
Abstract
Forensic medicine defines the unexplained sudden death as a death with a non-conclusive diagnosis after autopsy. Molecular diagnosis is being progressively incorporated in forensics, mainly due to improvement in genetics. New genetic technologies may help to identify the genetic cause of death, despite clinical interpretation of genetic data remains the current challenge. The identification of an inheritable defect responsible for arrhythmogenic syndromes could help to adopt preventive measures in family members, many of them asymptomatic but at risk of sudden death. This multidisciplinary translational research requires a specialized team.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Negative autopsy
An autopsy is the examination of a body after death, which may reveal aspects such as cause of death and the time since death. The autopsy is considered to be negative when all protocol efforts fail to reveal the cause of death. Thus, unexplained death is defined as the death that remains unsolved after a thorough autopsy of an individual without previous cardiac history and who has been seen alive within the previous 12 h of the death [1]. In a typical medical examiner practice, approximately 50 % of the deaths are natural, 5–10 % is unexplained after a gross autopsy and 1–5 % is negative after extensive autopsy (gross and microscopic examination, toxicological analysis and laboratory investigations) [2]. Recent studies suggest that around 5–10 % of the autopsy cases remain unexplained in Europe, (the so-called unexplained sudden death or “mors sine materia”), suggesting a cardiac death due to electric inherited disorders without heart structural alterations [3]. The Heart Rhythm Society and the European Heart Rhythm Association provided an expert consensus statement providing guidelines for post-mortem genetic testing in cases of unexplained sudden death [4]. Despite this fact, post-mortem genetic testing (or molecular autopsy) has not yet been included into a routine of the conventional autopsy when the coroner, medical examiner, or forensic pathologist is faced with a negative autopsy with suspicious arrhythmogenic cause of death.

Overlapping of genes associated with channelopathies. BrS Brugada syndrome, LQT Long QT syndrome, SQT Short QT syndrome, CPVT Catecholaminergic polymorphic ventricular tachycardia. SCN1B associated with BrS is an unpublished manuscript, currently under review

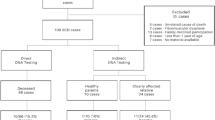
Flow chart of a multidisciplinary cooperation in cases of SUD/SCD
In infancy population, sudden infant death syndrome (SIDS)—the incidence of sudden unexpected deaths without conclusive diagnose after autopsy range between 40 and 80 % [5]. A case of definite SIDS is generally accepted to require a complete post-mortem examination including a negative autopsy. Thus, SIDS is a diagnosis of exclusion, remaining the leading cause of death in the first 6 months after birth in the industrialized world. Other special kind of sudden death is one that occurs in a patient previously diagnosed with epilepsy—sudden unexpected death in epilepsy (SUDEP). The autopsy did not reveal any anatomical nor toxicological cause of death, excluding status epilepticus and traumatic death secondary to a seizure. SUDEP is the most important cause of death in young epileptic patients [6].
Reinterviewing family members may reveal helpful information not discovered in the initial research. In addition, consultation with cardiac pathologists, cardiologists, electrophysiologists and geneticists may be useful. Unfortunately, collaboration between medical genetics departments and cardiology only exists for around 20 % of respondents [7]. The link between the post-mortem forensic investigation of a victim and the clinical investigation of the surviving family members is difficult to establish. This difficulty may result from legal restrictions. Resolving cause of death in natural deaths, particularly when it occurred suddenly, unexpectedly or in the young, is a main part of current forensic autopsy practice in order to assist the relatives of the deceased to deal with the death.
Sudden cardiac death
Sudden death (SD) is defined as an unexpected natural death in apparently healthy individuals that takes place during the first hour after onset of symptoms. Almost 85 % of all SD are of cardiac origin (sudden cardiac death, SCD), being a leading cause of death in Western countries, responsible for around 30–200/100,000 every year [8]. The main difficulty of calculating the effect lies in the definition of the SCD; although also medical reports are not accessible in all cases, no autopsy is performed or the cause of death given on the death certificate is not specific [9]. As a consequence, there are a large proportion of SCD that remains unresolved, without a definitive diagnosis. Despite these facts, it is established that in individuals over 55 years old, 80 % of SCD cases are consequence of coronary heart disease, but in the younger population (<35 years), the SCD cause is mainly due to inherited genetic variations [10]. A recent retrospective study performed in a cohort of children and adolescents shows that death in the very young is often caused by arrhythmic syndromes, and death during effort is typically seen in those with structural heart disease [11].
The post-mortem genetic testing has the goal to increase diagnostic yield, facilitating the cascade screening of family members with more focused testing [12]. Thus, it was stated that molecular autopsy should be considered as a part of the comprehensive medicolegal investigation in SCD cases without structural heart alterations. However, post-mortem genetic testing of the proband was reported in only 10 % of SCD patients with no structural alterations. It occurs mainly because forensic centres do not have the economic resources to perform genetic testing, and/or do not collect samples for genetic screening due to currently legal restrictions involved with the sampling and storage of DNA [7].
To this end, we need to remember that, due to the ease of storing and transporting formalin fixed–paraffin-embedded tissue (FF-PET), the vast majority of forensic and general pathologists archive it as the only source available for DNA procurement [13]. DNA from FF-PET can be used in clinical diagnosis, such as cancer testing, but usually DNA is degraded by formalin and FF-PETs constitute suboptimal sources for post-mortem genetic testing [14]. In contrast, blood collected in ethylenediaminetetraacetic acid (EDTA; purple-top tube) or frozen heart, liver or spleen provides the greatest source of intact DNA, permitting the successful performance of post-mortem cardiac channel genetic testing. Moreover, at least 10 ml of EDTA blood or 5 g of fresh tissue should be obtained at autopsy. The tissue should be stored at −80 °C, until DNA can be extracted. Alternatively, 50 to 100 μl of whole blood on filter paper can be used for a molecular autopsy. However, this tends to provide a very limited amount of DNA [13].
But DNA is not the only source for post-mortem studies. Considering the important role of ion channels and their function or malfunction in several heritable and acquired channelopathies, post-mortem mRNA expression analysis on tissue from pathologic and non-pathologic hearts could be a very useful source to investigate the expression of Na+ and K+ channels [15]. Taking into account all these information, it is of extreme importance that guidelines central to the procurement of DNA/mRNA-friendly sources should be added to the standard of care for the post-mortem analysis of SCD.
Genetics
The role of inheritance in SCD cases has been assessed by several epidemiological and comprehensive genetic studies. All these studies reveal that genetic testing of SD victims is useful for clinical and forensic post-mortem investigation [16]. It has been published that between 5 and 40 % of the so-called autopsy-negative SDs are believed to be due to sudden arrhythmic death syndrome, and genetic testing can be used to confirm the presence of a disease-causing mutation. Despite the fact that some significant overlaps have been reported (Fig. 1), inherited cardiac diseases associated with SCD can be classified into two main groups: (1) Channelopathies, in which the arrhythmogenic substrate is found in the electrical properties of the heart because mutations occur in genes that encode ion channels proteins (Table 1). This group is characterized by negative autopsy with no structural heart alterations and includes Long QT syndrome (LQTS), Brugada syndrome (BrS), Short QT syndrome (SQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) [17]; (2) Cardiomyopathies in which structural abnormalities such as hypertrophy, dilatation or fatty and/or fibrotic infiltration are responsible for inducing arrhythmias. Research has shown that it can be caused by mutations in genes mainly encoding three types of proteins: sarcomeric, which cause hypertrophic cardiomyopathy (HCM); cytoskeletal, which cause dilated cardiomyopathy (DCM); and desmosomal, which cause arrhythmogenic right ventricular cardiomyopathy (ARVC) [18].
A hallmark of SCD-related diseases is the incomplete penetrance and variable expressivity. Despite channelopathies has been define as monogenic diseases, it is difficult to attribute the marked incomplete penetrance and variable expressivity observed in Mendelian/monogenic disorders solely to the interaction between a pathogenic mutation [19]. However, so far, the identification of a pathogenic mutation associated with one of these diseases in a post-mortem case supposes that family members may be at risk of SCD [20] because despite many of them may remain asymptomatic, the first manifestation of disease could be the syncope/death. In the first group, autopsy inconclusive after macro/microscopic study, and negative toxicological analysis, is a major health problem. The investigation of an unexplained sudden death is extremely complicated in the families of the victims, especially when the victim is a child (SIDS) [21].
In recent years, advances in genetics of SCD syndromes are being incorporated as a clinical tool for patients and family member’s diagnosis [22]. It has been recommended that all relatives of unexplained SD victims undergo evaluation by a multidisciplinary team of cardiologists, forensic pathologists and geneticists [23] (Fig. 2). Unfortunately, nowadays, a genetic investigation in unexplained SD is not yet performed as part of the standard forensic investigation although recent legal and ethical issues of post-mortem genetic testing in the forensic context propose that the genetic analysis should be considered [24].
Channelopathies
This group includes familial arrhythmogenic syndromes caused by mutations in genes encoding ion channels or associated proteins [25]. Cardiac channelopathies are clinically identified mainly by characteristic ECG abnormalities because they are not accompanied by morphological heart defects. However, incomplete penetrance and variable expressivity in inherited arrhythmogenic disorders imply that the distinctive ECG patterns that characterize these disorders may be masked. Interestingly, the absence of the ECG markers of the disease is not an indicator of favourable outcome [26]. Genetic diagnosis helps to identify both the pathogenic variation and genetic carriers although it does not always occur. Despite this fact, their diagnosis, prevention and treatment may be expected to improve greatly with the availability of genetic tests.
Numerous mutation-related arrhythmogenic diseases, affecting sodium (Na+), potassium (K+) or calcium (Ca2+) ion currents, have been reported affecting either the generation of the action potential or calcium homeostasis. Thus, depending on which ion channel is affected, different syndromes will be present [27]. However, knowledge is not limited to the familial form, as it opens up new hypotheses as to how the gene interacts with the environment, drugs and damaged muscle, and how arrhythmias arise in acquired or non-inherited forms [17].
Brugada syndrome
This arrhythmogenic syndrome was described 20 years ago by Pedro and Josep Brugada [28]. It is characterized by an ECG pattern consisting of coved-type ST-segment elevation in atypical right-bundle branch block in leads V1 to V3 (often referred to as type-1 Brugada ECG pattern), and an increased risk for SCD resulting from episodes of polymorphic ventricular tachyarrhythmia [29]. The prevalence of the disease is 4–12 % of all SCD causes. The penetrance and expressivity of the disorder are highly variable, although it is considered a disorder involving mainly young male adults (30–40 years old), and SCD typically occurring during sleep. Curiously, in 2002, sudden unexpected nocturnal death syndrome (SUNDS) was related to the most common cause of natural death in young Asians [30].
To date, more than 200 pathogenic mutations has been identified in 16 genes (SCN5A, GPD1-L, SCN1B, SCN3B, KCNE3, KCNE5, KCNJ8, KCND3, CACNA1C, CACNB2b, CACNA2D1, RANGRF, HCN4, SLMAP and TRPM4) [31]. Recently, our group has identified the last gene associated with the disease, the SCN2B gene [32]. Despite all these genes have been associated with BrS, an exhaustive genetic test only identifies the pathogenic cause in nearly 35–40 % of clinically diagnosed cases. Approximately 25–30 % of all these patients carry a pathogenic mutation in the SCN5A gene (BrS type 1) [33]. Therefore, 60–65 % of BrS patients remain without a pathogenic genetic cause identified. Following these genetic data, analysing the SCN5A gene should be the most cost-effective option in post-mortem cases of families showing no history of cardiac events, and with suspicions of BrS as cause of death.
Long QT syndrome
LQTS is a channelopathy characterized for a prolongation of the QT interval in the ECG (QTc > 480 ms) [34]. The clinical presentation can be variable, ranging from asymptomatic patients to episodes of syncope and SCD due to ventricular tachyarrhythmia (torsade de pointes) in the setting of a structurally normal heart, and with a prevalence of 1/2,500 individuals [35]. However, the penetrance of the disease is not 100 % and therefore individuals and family members at risk with a normal ECG may be identified through genetic testing. Due to the ability to identify the individuals at risk through ECG analysis, and that LQTS is one of the leading causes of SCD among young people, massive population screening by ECG has been performed in certain regions, with success of lowering rates of SD among infants and athletes [36].
Currently, pathogenic single mutations and splice-site altering mutations have been identified in 15 genes (KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1, RYR2 and KCNJ5) [31]. The last gene associated with LQTS is SCN1B, reported recently by our group and Riuro et al., Heart Rhythm 2013 (under review). Approximately, all genes together are responsible for nearly 80–85 % of all LQT cases [37]. Concretely, a 70–75 % of LQT cases are caused by pathogenic mutations only in three genes: KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3). The most common form of LQT (type 1) is caused by mutations in KCNQ1 and it is responsible for around 35 % of the cases of prolonged QT interval. Several mutations in KCNH2 (hERG, human-ether-a-go-go-related) that codifies the α-subunit of Ikr were also identified. The mutations in this gene were supposed to be around 30 % of cases (type 2). Gain of function mutations in SCN5A (inappropriate prolonged entry of Na+ into the myocytes), the third gene most prevalent in LQTS (around 10 %), has been also identified. Therefore, genetic analysis of these three genes is the cost-effective decision in post-mortem cases of families showing no history of cardiac events, and with suspicions of LQTS as cause of death [38].
Short QT syndrome
This rare syndrome was described in 2000 [39]. It is a highly lethal arrhythmic disease characterized by a short QT interval in ECG (<330 ms), with a high sharp T wave and a short interval between the peak and the end of the T wave, leading some clinical manifestations from lack of symptoms to atrial fibrillation (AF), recurrent syncope, and SCD. Clinical manifestations may appear as early as childhood, and it is considered one of the main causes of SIDS [40].
The genetic origin has been reported with an autosomal dominant pattern of inheritance and high penetrance. To date, several mutations related to SQTS has been identified in six genes: three of them (KCNQ1, KCNJ2 and KCNH2) encoding potassium channels and three more (CACNA1C, CACNB2B and CACNA2D1) encodes calcium channels [31]. All these six genes enclose nearly 50 % of clinically diagnosed SQT cases. However, pathogenic mutations in potassium channels are the most prevalent, mainly in KCNH2. Type 1 SQTS has been associated with pathogenic mutations in the KCNH2 gene (hERG). In general, cardiac events are associated with adrenergic situations such as noise or exercise, although they have also been reported at rest. Type 2 SQTS has been linked, so far to two pathogenic mutations in the KCNQ1 gene. There is a particular entity caused by pathogenic mutation in this same gene which is manifested in utero as bradycardia and which is diagnosed as AF and short QT in the neonatal period. Type 3 SQTS is related to pathogenic mutations in the KCNJ2. Pathogenic mutations in CACNA1C, CACNB2B have been associated to patients affected by an overlap of BrS and shortened QT [41]. Last year, other pathogenic mutation in CACNA2D1 was associated to short QT interval in one patient [42]. Therefore, genetic test of the KCNH2 gene should be the best cost-effective option in post-mortem cases of families showing no history of cardiac events, and with suspicions of SQTS as cause of death.
Catecholaminergic polymorphic ventricular tachycardia
CPVT is an inherited disorder characterized by a two-way polymorphic ventricular tachycardia, trigger by an adrenergic stimulus. It is an arrhythmogenic cardiac disease associated to high mortality (around 30 % by the age of 30 years) [43]. Usually, the first manifestation of CPVT is the death of the patient. This fact supposes that identification of family members at risk is crucial to avoid new cases of SCD.
To date, five genes have been related to the disease, being responsible for nearly 60 % of all clinically diagnosed cases (RyR2, CASQ2, KCNJ2, TRDN and CALM1) [31]. The main gene responsible for CPVT is RYR2, that encodes the ryanodine receptor (autosomal dominant)(type 1). It is responsible for nearly 50 % of all cases. All other genes together are responsible for nearly 10 % of CPVT cases. The second gene is CASQ2, that encodes calsequestrin protein (autosomal recessive)(type 2) [44]. Both RyR2 and CASQ2 are implicated in regulating intracellular calcium, and pathogenic mutations in both lead to increased function of encoded proteins, and consequently increasing outflow of calcium from the sarcoplasmatic reticulum. Recently, CPVT type 3, type 4 and type 5 have been described. Type 3 is due to a de novo KCNJ2 pathogenic mutation associated with classic phenotypic features of Andersen-Tawil syndrome (type 7) and CPVT mimicry [45]. Type 4 is caused by a pathogenic mutation in TRDN with a recessive autosomic pattern that encodes triadin protein [46]. The last gene associated with CPVT is CALM1, (type 5) that encodes calmodulin protein, involved in calcium intracellular pathways [47]. Genetic test of the RyR2 gene should be the best cost-effective option in post-mortem cases of families showing no history of cardiac events, and with suspicions of CPVT as cause of death.
Next-generation sequencing
Current genetic studies focused on inherited channelopathies associated with SCD are performed using Sanger sequencing method. This genetic analysis method is called after its creator, Frederick Sanger, who in 1975 developed the process by dideoxynucleotide sequencing or Sanger method. Despite slow and expensive, this technique remains as the gold standard method. As mentioned before, several genes have been associated to lethal arrhythmias, and all of them should be tested in a genetic analysis, the best cost-effective option would be to analyse only four genes (SCN5A, KCNQ1, KCNH2 and RyR2). It is according to the key recommendations of the Heart Rhythm Society/European Heart Rhythm Association guidelines [4]. However, in the last 10 years, new technologies has been developed to sequence many genes at once (even an entire exome or genome), in a few hours and at low cost. This new genetic technology has been called next-generation sequencing (NGS).
Despite NGS technology shows great advantages compared to Sanger method, there are some limitations, such as the high specificity of purity and integrity of DNA in the process of sample preparation. Another problem of NGS technology is the amount of data provided. To manage this problem, new bioinformatic tools appear continuously. However, the biggest current challenge is distinguishing between pathogenic mutations and benign rare variants, and the clinical interpretation of all this genetic data [48]. Despite in silico predictions of pathogenicity, in vitro studies of selected variants, genotype-phenotype segregation in relatives and global database projects including frequency of genetic variants of unknown significance (GVUS), most part of these GVUS remains unclassified, without a solid pathogenicity. In addition, other numerous rare variants are not catalogued yet. The most accurate solution should be to sequence large control cohorts of different ethnic origins that have very accurate cardiovascular phenotypes.
Despite it is not included in current forensics guidelines, some centres have incorporated Sanger technology in unsolved cases after autopsy. Recent studies using the Sanger method identify 25 % of cases carrying a genetic defect that could explain the sudden death [38, 49]. These studies analyse only between five and ten genes associated with lethal arrhythmias and SCD. So far, any study using NGS technology in forensic cases suspicious of SCD has not been reported. To our acknowledgement, our group has published the first study in a SIDS case using a target resequencing panel (Campuzano et al., (under review)).
We believe that the use of NGS technology in forensics may help to identify more genetic defects in non-conclusive autopsies. It could explain the cause of death in a high number of cases, in a shorter amount of time and less cost, being a cost-effective option in the near future. The molecular autopsy should be covered by the same institution that pays for toxicological and histological testing, either the hospital or the legal institution when the death remains unsolved after a complete autopsy.
Conclusions
The progressive incorporation of genetic testing in non-diagnosed forensic cases is allowing identifying the genetic alteration that could be recognized as the probable cause of death in a high percentage of cases. Positive identification of the genetic cause not only gives an answer to the family about the most probable cause of death of the family member that died suddenly, but especially helps to identify relatives who are carriers of the genetic alteration at that may be at risk of sudden death. Therefore, we believe that the hospital or legal institution should cover the cost of molecular autopsy because it is a measure of prevention. For all these reasons, in agreement with previous studies [50], we recommend genetic study in post-mortem cases with a non-conclusive autopsy, as well as the introduction of this process as a routine part of the autopsy protocol. To this end, a close interdisciplinary collaboration between the forensic pathologist, the clinical geneticist and the cardiologist is necessary for the best management of the families involved in these cases. Forensic pathologists play here a crucial role, since genetic analysis is an important tool to ascertain the cause of death and is prospectively lifesaving of those left behind. Finally, we need to remember that due to the issues associated with incomplete penetrance and variable expressivity, the genetic test results must be interpreted cautiously and incorporated into the overall diagnostic evaluation for these disorders. Even when a genetic variant has been published previously as a putative pathogenic mutation, assignment of a specific genetic variant as a true pathogenic disease-causing mutation still requires vigilant scrutiny. To be sure, post-mortem genetic tests are fundamentally probabilistic tests rather than absolutely deterministic ones.
References
Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M, Thiene G, van der Wal A (2008) Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch 452(1):11–18
Lawler W (1990) The negative coroner’s necropsy: a personal approach and consideration of difficulties. J Clin Pathol 43(12):977–980
Oliva A, Brugada R (2010) D’Aloja E, Boschi I, Partemi S, Brugada J. State of the art in forensic investigation of sudden cardiac death. Am J Forensic Med Pathol, Pascali VL
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R et al (2011) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8(8):1308–1339
Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T (2011) Cardiac channelopathies and sudden infant death syndrome. Cardiology 119(1):21–33
Devinsky O (2011) Sudden, unexpected death in epilepsy. N Engl J Med 365(19):1801–1811
Michaud K, Mangin P, Elger BS (2011) Genetic analysis of sudden cardiac death victims: a survey of current forensic autopsy practices. Int J Legal Med 125(3):359–366
Pachon M, Almendral J (2011) Sudden death: managing the patient who survives. Heart 97(19):1619–1625
Adabag AS, Luepker RV, Roger VL, Gersh BJ (2010) Sudden cardiac death: epidemiology and risk factors. Nat Rev Cardiol 7(4):216–225
Arzamendi D, Benito B, Tizon-Marcos H, Flores J, Tanguay JF, Ly H, Doucet S, Leduc L, Leung TK, Campuzano O et al (2011) Increase in sudden death from coronary artery disease in young adults. Am Heart J 161(3):574–580
Pilmer CM, Kirsh JA, Hildebrandt D, Krahn AD, Gow RM: Sudden cardiac death in children and adolescents between 1 and 19 years of age. Heart Rhythm 2013
Tester DJ, Ackerman MJ (2012) The molecular autopsy: should the evaluation continue after the funeral? Pediatr Cardiol 33(3):461–470
Basso C, Carturan E, Pilichou K, Rizzo S, Corrado D, Thiene G (2010) Sudden cardiac death with normal heart: molecular autopsy. Cardiovasc Pathol 19(6):321–325
Carturan E, Tester DJ, Brost BC, Basso C, Thiene G, Ackerman MJ (2008) Postmortem genetic testing for conventional autopsy-negative sudden unexplained death: an evaluation of different DNA extraction protocols and the feasibility of mutational analysis from archival paraffin-embedded heart tissue. Am J Clin Pathol 129(3):391–397
Partemi S, Berne PM, Batlle M, Berruezo A, Mont L, Riuro H, Ortiz JT, Roig E, Pascali VL, Brugada R et al: Analysis of mRNA from human heart tissue and putative applications in forensic molecular pathology. Forensic Sci Int 2010
Elger BS, Michaud K, Fellmann F, Mangin P (2010) Sudden death: ethical and legal problems of post-mortem forensic genetic testing for hereditary cardiac diseases. Clin Genet 77(3):287–292
Roberts JD, Gollob MH (2010) The genetic and clinical features of cardiac channelopathies. Futur Cardiol 6(4):491–506
Jacoby D, McKenna WJ (2012) Genetics of inherited cardiomyopathy. Eur Heart J 33(3):296–304
Giudicessi JR, Ackerman MJ (2013) Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Translational research. J Lab Clin Med 161(1):1–14
Hendrix A, Borleffs CJ, Vink A, Doevendans PA, Wilde AA, van Langen IM, van der Smagt JJ, Bots ML, Mosterd A (2012) Cardiogenetic screening of first-degree relatives after sudden cardiac death in the young: a population-based approach. Europace 13(5):716–722
Klaver EC, Versluijs GM, Wilders R (2011) Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol 152(2):162–170
Brion M, Quintela I, Sobrino B, Torres M, Allegue C, Carracedo A: New technologies in the genetic approach to sudden cardiac death in the young. Forensic Sci Int 2010
Behr E, Wood DA, Wright M, Syrris P, Sheppard MN, Casey A, Davies MJ, McKenna W (2003) Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet 362(9394):1457–1459
Ormondroyd E, Moynihan C, Watson M, Foster C, Davolls S, Ardern-Jones A, Eeles R (2007) Disclosure of genetics research results after the death of the patient participant: a qualitative study of the impact on relatives. J Genet Couns 16(4):527–538
Abriel H, Zaklyazminskaya EV (2013) Cardiac channelopathies: genetic and molecular mechanisms. Gene 517(1):1–11
Napolitano C, Bloise R, Monteforte N, Priori SG (2012) Sudden cardiac death and genetic ion channelopathies: long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation 125(16):2027–2034
Campuzano O, Beltran-Alvarez P, Iglesias A, Scornik F, Perez G, Brugada R (2010) Genetics and cardiac channelopathies. Genet Med 12(5):260–267
Brugada P, Brugada J (1992) Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 20(6):1391–1396
Berne P, Brugada J (2012) Brugada syndrome 2012. Circ J 76(7):1563–1571
Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, Nademanee K, Brugada R, Brugada J, Veerakul G et al (2002) Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet 11(3):337–345
Campuzano O, Allegue C, Brugada R: [Genetics of sudden unexplained death.]. Medicina clinica 2013
Riuro H, Beltran-Alvarez P, Tarradas A, Selga E, Campuzano O, Verges M, Pagans S, Iglesias A, Brugada J, Brugada P et al (2013) A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 34(7):961–966
Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C et al (2010) An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7(1):33–46
Roden DM (2008) Clinical practice. Long-QT syndrome. N Engl J Med 358(2):169–176
Schwartz PJ, Crotti L, Insolia R (2012) Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 5(4):868–877
Wilders R (2012) Cardiac ion channelopathies and the sudden infant death syndrome. ISRN Cardiol 2012:846171
Tester DJ, Benton AJ, Train L, Deal B, Baudhuin LM, Ackerman MJ (2010) Prevalence and spectrum of large deletions or duplications in the major long QT syndrome-susceptibility genes and implications for long QT syndrome genetic testing. Am J Cardiol 106(8):1124–1128
Winkel BG, Larsen MK, Berge KE, Leren TP, Nissen PH, Olesen MS, Hollegaard MV, Jespersen T, Yuan L, Nielsen N et al (2012) The prevalence of mutations in KCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases. J Cardiovasc Electrophysiol 23(10):1092–1098
Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P (2000) Idiopathic short QT interval: a new clinical syndrome? Cardiology 94(2):99–102
Patel U, Pavri BB (2009) Short QT syndrome: a review. Cardiol Rev 17(6):300–303
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B et al (2007) Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115(4):442–449
Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D et al (2011) Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J 32(9):1077–1088
Ylanen K, Poutanen T, Hiippala A, Swan H, Korppi M (2010) Catecholaminergic polymorphic ventricular tachycardia. Eur J Pediatr 169(5):535–542
Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E (2001) Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation 103(23):2822–2827
Barajas-Martinez H, Hu D, Ontiveros G, Caceres G, Desai M, Burashnikov E, Scaglione J, Antzelevitch C (2011) Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ Cardiovasc Genet 4(1):51–57
Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A et al: Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet 2012
Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G et al (2012) Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet 91(4):703–712
Raffan E, Semple RK (2011) Next generation sequencing—implications for clinical practice. Br Med Bull 99:53–71
Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ (2012) Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc Mayo Clin 87(6):524–539
Kauferstein S, Kiehne N, Jenewein T, Biel S, Kopp M, Konig R, Erkapic D, Rothschild M, Neumann T (2013) Genetic analysis of sudden unexplained death: a multidisciplinary approach. Forensic Sci Int 229(1–3):122–127
Acknowledgments
This work was supported by the “La Caixa” Foundation.
Conflict of interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Campuzano, O., Allegue, C., Partemi, S. et al. Negative autopsy and sudden cardiac death. Int J Legal Med 128, 599–606 (2014). https://doi.org/10.1007/s00414-014-0966-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-014-0966-4