutations cluster on some exons, many laboratories restrict the analysis to these exons. We report the first missense mutation involving exon 24 and causing CADASIL in a 64-year-old man. The patient was admitted to the hospital for a loss of consciousness accompanied by profuse sweating. On examination, some parkinsonian features were present. Over the last 4 years, he had developed postural instability and gait disturbances with repeated falls, behavioral disorders, and cognitive impairment. A diagnostic hypothesis of atypical parkinsonism had been advanced. The presence of multiple subcortical lacunar infarcts and leukoencephalopathy extended to the external capsule on cerebral MRI suggested the presence of CADASIL. The diagnosis was confirmed by finding a heterozygous mutation leading to a cysteine substitution on exon 24 of the NOTCH3 gene. One proband’s brother, who had progressive gait disturbances, unilateral action tremor and bradykinesia, and an asymptomatic niece also resulted affected. This report underlines that when CADASIL is suspected the genetic analysis should be performed on all the NOTCH3 exons coding for EGF-like repeats including exon 24 and confirms that CADASIL may have heterogeneous phenotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [MIM 125310] is an inherited autosomal dominant small vessel disease caused by point mutations of the NOTCH3 gene localized on chromosome 19p13.1 [1]. The disease is clinically characterized by a variable combination of migraine, recurrent transient ischemic attacks or strokes, cognitive impairment, and psychiatric disturbances [2]. Less frequent manifestations of the disease are epilepsy, transient disturbances of consciousness, visual impairment, and hemorrhagic strokes [2–5]. CADASIL has a progressive course, and severe disability and dementia develop in about three-fourths of the patients at the end stage of the disease [2, 6]. Cerebral magnetic resonance imaging (MRI) abnormalities in CADASIL include diffuse white matter hyperintensities, typically extending to the anterior portion of the temporal lobe and the external capsule, subcortical lacunar infarcts, and microbleeds [2, 7–10]. The clinical–radiological phenotype of the disease is, however, highly variable [2, 6, 11].
The disease is caused by mutations in any of the 23 exons of the NOTCH3 gene coding for the EGF-like repeat domains of the extracellular portion of the NOTCH3 receptor. The vast majority are missense point mutations leading to a cysteine substitution [1, 2, 12–14]. So far, about 170 mutations have been reported [14–16]. Other less common types of mutations include deletions, insertions, splice sites, frameshift mutations and duplications [14, 15]. Mutation screening covering the whole region coding for EGF repeats (exons 2–24) is time consuming and expensive. Thus, many laboratories analyze only those exons that, according to the previous reports, harbor the majority of the mutations, mainly exons 3, 4, 5 and 8 (covering 62% of all the reported pathogenic NOTCH3 mutations) and less frequently exons 2, 6, 11 and 18 (to cover 80% of cases) [2, 15–17]. This strategy has turned out to be unreliable in our experience. In fact, at least in Italy, the distribution of mutations is more spread among exons [12]. Thus, in the last few years, we started to examine all the exons codifying for EGF-like repeats in suspected patients, and more recently, our laboratory has started analyzing also exon 24 where, to the best of our knowledge, mutations have never been found.
Here, we report the first missense point mutation on exon 24 of the NOTCH3 gene responsible for CADASIL in a patient with unusual clinical manifestations of the disease.
Case report
A 64-year-old man (proband) presented to the emergency department for a transient loss of consciousness with fall accompanied by profuse sweating. No involuntary movement or urinary incontinence occurred. The episode was not preceded by dyspnea or dysautonomic prodromal symptoms such as nausea and vomiting. A brain CT-scan excluded hemorrhage and revealed diffuse leukoencephalopathy, multiple lacunar infarcts, and cortical atrophy. Blood tests showed normal values of hemoglobin, electrolytes, and elevated glycemia (262 g/L). The ECG recorded no abnormalities. The patient was then admitted to the neurology ward. Salient findings of his neurologic examination included: spatial and temporal disorientation, facial hypomimia (with absent blinking), bradykinesia, festination, start hesitation and freezing of gait with decreased arm swing. Muscle tone was increased, with more marked axial than limb rigidity. Tremor was absent and ocular movements were preserved. Hypokinetic dysarthria and mild left facial paralysis without dysphagia were present.
The patient had a history of diabetes mellitus, hypertension, and myocardial infarction treated with percutaneous coronary angioplasty. He suffered from postural hypotension and, over the previous 4 years, he had progressively developed postural instability, rigidity, bradykinesia and gait disturbances with repeated falls, usually accompanied by abundant sweating. Constipation, urinary incontinence or sexual dysfunction were not reported. His relatives reported behavioral disturbances, in particular aggressiveness and apathy, and cognitive impairment characterized by progressive difficulty in complex activities (such as playing the flute, that had been his favorite hobby for years) and short-term memory disturbances. Over the last 2 years, he had presented repeated episodes of acute mental confusion with temporal and spatial disorientation. He had never had hallucinations, psychotic features, sleep disturbances or vivid nightmares. No stroke had occurred. He had become gradually dependent in the activities of daily living and had started therapy with levo-dopa without benefits. For severe agitation quetiapine was also started. A diagnosis of atypical parkinsonism (Lewy body disease, multiple system atrophy, progressive supranuclear palsy or vascular parkinsonism) was thus advanced. Cerebro-spinal fluid examination was normal and ruled out tau, beta-amyloid or protein 14.3.3 level abnormalities. Instrumental evaluations revealed reversed circadian rhythm of blood pressure and orthostatic hypotension; the standard monitoring ECG and the head up tilt test were normal. Neck vessels and transcranial Doppler sonography excluded extracranial and intracranial arteries stenoses. On EEG, diffuse slowing without stereotyped sharp wave discharges was present. A neuropsychological evaluation confirmed the presence of moderate cognitive impairment (MMSE = 17/30) involving several domains, in particular memory, attention and executive functions, abstraction capacity, impulse control, set-shifting, and psychomotor slowing. A cerebral MRI showed severe white matter lesions, with multiple subcortical lacunar infarcts in the basal ganglia, thalami, pons and cerebellum, and cortical, subcortical and medial temporal atrophy. Atrophy of the infratentorial middle cerebellar peduncle or midbrain tegmentum were not present. SPECT-scan showed reduction of DAT sites in the right striatum in comparison with contralateral side deemed to be a consequence of the ischemic lesions detected by MRI.
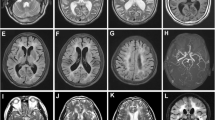
The presence of severe white matter lesions with involvement of the external capsule (despite the lack of extension to temporal poles), associated with the presence of multiple subcortical lacunar infarcts (Fig. 1), brought to suspect CADASIL. The family history was then specifically assessed. The patient’s father had died from a brain hemorrhage at the age of 62 years, the paternal grandfather had repeated strokes in midlife, a paternal uncle was reported to have had gait disturbances, and a 64-year-old brother had gait and behavioral disturbances. After consent, the patient was subjected to the genetic test that revealed the heterozygous mutation c.3944G>A on exon 24 of the NOTCH3 gene responsible for the replacement of a cysteine at position 1315 with a tyrosine (p.Cys1315Tyr); CADASIL was thus diagnosed.

Cerebral MRI (proband). Diffuse, symmetric, and confluent hyperintense lesions in the periventricular and deep white matter with extension of to the external capsules on an axial FLAIR image (a and b). Cerebral MRI (proband’s brother). Diffuse, symmetric, and confluent hyperintense lesions in the periventricular and deep white matter on a FLAIR images (c). Multiple lacunar infarcts in the thalami and basal ganglia in the proband’s brother on a T1-weighted image (d)
Other family members then required genetic test. The same mutation was found in the proband’s brother who had a 4-year history of progressive gait disturbances and right hand action tremor associated with bradykinesia. For these disturbances, levo-dopa had been started and then stopped for lack of efficacy and the presence of side effects. He also had some memory disturbances and behavioral disorders, mainly excessive irritability. Nevertheless, he was still able to work as computer engineer. No urinary incontinence or history of depression was reported. In the past, he suffered from migraine without aura. His cerebral MRI revealed moderate periventricular leukoencephalopathy (with minimal involvement of the temporal poles) associated with multiple subcortical lacunar infarcts in the thalami, corona radiata, and pons, in addition to cortical atrophy (Fig. 1). Like the proband, he had several vascular risk factors (diabetes mellitus, hypertension, smoking, hypercholesterolemia, and obesity). The genetic test was performed also in his two children. The first was a 30-year-old man who suffered from migraine but did not have history of cerebrovascular events, epilepsy or mood disorders. The 27-year-old daughter did not present any CADASIL typical disturbance. The automated sequencing of exon 24 revealed the 3944G>A mutation only in the daughter. The other 62-year-old proband’s brother refused the genetic investigation (Fig. 2).

Family pedigree. The proband is indicated by the arrow. Symptomatic subjects for CADASIL are represented in black filled symbols. When known, cause of death are reported
DNA analysis
Following informed consent, total genomic DNA was extracted from peripheral blood leucocytes using standard procedures. Polymerase chain reaction (PCR) was performed with primers (comprising intron–exon boundaries) specific for exons 2–24 of the NOTCH3 gene (primers and PCR conditions available on request). Following purification of PCR products, sequencing was performed using the automated sequencer ABI 3730 (Applied Biosystems, Foster City, CA, USA).
Discussion
We have reported for the first time a family with a pathogenic mutation responsible for CADASIL located on exon 24 of the NOTCH3 gene. Despite EGF-like repeats are coded also by exon 24, mutations on this exon had never been found. In fact, the entire NOTCH3 gene has a high GC content (about 65% in the coding region) and exon 24, with a 77% GC composition, is difficult to amplify by the PCR [18]. In our laboratory, a variety of additives agents including dimethyl sulfoxide, betaine, formamide, glycerol, non-ionic detergents and their combinations were used until we were able to obtain a sufficient quantity and a good quality of DNA amplification.
Considering the previously published pathogenic mutations, about half of them have been located on exons 2–4 and the other half on exons 5–23 [2, 14]. Also, the mutation found in this family leads to a cysteine substitution within the EGF-like repeat 33 of the NOTCH3 receptor following the stereotyped nature of mutations typical of CADASIL, thus supporting the pathogenic role of this mutation in the disease. Our report further expands the genetic spectrum of CADASIL and confirms the disperse distribution of NOTCH3 mutations on the different exons. This finding points out the need of including the analysis of exon 24 when CADASIL is suspected. This extended analysis seems in order to avoid an underdiagnosis of the disease. Another aspect to consider is that there could be a perpetuating erroneous knowledge of the distribution of the mutations across the NOTCH3 exons if the analysis is limited to a few exons.
Another point of interest concerns the clinical features of the patient. The proband and his brother presented some peculiar phenotypic aspects, specifically a prominent parkinsonian syndrome [19–21], and absence of some typical aspects of CADASIL, namely stroke and migraine. This phenotype could be regarded as a case of vascular parkinsonism [22]. According to some authors, this is characterized by symmetric parkinsonian syndrome with gait disorders, specifically freezing, rigidity (prevalent in the lower limbs), cognitive impairment, and finally pseudobulbar palsy; resting tremor is usually absent [22]. This picture is poorly responsive to dopaminergic treatment and is attributed to diffuse subcortical vascular lesions disrupting basal ganglia/motor cortex connections [22]. One case of vascular parkinsonism diagnosed as CADASIL was reported in a 55-year-old male patient with functional integrity of the nigrostriatal dopaminergic system in whom the disease was diagnosed by skin biopsy [23]. Another CADASIL patient was reported with a clinical presentation compatible with severe progressive supranuclear palsy [24]. Our case confirms that the parkinsonian phenotype may represent one of the manifestations of CADASIL and suggests that the disease should be considered in patients with atypical parkinsonism, particularly vascular parkinsonism. Of note, the MRI picture of our proband lacked the most typical CADASIL features (i.e., white matter hyperintensities in the anterior temporal lobe). In this sense, our report confirms the heterogeneity of the clinical–radiological phenotypic expression in CADASIL [2]. However, also the possible coexistence of CADASIL and other neurodegenerative disorders should be taken into account in this regard as recently pointed out [25].
The proband had a history of coronary artery disease which is not listed among the typical CADASIL manifestations. However, one study showed that myocardial infarction is not infrequent in CADASIL patients and is possibly caused by the presence of coronary microvascular alterations typical of the disease [26]. It is to be considered that our patient, like his brother, presented with a high vascular risk profile.
Because the genotype–phenotype correlation in CADASIL has not been clarified and only few data support its existence [2, 11, 12, 27], it is hard to speculate that the novel mutation found in our patient has a specific effect on the expressivity of the disease. In CADASIL, the phenotypic variability and atypical features may be attributable for example to the influence of vascular risk factors [28, 29] or to other unexplored factors. Even within a single family, the age of onset and the disease severity and progression can vary significantly. Being this the first reported family, it is not clear whether also other mutations on exon 24 may cause different phenotype expression leading to unusual clinical pictures and whether this might have led, along with the mentioned laboratory difficulties in analyzing this exon, to the uncovered role of exon 24 in CADASIL.
In summary, this case report underlines that when CADASIL is suspected, analysis should be performed on all the NOTCH3 exons coding for EGF-like repeats extending the investigation also to exon 24 and that the parkinsonian syndrome might represent one of the potential manifestation of CADASIL. Moreover, this report confirms the heterogeneity of the clinical–radiological phenotypic expression in CADASIL.
References
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383:707–710
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG (2009) CADASIL. Lancet Neurol 8:643–653
Singhal S, Bevan S, Barrick T, Rich P, Markus HS (2004) The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain 127:2031–2038
Rufa A, De Stefano N, Dotti MT, Bianchi S, Sicurelli F, Stromillo ML, D’Aniello B, Federico A (2004) Acute unilateral visual loss as the first symptom of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Arch Neurol 61:577–580
Choi JC, Kang SY, Kang JH, Park JK (2006) Intracerebral hemorrhages in CADASIL. Neurology 67:2042–2044
Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M (2004) Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain 127:2533–2539
Chabriat H, Levy C, Taillia H, Iba-Zizen MT, Vahedi K, Joutel A, Tournier-Lasserve E, Bousser MG (1998) Patterns of MRI lesions in CADASIL. Neurology 51:452–457
Pantoni L, Pescini F, Nannucci S, Sarti C, Bianchi S, Dotti MT, Federico A, Inzitari D (2010) Comparison of clinical, familial, and MRI features of CADASIL and NOTCH3-negative patients. Neurology 74:57–63
O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS (2001) MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 56:628–634
Singhal S, Rich P, Markus HS (2005) The spatial distribution of MR imaging abnormalities in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and their relationship to age and clinical features. AJNR Am J Neuroradiol 26:2481–2487
Desmond DW, Moroney JT, Lynch T, Chan S, Chin SS, Mohr JP (1999) The natural history of CADASIL: a pooled analysis of previously published cases. Stroke 30:1230–1233
Dotti MT, Federico A, Mazzei R, Bianchi S, Scali O, Conforti FL, Sprovieri T, Guidetti D, Aguglia U, Consoli D, Pantoni L, Sarti C, Inzitari D, Quattrone A (2005) The spectrum of Notch3 mutations in 28 Italian CADASIL families. J Neurol Neurosurg Psychiatry 76:736–738
Federico A, Bianchi S, Dotti MT (2005) The spectrum of mutations for CADASIL diagnosis. Neurol Sci 26:117–124
Tikka S, Mykkänen K, Ruchoux MM, Bergholm R, Junna M, Pöyhönen M, Yki-Järvinen H, Joutel A, Viitanen M, Baumann M, Kalimo H (2009) Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 132:933–939
Markus HS, Martin RJ, Simpson MA, Dong YB, Ali N, Crosby AH, Powell JF (2002) Diagnostic strategies in CADASIL. Neurology 59:1134–1138
Escary JL, Cécillon M, Maciazek J, Lathrop M, Tournier-Lasserve E, Joutel A (2000) Evaluation of DHPLC analysis in mutational scanning of Notch3, a gene with a high G-C content. Hum Mutat 16:518–526
Bianchi S, Rufa A, Ragno M, D’Eramo C, Pescini F, Pantoni L, Cappelli A, Perretti A, Zicari E, Zolo P, Inzitari D, Dotti MT, Federico A (2010) High frequency of exon 10 mutations in the NOTCH3 gene in Italian CADASIL families: phenotypic peculiarities. J Neurol 257:1039–1042
Kieleczawa J (2006) Fundamentals of sequencing of difficult templates. An overview. J Biomol Tech 17:207–217
Bhidayasiri R, Ling H (2008) Multiple system atrophy. Neurologist 14:224–237
McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124
Williams DR, Lees AJ (2009) Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 8:270–279
Benamer HT, Grosset DG (2009) Vascular Parkinsonism: a clinical review. Eur Neurol 61:11–15
Wegner F, Strecker K, Schwarz J, Wagner A, Heinritz W, Sommerer F, Thal DR, Schneider JP, Kendziorra K, Sabri O (2007) Vascular Parkinsonism in a CADASIL case with an intact nigrostriatal dopaminergic system. J Neurol 254:1743–1745
Van Gerpen JA, Ahlskog JE, Petty GW (2003) Progressive supranuclear palsy phenotype secondary to CADASIL. Parkinsonism Relat Disord 9:367–369
Rice CM, McGuone D, Kurian KM, Love S, Renowden SA, Giffin NJ (2011) Autopsy-confirmed, co-existent CADASIL and multiple system atrophy. Parkinsonism Relat Disord [Epub ahead of print]
Lesnik Oberstein SA, Jukema JW, Van Duinen SG, Macfarlane PW, van Houwelingen HC, Breuning MH, Ferrari MD, Haan J (2003) Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Medicine (Baltimore) 82:251–256
Pescini F, Bianchi S, Salvadori E, Poggesi A, Dotti MT, Federico A, Inzitari D, Pantoni L (2008) A pathogenic mutation on exon 21 of the NOTCH3 gene causing CADASIL in an octogenarian paucisymptomatic patient. J Neurol Sci 267:170–173
Pantoni L, Sarti C, Pescini F, Bianchi S, Bartolini L, Nencini P, Basile AM, Lamassa M, Kalaria RN, Dotti MT, Federico A, Inzitari D (2004) Thrombophilic risk factors and unusual clinical features in three Italian CADASIL patients. Eur J Neurol 11:782–787
Adib-Samii P, Brice G, Martin RJ, Markus HS (2010) Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype: study in 200 consecutively recruited individuals. Stroke 41:630–634
Conflict of interest
The authors declare that they have no conflict of interest in relation to this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Valenti, R., Bianchi, S., Pescini, F. et al. First report of a pathogenic mutation on exon 24 of the NOTCH3 gene in a CADASIL family. J Neurol 258, 1632–1636 (2011). https://doi.org/10.1007/s00415-011-5983-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-011-5983-3