
Abstract
There is evidence from in vitro and animal experiments that oral creatine (Cr) supplementation might prevent or slow down neurodegeneration in Huntington’s disease (HD). However, this neuroprotective effect could not be replicated in clinical trials, possibly owing to treatment periods being too short to impact on clinical endpoints. We used proton magnetic resonance spectroscopy (1H-MRS) as a surrogate marker to evaluate the effect of Cr supplementation on brain metabolite levels in HD.
Twenty patients (age 46±7.3 years, mean duration of symptoms 4.0±2.1 years, number of CAG repeats 44.5±2.7) were included. The primary endpoint was metabolic alteration as measured by 1H-MRS in the parieto-occipital cortex before (t1) and after 8–10 weeks (t2) of Cr administration. Secondary measures comprised the motor section of the Unified Huntington’s Disease Rating Scale and the Mini Mental State Examination.
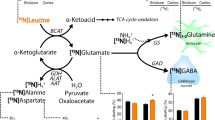
1H-MRS showed a 15.6% decrease of unresolved glutamate (Glu)+glutamine (Gln; Glu+Gln=Glx; p<0.001) and a 7.8% decrease of Glu (p<0.027) after Cr treatment. N-acetylaspartate trended to fall (p=0.073) whereas total Cr, choline-containing compounds, glucose, and lactate remained unchanged. There was no effect on clinical rating scales.
This cortical Glx and Glu decrease may be explained by Cr enhancing the energy-dependent conversion of Glu to Gln via the Glu-Gln cycle, a pathway known to be impaired in HD. Since Glu-mediated excitotoxicity is presumably pivotal in HD pathogenesis, these results indicate a therapeutic potential of Cr in HD. Thus, longterm clinical trials are warranted.
Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
References
Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF (2001) Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol Dis 8:479–491
Andreassen OA, Jenkins BG, Dedeoglu A, Ferrante KL, Bogdanov MB,Kaddurah-Daouk R, Beal MF (2001) Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J Neurochem 77:383–390
Balsom PD, Soderlund K, Ekblom B (1994) Creatine in humans with special reference to creatine supplementation. Sports Med 18:268–280
Beal MF (1992) Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol 31:119–130
Beal MF, Ferrante RJ, Swartz KJ, Kowall NW (1991) Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci 11:1649–1659
Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125:1908–1922
Bellocchio EE, Reimer RJ, Fremeau RT Jr, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957–960
Brewer GJ, Wallimann TW (2000) Protective effect of the energy precursor creatine against toxicity of glutamate and beta-amyloid in rat hippocampal neurons. J Neurochem 74:1968–1978
Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41:646–653
Brustovetsky N, Brustovetsky T, Dubinsky JM (2001) On the mechanisms of neuroprotection by creatine and phosphocreatine. J Neurochem 76:425–434
Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ (1996) Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med 2:347–350
Butterworth J, Yates CM, Reynolds GP (1985) Distribution of phosphate-activated glutaminase, succinic dehydrogenase, pyruvate dehydrogenase and gamma-glutamyl transpeptidase in post-mortem brain from Huntington’s disease and agonal cases. J Neurol Sci 67:161–171
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695
Coyle JT, Schwarcz R (1976) Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 263:244–246
Daikhin Y, Yudkoff M (2000) Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr 130:1026S–1031S
Dechent P, Pouwels PJ, Wilken B, Hanefeld F, Frahm J (1999) Increase of total creatine in human brain after oral supplementation of creatine-monohydrate. Am J Physiol 277:R698–R704
Dedeoglu A, Kubilus JK, Yang L, Ferrante KL,Hersch SM, Beal MF, Ferrante RJ (2003) Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington’s disease transgenic mice. J Neurochem 85:1359–1367
Erecinska M, Silver IA (1990) Metabolism and role of glutamate in mammalian brain. Prog Neurobiol 35:245–296
Erecinska M, Zaleska MM, Nissim I, Nelson D, Dagani F, Yudkoff M (1988) Glucose and synaptosomal glutamate metabolism: studies with [15N]glutamate. J Neurochem 51:892–902
Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF (2000) Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci 20:4389–4397
Gramsbergen JB, Veenma-Van der Duin L, Venema K, Korf J (1986) Cerebral cation shifts and amino acids in Huntington’s disease. Arch Neurol 43:1276–1281
Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH (1996) Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39:385–389
Guyot MC, Hantraye P, Dolan R, Palfi S, Maziere M, Brouillet E (1997) Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 79:45–56
Harms L, Meierkord H, Timm G, Pfeiffer L, Ludolph AC (1997) Decreased Nacetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington’s disease: a proton magnetic resonance spectroscopy study. J Neurol Neurosurg Psychiatry 62:27–30
Huntington Study Group (1996) Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord 11:136–142
Jan GG, Veldink JH, van dT, I, Kalmijn S, Beijer C, de Visser M,Wokke JH, Franssen H, van den Berg LH (2003) A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol 53:437–445
Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR (1993) Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 43:2689–2695
Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, MacDonald M, Rosen BR, Beal MF, Koroshetz WJ (1998) 1H NMR spectroscopy studies of Huntington’s disease: correlations with CAG repeat numbers. Neurology 50:1357–1365
Juhn MS, Tarnopolsky M (1998) Oral creatine supplementation and athletic performance: a critical review. Clin J Sport Med 8:286–297
Kanner BI, Schuldiner S (1987) Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem 22:1–38
Kay L, Nicolay K, Wieringa B, Saks V, Wallimann T (2000) Direct evidence for the control of mitochondrial respiration by mitochondrial creatine kinase in oxidative muscle cells in situ. J Biol Chem 275:6937–6944
Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF (1999) Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med 5:347–350
Lawler JM, Barnes WS, Wu G, Song W, Demaree S (2002) Direct antioxidant properties of creatine. Biochem Biophys Res Commun 290:47–52
Li H, Li SH, Johnston H, Shelbourne PF, Li XJ (2000) Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet 25:385–389
Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP (2001) Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis 8:807–821
Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF (1999) Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol 157:142–149
Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF (1998) Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci 18:156–163
Mazzola JL, Sirover MA (2002) Alteration of nuclear glyceraldehyde-3-phosphate dehydrogenase structure in Huntington’s disease fibroblasts. Brain Res Mol Brain Res 100:95–101
O’Gorman E, Beutner G, Dolder M, Koretsky AP, Brdiczka D, Wallimann T (1997) The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Lett 414:253–257
Perry TL, Hansen S (1990) What excitotoxin kills striatal neurons in Huntington’s disease? Clues from neurochemical studies. Neurology 40:20–24
Reynolds GP, Pearson SJ (1987) Decreased glutamic acid and increased 5-hydroxytryptamine in Huntington’s disease brain. Neurosci Lett 78:233–238
Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG (1999) In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate-glutamine neurotransmitter cycle and functional neuroenergetics. Philos Trans R Soc Lond B Biol Sci 354:1165–1177
Schiefer J, Landwehrmeyer GB, Luesse HG, Sprunken A, Puls C, Milkereit A, Milkereit E, Kosinski CM (2002) Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov Disord 17:748–757
Shear DA, Haik KL, Dunbar GL (2000) Creatine reduces 3-nitropropionicacid-induced cognitive and motor abnormalities in rats. Neuroreport 11:1833–1837
Shoulson I (1981) Huntington disease: functional capacities in patients treated with neuroleptic and antidepressant drugs. Neurology 31:1333–1335
Tabrizi SJ, Blamire AM, Manners DN, Rajagopalan B, Styles P, Schapira AH, Warner TT (2003) Creatine therapy for Huntington’s disease: Clinical and MRS findings in a 1-year pilot study. Neurology 61:141–142
Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH (1999) Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol 45:25–32
Tarnopolsky MA, Beal MF (2001) Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol 49:561–574
Taylor-Robinson SD, Weeks RA, Bryant DJ, Sargentoni J, Marcus CD, Harding AE, Brooks DJ (1996) Proton magnetic resonance spectroscopy in Huntington’s disease: evidence in favour of the glutamate excitotoxic theory. Mov Disord 11:167–173
Verbessem P, Lemiere J, Eijnde BO, Swinnen S, Vanhees L, Van Leemputte M, Hespel P, Dom R (2003) Creatine supplementation in Huntington’s disease: A placebo-controlled pilot trial. Neurology 61:925–930
Vielhaber S, Kaufmann J, Kanowski M, Sailer M, Feistner H, Tempelmann C, Elger CE, Heinze HJ, Kunz WS (2001) Effect of creatine supplementation on metabolite levels in ALS motor cortices. Exp Neurol 172:377–382
Xu CJ, Klunk WE, Kanfer JN, Xiong Q, Miller G, Pettegrew JW (1996) Phosphocreatine-dependent glutamate uptake by synaptic vesicles.A comparison with atp-dependent glutamate uptake. J Biol Chem 271:13435–13440
Yudkoff M, Nissim I, Daikhin Y, Lin ZP, Nelson D, Pleasure D, Erecinska M (1993) Brain glutamate metabolism: neuronal-astroglial relationships. Dev Neurosci 15:343–350
Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA (2001) Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci 17:41–53
Author information
Authors and Affiliations
Corresponding author
Additional information
* Drs Bender and Auer both contributed equally.
Rights and permissions
About this article
Cite this article
Bender*, A., Auer*, D.P., Merl, T. et al. Creatine supplementation lowers brain glutamate levels in Huntington’s disease. J Neurol 252, 36–41 (2005). https://doi.org/10.1007/s00415-005-0595-4
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00415-005-0595-4