
Abstract
Families with autosomal dominant frontotemporal dementia and amyotrophic lateral sclerosis (FTD/ALS) have previously been linked to a locus on chromosome 9p21. We describe the clinical phenotype and pathology of a large family with autosomal dominant FTD/ALS with nine affected members originating from Gwent in South Wales, UK. We also further refine the locus on chromosome 9p21 using a haplotype sharing approach and assess heterogeneity in 9p21 linked families. Within this family, affected individuals present with either FTD or ALS or both diseases simultaneously. In addition there was marked phenotypic variation including ataxia, Parkinsonism, psychosis and visuo-spatial cognitive deficits. The pathological features of the three cases described were consistent with type 2 FTD pathology, as previously reported in similar families. However, we also report distinctive cerebellar and glial pathology and a significant proportion of TDP-43 negative inclusions. No mutations in known genes for FTD or ALS were found. We identified a large 4.8-megabase haplotype on chromosome 9p21, which was shared by all affected family members. This haplotype overlaps and limits the previously reported FTD/ALS linkage region on chromosome 9p21. Sequencing of this region did not identify any evidence of a pathogenic exonic mutation. This suggests that the pathogenic change affects non-coding DNA and that the disease is caused by variation in gene or protein expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Frontotemporal lobar degeneration (FTLD) is the third most common neurodegenerative cause of dementia, and is a particularly common cause of dementia in those under the age of 65 years [1]. A number of lines of evidence indicate substantial overlap between FTLD and motor neuron disease/amyotrophic lateral sclerosis (ALS). In one study, 50% of cases with ALS were found to have cognitive impairment, with 15% fulfilling the criteria for FTLD and 23% meeting the criteria for dementia [2, 3]. Conversely, 14% of cases with FTLD have a diagnosis of ALS, while 36% have some suggestive clinical features [4]. This clinical overlap has been underlined by the recent finding that TAR-DNA binding protein-43 (TDP-43), encoded by the TAR-DNA binding protein gene (TARDBP), is a major component of the pathological features of both diseases [5]. Recent consensus criteria classify these diseases according to the type of pathology together with the known genetic aetiology [6]. Cases with FTLD whose pathology is characterised by ubiquitin inclusions and lack hippocampal sclerosis are designated FTLD-U. These intra-neuronal and glial inclusions are negative for tau and α-synuclein but stain positively for TDP-43 in most cases of sporadic and familial FTLD-U, with and without clinical ALS, as well as in sporadic ALS and some familial ALS [7]. TDP-43 is a nuclear protein, but in the pathological state the distribution of TDP-43 changes such that it is sequestered into neuronal cytoplasmic inclusions (NCI), neuronal intranuclear inclusions (NII) and dystrophic neurites (DN). Depending on the predominant inclusion present, the pathological appearances of FTLD-U are subdivided into types 1–4, where type 2 pathology is associated with cases of FTD/ALS linked to chromosome 9p [6, 7]. This supports a potential aetiological link between FTD and ALS.
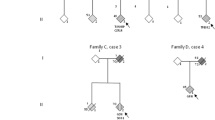
We describe the clinical, pathological and genetic investigation of a family originating in the Gwent region of South Wales, UK, with nine affected members (Fig. 1) originally reported in 2003 [8], in which clinical FTLD and ALS are alternative presentations of the same disease.

Gwent kindred pedigree. Numbers above individuals indicate case number. Numbers below individuals denote age at onset. Asterisk indicates cases with DNA available
Methods
Clinical study
Following informed consent, family members were interviewed and examined, and DNA was stored. Case notes and genealogical records of affected individuals were reviewed. Age at onset was defined using the time of onset of the first symptom attributable to the disease. Clinical and genetic analysis was carried out under ethical approval from the Multi-Centre Research Ethics Committee for Wales.
Pathological study
The brains and spinal cords were removed from cases 5, 7 and 9, weighed and processed for neuropathological examination. Immuno-histochemistry using dehydrated paraffin wax sections was performed using rabbit anti-human tau (1/500, DAKO), anti-ubiquitin (1/250, DAKO), anti-α-synuclein (1/80, Novocastra), anti-glial fibrillary acidic protein (GFAP) (1/1500, DAKO) and two types of anti-neurofilament (Sigma: 200 kDa, phosphorylated and non-phosphorylated 1/800; ICN Pharmaceuticals: 70 and 200 kDa 1/160) and mouse anti-human monoclonal A-beta (Aβ4 at 1/100, pre-treated with formic acid). Antibodies were diluted in phosphate-buffered saline, and antigen retrieval was carried out with microwaving for 5 min in citrate buffer in each case. Primary antibody visualisation used the DAKO™ Envison system. Rabbit anti-human polyclonal antibody TDP-43 (Proteintech group, 10782-1-AP 1/500) was used with an antigen retrieval method following Namimatsu and colleagues [9]. The primary antibody was visualised using an avidin biotin complex kit with 3,3′-diaminobenzidine. Professor J.W. Ironside, University of Edinburgh kindly performed staining with anti-prion protein antibodies on cases 5 and 7.
Genetic analysis
Sequence analysis was carried out with BigDye terminator chemistry analysed using an ABI 3100, using previously published primers [10]. Following quantification by PicoGreen (Molecular Probes, Eugene, OR, USA), the DNA for all samples was genotyped using the Illumina HumanHap550 BeadChip (Illumina, San Diego, CA, USA) with genotype calling using BeadStudio according to the standard protocols defined by Illumina. PLINK [11] was then used to ensure that all samples included in our analysis passed a standard single-nucleotide polymorphism (SNP)-based quality control (QC) procedure, only including SNPs with call rate >0.99 and samples with call rate >0.95.
For accurate estimations of allele frequency and linkage disequilibrium (LD), the genotypes of the Gwent family were first merged with genotypes of 1,000 unrelated UK population controls that had also been genotyped on the Illumina HumanHap550 array and subjected to rigorous SNP-based QC [SNP call rate >0.99, sample call rate >0.95, no related or duplicate samples (proportion identical by descent [IBD] <0.03), no potentially contaminated samples (proportion IBD <0.03 with multiple samples) and ethnically homogeneous according to multidimensional scaling plots]. Using this merged dataset all markers with minor allele frequency >0.05 were then pruned for LD using the “indep-pairwise” function of PLINK by defining a window of 50 SNPs, a step of 5 SNPs and an intermarker r 2 threshold of 0.2. This procedure generated a set of 62,618 markers that were then used to identify any extended haplotype longer than 1 Mb shared by any pair of samples. All haplotypes shared by affected members of the Gwent family were identified. Then, based on an autosomal dominant model, we manually excluded haplotypes also carried by any elderly unaffected family member who had survived for more than 10 years beyond the oldest known age at onset in the family. We used Human Genome 18 build for all genomic numbering.
Results
Clinical features
Mean age at disease onset was 42.7 years (range 31–52 years), with mean disease duration of 3.6 years (0.5–12 years) and mean age at death of 45.8 years (40–54 years). Before the definitive pathological studies, clinical diagnoses in this family included Parkinsonism, Creutzfeld–Jakob disease, progressive muscular atrophy and ALS. There was wide variation in the clinical phenotype (Tables 1, 2). Five cases (62.5%) had ALS at presentation (cases 2–4, 6 and 9). Of these cases, three presented with both bulbar and limb involvement and two with purely limb onset with one of these cases later developing bulbar involvement. Case 3 had features of both ALS and behavioral variant frontotemporal dementia (bvFTD) at presentation, and cases 2 and 6 later developed features of bvFTD. Three cases (37.5%) presented primarily with executive dysfunction, behavioural change and cognitive impairment consistent with bvFTD without ALS (cases 5, 7 and 8). Case 7 had prominent psychosis and personality change at presentation, and case 8 has also developed psychosis with hallucinations and delusions. All three of the cases who presented with bvFTD developed features of ALS later in their disease course. Cases 2, 5 and 8 had extrapyramidal signs, and case 7 had prominent cerebellar ataxia. Cases 6, 7 and 8 had neuropsychological assessments, and all had significant executive dysfunction and impaired visuo-spatial skills with relatively less impaired memory and language. Neuroimaging showed that case 6 had predominantly parietal and occipital cortical atrophy, case 8 had frontotemporal cortical atrophy, and the imaging in case 9 was normal (Online Resource 1). Detailed case histories of these cases can be found in the supplementary material (Online Resource 2).
Pathology
Case 5
The brain weighed 800 g and demonstrated atrophy of the frontal lobes and cerebellar hemispheres but not brain stem or basal ganglia. The spinal cord was atrophic, as were the individual spinal nerves. Frontal cortex (Brodmann area 9), but not temporal or parietal cortices showed GFAP positive gliosis and vacuolation. There were no tau, α-synuclein or β-amyloid inclusions in brain or spinal cord. There were intra-neuronal ubiquitinated inclusions in the spinal cord and frontal cortex with equal numbers of TDP-43 positive neuronal cytoplasmic inclusions (NCI) in laminae III and IV. Numerous ubiquitinated neurons were present in the dentate gyrus and an occasional TDP-43 positive NCI was seen (Fig. 2e, f). Hippocampal sclerosis was not present. The medulla contained ubiquitinated, TDP-43 positive, skein-like inclusions in the hypoglossal nucleus, again best described as NCI found in type 2 FTLD (Fig. 3e). No inclusions were present in the olivary nucleus. There was no neuronal loss from the substantia nigra. The putamen and globus pallidus contained occasional TDP-43 and ubiquitin positive NCIs. The cerebellum demonstrated focal Purkinje cell loss, but did not contain inclusions in the dentate nucleus. TDP-43 positive staining threads were present in the white matter of the cerebellar peduncle and internal capsule, and some had the appearance of intraglial inclusions (Fig. 2 c, d). The spinal cord anterior horn cells were reduced in number, but the surviving neurons contained intra-neuronal ubiquitin inclusions and “skein-like” TDP-43 positive NCIs (Fig. 3a, b). Myelin staining in the lateral corticospinal tracts was reduced.

Neocortex and hippocampus pathology. Case 7: frontal cortex TDP-43 a and ubiquitin positive b NCI. Case 5: internal capsule white matter TDP-43 positive glia c and threads d; dentate gyrus neurons showing more ubiquitin e than TDP-43 f staining NCI. Scale bar 40 µm

TDP-43 pathology in brain stem and spinal cord. Case 5, 9 a, b: spinal cord, anterior horn cell NCI. Case 9 c, d: hypoglossal nucleus, “skein-like” NCI and dendritic staining. Case 5 e: hypoglossal nucleus, NCI and dendritic processes. Case 7 f: substantia nigra, NCI. Scale bar 20 µm
Case 7
Brain weight was 1,350 g, with no evidence of cortical or cerebellar atrophy. The frontal cortex was vacuolated and gliotic in laminae II/III. The entorhinal, insular, parietal and temporal cortex did not contain vacuolation. Frontal cortex contained TDP-43 positive NCIs and ubiquitin inclusions in equal numbers (Fig. 2a, b). Positive TDP-43 staining was present in the white matter and interpreted as intraglial inclusions. The dentate gyrus contained both ubiquitin and TDP-43 positive NCIs, but no hippocampal sclerosis. In the medulla, ubiquitin and TDP-43 positive NCIs were present in the hypoglossal nucleus but not in the olivary nuclei. Numerous TDP-43 positive NCIs were present in the substantia nigra, but there was no significant neuronal loss (Fig. 3f). There was extensive Purkinje neuron loss with Bergman gliosis as shown by GFAP staining. Bielschowsky positive torpedoes were present in the granular layer (Fig. 4a, b). An occasional ubiquitin inclusion and TDP-43 positive NCI was present within Purkinje neurons (Fig. 4c–e). Anti-neurofilament positive aggregates contiguous with surviving Purkinje neurons were present in the granular layer (Fig. 4f). The dentate nucleus contained ubiquitin and TDP-43 positive inclusions. There was no neuronal loss or loss of myelin staining in the spinal cord, and no TDP-43 positive inclusions were present in the anterior horn cells.

Case 7, cerebellar pathology: Purkinje neuron with Bielschowsky staining dystrophic granular layer dendrites a and “torpedo” b. Purkinje neurons with ubiquitin staining NCI c, TDP-43 staining NCI d, e and anti-neurofilament antibody staining dystrophic dendrites within the granular layer f. Scale bar 40 µm
Case 9
Brain weight was 1,300 g, and there was mild frontal cortical and superior temporal gyrus atrophy. The frontal cortex contained gliosis and neuronal loss, but only minimal vacuolation. Frontal cortical areas contained ubiquitin and TDP-43 positive neurons. In the cingulate cortex, ubiquitin positive NCIs were more numerous than TDP-43 positive inclusions. In the medulla there were ubiquitin and TDP-43 positive skein-like inclusions in the hypoglossal nucleus (Fig. 3c, d), but no inclusions in the olivary nuclei. The substantia nigra did not demonstrate neuron loss, but TDP-43 NCIs were present in the pars compacta neurons. Only an occasional TDP-43 and ubiquitin positive NCI were present in the putamen and globus pallidus. The dentate gyrus contained ubiquitin and TDP-43 positive inclusions with neither gliosis nor sclerosis. There was no loss of Purkinje neurons and no NCI. There were ubiquitin positive neurons in the dentate nucleus. TDP-43 inclusions were present in the white matter and interpreted as intraglial inclusions. The spinal cord showed loss of anterior horn cells and lateral corticospinal white matter loss. Ubiquitin and TDP-43 positive and “skein-like” NCIs were present in surviving anterior horn cells (Fig. 3b). The TDP-43 staining neuritic processes were regarded as short dystrophic neurites as found in type 2 FTLD [7].
Genetics
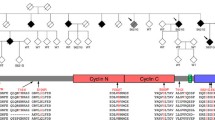
DNA was available from a total of four affected and eight unaffected family members. Sequence analysis of TARDBP, progranulin, SOD-1, FUS, and MAPT failed to identify any exonic or splice site variants that segregated with disease. Given the relatively close relationships of the available affected DNA samples (three siblings and one first cousin), traditional logarithm of the odds (LOD) score based linkage analysis on this pedigree is statistically limited. Using a haplotype sharing approach we identified seven candidate regions, six of which did not span any of the previously identified FTD or ALS genes nor were located in other known FTLD or ALS linkage regions (chr2:14263242-22765654, chr6:6219083-28445748, chr8:100832853-128526872, chr10:30318242-50121673, chr12:42688707-59146076, chr18:32478457-44657016). The final candidate region was defined by a large 4.8-Mb haplotype shared by all affected family members which mapped to chromosome 9p21. The region spans chr9:25406556-30247784, corresponds to chromosomal bands 9p21.2–9p21.1 and overlaps and limits the region previously reported to show linkage to families with FTD/ALS [12–18] (Fig. 5). Given the clinical and pathological similarities with these families (Table 3) we feel that the 4.8-Mb region remains the most likely location for the FTD/ALS gene. However the current analysis is unable to exclude the possibility that the haplotype surrounding the disease gene is less than 1 Mb or that it lies within any of the other six regions. Copy number variants (CNVs) were determined from the Illumina SNP data using PennCNV [19] defined by at least ten SNPs spanning a minimum of 50 kb. This failed to reveal any CNVs present at the chromosome 9 locus in any members of the pedigree. SNP arrays can have limited resolution in detecting small CNVs, therefore it remains possible that a CNV smaller than 50 kb is present at this locus. The region contains only eight protein coding genes: TUSC1, PLAA, IFT74, LRRC19, TEK, MOBKL2B, IFNK and LINGO2. The exons of each of these genes were sequenced in both affected and unaffected members. However this failed to identify any evidence of a protein coding mutation that segregated with disease.

The chromosome 9p21 shared haplotype: a the previous linkage regions for FTD/ALS families on chromosome 9 shown with the Gwent kindred shared haplotype; b the Gwent shared haplotype with the genes contained within the region
Comment
We report marked clinical heterogeneity in a family with FTD and ALS, likely to have a single disease caused by a mutation in the chromosome 9 FTD/ALS linkage region. As well as typical bvFTD and ALS, members of this family have ataxia, psychosis, visuo-constructional dysfunction and Parkinsonism. Clinical follow-up of these cases indicated that, despite striking variation in presentation, most cases developed mixed clinical syndromes with features of both ALS and bvFTD. Interestingly, some members of this family developed overt psychosis (7 and 8) and visuo-constructional dysfunction (6, 7 and 8), indicating that the cognitive and behavioural aspects of this disease are not limited to frontal dysfunction. Four of the cases had clinical features of extrapyramidal disease characterised by Parkinsonism (1, 2, 5 and 8).
This family adds to the growing number of reported families with inherited FTLD and ALS. Linkage to chromosome 9 was originally reported in one Dutch and one Scandinavian family with FTD/ALS and ubiquitin pathology (FTLD-U) [16, 18]. The inclusion pathology in these families has subsequently been shown to be TDP-43 positive with type 2 distribution, characterised by predominant NCIs with short DNs [7]. Subsequently, several other FTD/ALS families have been published with chromosome 9 linkage (Table 3) [12–15, 17]. In all published families there has been phenotypic variability. Individuals may present with features of FTD and ALS simultaneously, with one disease followed by the other, or alternatively with FTD or ALS alone. Compared with the chromosome 9p21 linked families, the Gwent family has a higher proportion of cases developing both conditions (67% versus 22%) and younger age at onset (42.2 versus 55.7 years). The average disease duration of 3.6 years in the Gwent family is similar to that of other families (4.6 years). Prominent cerebellar ataxia as seen in case 7 has not previously been reported in any case with familial FTD/ALS. In this family, clinical ataxia was accompanied by marked cerebellar Purkinje cell loss at post mortem (Fig. 4). Psychosis as observed in our family has been described in five members of four other families [12, 13, 18, 20]. Visuo-spatial dysfunction has also been noted in a small number of cases in two families [13, 16]. Two cases that had clinical FTD and prominent visuo-constructional dysfunction had a contrasting distribution of atrophy: parieto-occipital in case 6 and frontotemporal in case 8. However, in case 9, who had pure ALS, the scan was normal (Online Resource 1). In other FTD/ALS families where imaging findings have been reported, imaging has revealed predominantly frontotemporal atrophy, although fronto/parieto/occipital atrophy has been described in one family with some members developing a cortico-basal syndrome [14].
The pathology in all these cases was consistent with type 2 FTLD-U pathology [6] consistent with the findings in chromosome 9 linked families and in other families with a similar phenotype but without proven linkage [21]. In all cases, ubiquitin and TDP-43 inclusions were consistently present in the frontal cortex, dentate gyrus and hypoglossal nucleus, with some TDP-43 positive pathology in the substantia nigra and basal ganglia. Pathological heterogeneity was seen, which corresponded with the clinical findings. Cases 5 and 9 demonstrated spinal cord involvement, whereas in case 7 there was cerebellar pathology with Purkinje neuronal loss, intra-neuronal neurofilament (ubiquitin positive and TDP-43 negative) and intra-neuronal cytoplasmic ubiquitin inclusions, but no spinal cord involvement. The findings in case 7 correlate with progressive ataxia described in this individual, whereas cases 5 and 9 both had clinical signs attributed to lower motor neuron changes in the spinal cord and cranial nuclei. Cerebellar involvement in FTLD has been reported, but to our knowledge the unusual neuropathological findings we describe in case 7 consisting of severe Purkinje cell loss, argyrophilic torpedoes and neurofilament staining have not been identified in other families. More abundant intra-neuronal ubiquitinated inclusions were noted than TDP-43 inclusions, especially in the dentate gyrus and neocortex in cases 5 and 9, raising the possibility of an additional unidentified component within the ubiquitinated inclusions apart from TDP-43. These findings are similar to the pathology in the VSM-20 family [14]. The number of ubiquitinated neuronal inclusions in the dentate gyrus and pyramidal neuron layers in the hippocampus from all three cases was higher than for TDP-43 NCI in the same areas. The presence of TDP-43 pathology in white matter threads and glia as noted in the three cases is in agreement with the observations made in two other families [12, 14], raising the possibility of a role for glia in the pathology of this disease.
Causative mutations for familial ALS have been identified in the SOD-1 [22] and FUS [23] genes, but in these families the pathology is distinct from sporadic ALS. The current family does not have mutations in FUS, SOD1 or TARDBP. A mutation in the IFT74 gene has been identified in one family thought to be chromosome 9p linked [10], but this has not been replicated in other families and is also absent in our family. Other groups have also failed to identify a non-polymorphic exonic mutation in this region, suggesting a non-protein coding pathogenic mechanism [12–18]. Given that RNA processing is emerging as a pathogenic mechanism in ALS, it is possible that the chromosome 9 region contains a pathogenic RNA encoding sequence [24].
References
Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58(11):1615–1621
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65(4):586–590. doi:10.1212/01.wnl.0000172911.39167.b6
Rippon GA, Scarmeas N, Gordon PH, Murphy PL, Albert SM, Mitsumoto H, Marder K, Rowland LP, Stern Y (2006) An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch Neurol 63(3):345–352. doi:10.1001/archneur.63.3.345
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59(7):1077–1079
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. doi:10.1126/science.1134108
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol 114(1):5–22. doi:10.1007/s00401-007-0237-2
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171(1):227–240
Polvikoski TM, Murray A, Harper PS, Neal JW (2003) Familial motor neurone disease with dementia: phenotypic variation and cerebellar pathology. J Neurol Neurosurg Psychiatry 74(11):1516–1520
Namimatsu S, Ghazizadeh M, Sugisaki Y (2005) Reversing the effects of formalin fixation with citraconic anhydride and heat: a universal antigen retrieval method. J Histochem Cytochem 53(1):3–11. doi:10.1369/jhc.4C6466.2005
Momeni P, Schymick J, Jain S, Cookson MR, Cairns NJ, Greggio E, Greenway MJ, Berger S, Pickering-Brown S, Chio A, Fung HC, Holtzman DM, Huey ED, Wassermann EM, Adamson J, Hutton ML, Rogaeva E, St George-Hyslop P, Rothstein JD, Hardiman O, Grafman J, Singleton A, Hardy J, Traynor BJ (2006) Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol 6:44. doi:10.1186/1471-2377-6-44
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. doi:10.1086/519795
Le Ber I, Camuzat A, Berger E, Hannequin D, Laquerriere A, Golfier V, Seilhean D, Viennet G, Couratier P, Verpillat P, Heath S, Camu W, Martinaud O, Lacomblez L, Vercelletto M, Salachas F, Sellal F, Didic M, Thomas-Anterion C, Puel M, Michel BF, Besse C, Duyckaerts C, Meininger V, Campion D, Dubois B, Brice A (2009) Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72(19):1669–1676. doi:10.1212/WNL.0b013e3181a55f1c
Luty AA, Kwok JB, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, Halliday GM, Schofield PR (2008) Pedigree with frontotemporal lobar degeneration—motor neuron disease and tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol 8:32. doi:10.1186/1471-2377-8-32
Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, Whitwell J, Foti D, McDade E, Molano J, Karydas A, Wojtas A, Goldman J, Mirsky J, Sengdy P, Dearmond S, Miller BL, Rademakers R (2010) Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp.2009.204081
Gijselinck I, Engelborghs S, Maes G, Cuijt I, Peeters K, Mattheijssens M, Joris G, Cras P, Martin JJ, De Deyn PP, Kumar-Singh S, Van Broeckhoven C, Cruts M (2010) Identification of 2 loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 67(5):606–616. doi:10.1001/archneurol.2010.82
Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH Jr (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66(6):839–844. doi:10.1212/01.wnl.0000200048.53766.b4
Valdmanis PN, Dupre N, Bouchard JP, Camu W, Salachas F, Meininger V, Strong M, Rouleau GA (2007) Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol 64(2):240–245. doi:10.1001/archneur.64.2.240
Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 129(4):868–876. doi:10.1093/brain/awl030
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M (2007) PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17(11):1665–1674. doi:10.1101/gr.6861907
Boxer RS, Kleppinger A, Brindisi J, Feinn R, Burleson JA, Kenny AM (2010) Effects of dehydroepiandrosterone (DHEA) on cardiovascular risk factors in older women with frailty characteristics. Age Ageing 39(4):451–458. doi:10.1093/ageing/afq043
Seelaar H, Schelhaas HJ, Azmani A, Kusters B, Rosso S, Majoor-Krakauer D, de Rijik MC, Rizzu P, ten Brummelhuis M, van Doorn PA, Kamphorst W, Willemsen R, van Swieten JC (2007) TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without progranulin mutations. Brain 130(Pt 5):1375–1385. doi:10.1093/brain/awm024
Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364(6435):362. doi:10.1038/364362c0
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. doi:10.1126/science.1165942
Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136(6):1001–1004. doi:10.1016/j.cell.2009.03.006
Acknowledgments
We are grateful to members of this family for participating in this research. We are grateful for the opportunity to review clinical notes made by Prof. P. Harper, Prof. A. Compston and Dr. T. Pickersgill and to Dr A. Liu (Consultant Neuroradiologist, University Hospital of Wales) for reviewing the radiology. This work was supported by the Motor Neuron Disease Assocaition (UK), the Medical Research Council (UK) Grant G0700943 and the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; project Z01 AG000951-06.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
415_2010_5815_MOESM1_ESM.pdf
Online Resource 1: Radiological features, coronal and axial T2 MRI showing parieto-occipital atrophy in case 6 (A–D), frontotemporal atrophy in case 8 (E–H) and normal imaging in case 9 (I–L). (PDF 148 kb)
Rights and permissions
About this article
Cite this article
Pearson, J.P., Williams, N.M., Majounie, E. et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol 258, 647–655 (2011). https://doi.org/10.1007/s00415-010-5815-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-010-5815-x