
Abstract
Recently, several authors described the observation that RNA degradation does not correlate with the postmortem interval (PMI), but rather with other parameters like environmental impact and the circumstances of death. Therefore, the question arose if the analysis of gene expression could be a valuable tool in forensic genetics to contribute to the determination of the cause of death. In our study, six human tissues obtained from six individuals with PMI varying between 15 and 118 h were used for total RNA extraction. Quantification was performed using a GAPDH real-time assay, and the quality of mRNA was checked by amplification of different fragment lengths of the GAPDH transcript. In our set of samples, nearly all tissues in all PMI revealed satisfactory results, while skeletal muscle, followed by brain and heart, gave the best results. No correlation between PMI and RNA degradation could be detected, as very good results were observed for all tissues from the individual with the longest PMI. The highly promising results obtained in this study raise hopes that in the near future several fields of forensic investigation may profit from additional information about gene expression patterns and their correlation with pathological findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Because DNA is the carrier of the genetic information for future generations, its relative stability is essential to assure the transfer of information from cells to their daughter cells. In contrast, a short half-life period of RNA is indispensable for gene regulation [1]. Ribonucleases (RNases), which are omnipresent, were found to be responsible for the fast in vivo degradation of RNA [2]. However, chemical and thermal processes leading to degradation can also take place immediately after death. Thus, the level of RNA degradation in postmortem tissues is dependent on environmental impact and storage conditions of the body. However, a significant correlation between postmortem interval and mRNA degradation could not be found for brain and reproductive tissue [3, 4].
Recently, several authors described successful gene expression analysis from postmortem human brain tissue (e.g. [5–7]). Additionally, Bauer et al. [8–10] reported successful RNA analysis from body fluids in crime scene stains. New technologies now enable a highly sensitive detection of mRNA. For example, real-time quantitative PCR is the method of choice for the detection of small numbers of target copies (e.g., [11]).
Therefore, the question arose whether examination of mRNA can give additional information on the circumstances of death. The aim of this study was to investigate which tissues are appropriate for postmortem mRNA analysis and whether the postmortem interval is a relevant factor for degradation of mRNA in these tissues.
Materials and methods
Samples
Tissue samples of six individuals (A–F) with postmortem intervals (PMI) varying from 15 to 118 h (Table 1) were collected from routine autopsies performed in the Freiburg Institute of Legal Medicine. This work was approved by the local ethics committee (No. 272/05 of the ethics committee, University of Freiburg). Criterion for sampling was no macroscopically visible signs of putrefaction. Samples were either snap-frozen in liquid nitrogen (samples B–F) or transferred to RNAlater solution (Ambion, Austin, TX) before storage at −80°C until further use. RNAlater was only used when no liquid nitrogen was available (sampling at weekend, sample A).
RNA extraction and cDNA synthesis
RNA was extracted from 500 mg of tissue using the RNAgents total RNA isolation systems (Promega, Madison, WI) in an elution volume of 100 μl. The RNA extract was treated with Turbo DNA-free (Ambion) in the presence of recombinant RNasin ribonuclease inhibitor (Promega). Samples were purified with the cleanup protocol of the RNeasy Mini Kit (Qiagen, Hilden, Germany). The quality and integrity was checked on a 1.5% denaturing agarose gel with ethidium bromide staining (example given in Supplementary Fig. 1b). Reverse transcription of RNA resulting in complementary DNA (cDNA) was performed with a random hexamer priming protocol using the cDNA Archive kit (Applied Biosystems, Darmstadt, Germany). cDNA was stored at −20°C.
Validation experiments
In a series of validation experiments, it was checked whether a post-extraction treatment of the RNA samples was necessary or not. Therefore, a comparison was made between samples with and without DNase treatment and with and without additional cleanup. DNase treatment and cleanup was performed as described above.
Quantitative real-time PCR
Quantitative real-time PCR was performed using a commercially available gene expression TaqMan assay for the endogenous control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The assay was chosen to span the boundary between exons 2 and 3 (according to RefSeq NM002046.2, assay position: 104; assay ID: Hs00266705_g1, Applied Biosystems). The assay was performed following the manufacturer’s recommendations except that the reaction volume was reduced to 10 μl after validation. As standard, commercially available total RNA from human skeletal muscle (Stratagene, La Jolla, Canada) was used in threefold serial dilutions. Real-time PCR was conducted on a SDS 7500 instrument (Applied Biosystems). All RNA extracts were applied in dilutions of 1:5 to obtain results within the range of the standard.
Amplification of GAPDH fragments
To check the quality of the extracted RNA and whether the quality decreases with increasing postmortem interval, primers for GAPDH cDNA were designed that generate fragments between 114 and 866 bp with one universal reverse primer and five varying forward primers (GAPDH_F1–GAPDH_F5). Primer design was carried out using primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and the RefSeq NM002046 (primer sequences given in Supplementary Table 1). Primers were chosen to span at least one exon/exon boundary. PCR was performed using 1 μl of each cDNA with 10× PCR buffer containing 15 mM MgCl2 and AmpliTaq Gold (Applied Biosystems), 100 mM each dNTP and 2.5 μg bovine serum albumin in a total volume of 25 μl. Cycling conditions for PCR using forward primers GAPDH_F1–F3 were as follows: 94°C (11 min); 30 cycles of 95°C (1 min), 52°C (1 min), 72°C (1 min); and a final elongation of 72°C (30 min). For GAPDH_F4/F5 the cycling conditions were as follows: 95°C (11 min) followed by three cycles of 95°C (1 min), 52°C (1 min), 72°C (1 min); three cycles of 95°C (1 min), 50°C (1 min), 72°C (1 min); and 25 cycles of 95°C (1 min), 48°C(1 min), 72°C (1 min) followed by a final elongation of 72°C for 30 min.
PCR products were electrophoretically separated in a 2% agarose gel and visualized by ethidium bromide staining.
Results
Validation experiments
Several validation experiments were conducted to find a reliable method for extracting maximum yields of high-quality RNA. First, it was checked whether it is necessary to treat the extracts with DNase to remove contaminating genomic DNA (gDNA). Therefore, cDNA from both untreated and treated RNA was used as template for PCR GAPDH_F1 (Fig. 1). Amplification of cDNA generated from untreated RNA (I) led to double bands indicating the simultaneous amplification of cDNA and gDNA, while cDNA is represented by a 114-bp band and gDNA by a 218-bp band where the intron (104 bp) is still present. When using cDNA generated from DNase-treated RNA (II), the amplification led to the expected cDNA band of 114 bp only. It was necessary to add RNase inhibitor to the DNase reaction to avoid RNase damage, which might be due to RNase that is still present in the extracted RNA (data not shown).

Amplification of GAPDH_F1. When cDNA from untreated RNA (I) is used, a second band (218 bp) is visible representing gDNA contamination. When cDNA from DNase-treated RNA is used (II), only the 114-bp band is visible representing the reverse transcribed mRNA. (N No-template control, P positive control)
Second, it was tested whether an additional cleanup using the RNeasy Mini kit (Qiagen) step is recommendable to eliminate PCR inhibitors present in the extract. As shown in Fig. 2a, amplification of the relatively large fragments of GAPDH_F4 and GAPDH_F5 was more successful for the sample treated with the cleanup protocol. Results of the real-time PCR (Fig. 2b) showed an increase of the C t value of approximately one cycle after cleanup. Thus, the loss of sample during the cleanup protocol is nearly 50%.

a PCR GAPDH_F4–F5. Results of cDNA samples obtained from untreated RNA and RNA with an additional cleanup step. The product of GAPDH_F5 is visible in the latter but not in the untreated sample, indicating the successful removal of PCR inhibitors by an additional cleanup step. b Real-time GAPDH assay. Results of the untreated (U) and of the cleaned (C) sample. The cleaned sample has a C t value of one additional cycle, indicating a huge loss of sample during the cleanup step
RNA yields from various human tissues
RNA was extracted from six different tissue types of six individuals with varying postmortem intervals. The amount of total RNA was estimated by real-time PCR (Supplementary Fig. 1a and Table 2). Skeletal muscle (M. iliopsoas) resulted in the earliest C t values and showed the lowest interindividual variance. Brain and heart also gave relatively early C t values, while the liver, kidney, and spleen resulted in C t values well over 20. In all tissues except the skeletal muscle, individual C with a PMI of 26 h showed the lowest C t and, thus, the highest RNA yields. Individual A with the shortest PMI (15 h) gave relatively low RNA yields. In Fig. 3, the C t values are given in relation to the PMI. No hint for a correlation between PMI and RNA yield could be found.

Diagram showing the PMI (X axis) and the C t values found in each tissue (Y axis). No tissue showed a correlation between PMI and C t value. Muscle showed the earliest C t values, while the RNA yields obtained from spleen seem to be the lowest
By amplifying different fragments of the GAPDH transcript, information on the quality of the extracted RNA were obtained (Supplementary Table 1). An example is given in the Supplementary Fig. 2. Subsequent sequencing of the amplicons confirmed the specificity of the amplified cDNA fragments (data not shown). It was possible to amplify fragments up to 453 bp (GAPDH_F3) in all tissues except for the spleen. The amplification of a 619-bp (GAPDH_F4) fragment was successful from the muscle and brain tissue of all individuals. GAPDH_F4 amplification was also successful in the heart except for individual A. PCR GAPDH_F5 was successful from all tissues except the liver of the individual with the longest postmortem interval (individual F), but failed in several other individuals with shorter PMI. The extent of RNA degradation was not found to be correlated with the postmortem interval: individual E with a PMI of 56 h and individual A with the shortest PMI (15 h) gave very good results for the longer amplicons, while in all other individuals, amplification of the longer fragments failed in several tissues (see Table 3). Additionally, amplification of all GAPDH fragments was possible in all tissue except liver in individual F, who had the longest PMI.
Discussion
RNA was successfully extracted from all investigated tissues. It was found to be necessary to treat the RNA extracts with DNAse to remove contaminating gDNA. The term “contaminating” in this context refers to genomic DNA of the same tissue sample and individual rather than cross-contamination between different tissues or individuals. If gDNA is still present, co-amplification of gDNA and cDNA during PCR takes place, thereby reducing the PCR efficiency of the target sequence. Considering this is extremely important when working with quantitative real-time PCR because the quantification is reliable only if the efficiency of the PCR reaction is optimal [12]. If the contaminating gDNA is amplified simultaneously, the efficiency of the amplification of cDNA targets is reduced. Hence, the quantities obtained by such an assay would fall short, if the TaqMan probe matches to cDNA only. But it is also possible that quantities are overstated if the TaqMan probe binds to a target present in both cDNA and gDNA. Thus, in this study, all extracts were DNase-treated in the presence of an RNase inhibitor. The latter was found to be necessary because otherwise remaining RNases are able to destroy RNA during the incubation time needed for DNase treatment.
Adding a cleanup step before reverse transcription was also found to be useful. Even though there is a remarkable loss of sample (resulting in an increased C t of one cycle, Fig. 2b), it could be shown that this step successfully removes PCR inhibitors (Fig. 2a). For further experiments, an improved cleanup method should be used to reduce loss of sample. The real-time PCR experiment shown in Fig. 2b did not contain an internal control. Thus, the presence of inhibiting factors could not be shown. It is therefore possible that the loss of sample is even greater because the reaction using untreated (U) sample was inhibited. On the other hand, PCR reactions of shorter amplicons (F1–F3) did not show inhibition (data not shown). Therefore, an inhibition of the quite short (120 bp) GAPDH amplicon of the real-time PCR experiment is not very likely.
Quantification of RNA extracts via real-time PCR showed that the highest total RNA yields were obtained from the muscle followed by the brain and liver. The use of GAPDH as target has pros and cons: the gene GAPDH is one of the typically used endogenous controls for gene expression studies because it is ubiquitously expressed. On the other hand, recent studies using real-time PCR gave hints that the expression level of GAPDH is not as invariant as thought from earlier Northern blot experiments [13]. Thus, the quantities obtained in our study have to be regarded as approximate values only. Several authors described 18sRNA as the best endogenous control for comparative quantification of RNA levels [14, 15]. On the other hand, 18sRNA does not represent mRNA levels and was, therefore, not suitable for the present study.
Results of the quantitative real-time PCR assay gave no hints of a correlation between RNA yields and postmortem interval. Individual C with a PMI of 26 h gave the overall earliest C t values. Individual A was not found to give the best results even though this could have been expected due to the shortest PMI. Figure 3 shows that the muscle gave the earliest C t values in all individuals followed by the brain and heart. In principle, even though no correlation between C t values and PMI can be observed, tissues show partially high variability in different individuals (Table 2). Thus, the postmortem RNA stability seems to be affected by other parameters. For example, Preece and Cairns [3] found an influence of gender and pH on the RNA stability of postmortem human brain tissue.
The quality and degree of degradation was tested by amplifying different lengths of the GAPDH cDNA (details given in Supplementary Table 1, results given in Table 3). All tissues except for the spleen gave satisfactory results in GAPDH_F1–F3. GAPDH_F4 failed at times in all individuals and all tissues, indicating that the template sequence for this amplicon was amiss. Amplification success of GAPDH_F5 was found in individuals A, E, and F. Best results were obtained from the brain, muscle and heart tissue. Surprisingly, in individual F, nearly all tissues including the spleen gave good results. Only RNA obtained from the liver seemed to be degraded because the amplification of the fragments F2 to F5 failed.
Thus, as far as the obtained results from as few as six individuals allow, it can be presumed that the state of degradation does not correlate with the postmortem interval. This is concordant with the findings of, e.g., Preece and Cairns [3] who reported no significant correlation between mRNA degradation and PMI in human brain tissue, and Fitzpatrick et al. [4] who found that total RNA and GAPDH yields were not changed significantly for up to 96 h PMI. Additionally, Phang et al. described the successful extraction of RNA from human heart and kidney postmortem tissue [16].
The determination of the early postmortem interval and the age of bloodstains is still one of the major topics in forensic science [17–20]. The findings of the present study lead to the conclusion that the estimation of a postmortem interval by means of mRNA degradation is not promising for the tissues selected for this study. In contrast, Bauer et al. [8, 9] reported a correlation between mRNA degradation and the age of bloodstains.
In conclusion, it can be stated that at least some human tissues, like the muscle, brain, and heart tissue, are suitable for postmortem mRNA analysis. The tissues where amplification of larger GAPDH fragments failed can also be used for mRNA analysis via quantitative real-time PCR, as long as the TaqMan assays span no more than 150 bp. Thus, theoretically all tissues are appropriate if they give good results for GAPDH_F2 spanning 233 bp. Of course, it has to be kept in mind that there is the possibility of changes in the mRNA pool due to the degradation processes. Some classes of mRNAs are known to be more stable than others because they are protected from ribonucleases [21]. Therefore, successful analysis of postmortem mRNA levels does not only depend on the postmortem interval or the extraction method used but also on the type of mRNA investigated.
Nevertheless, the results presented here give reason to expect valuable information for the forensic field. Recently, Ikematsu et al. [22] reported significant changes in the expression of four different genes as a supravital reaction to pressure on mouse skin as occurring during strangulation. Additionally, several causes of trauma may leave traces on gene expression levels. Another field of investigation can be changes in gene expression patterns due to alcohol and drug abuse [23].
References
Wilusz CJ, Wormington M, Peltz SW (2001) The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol 2:237–246
Catts VS, Catts SV, Fernandez HR, Taylor JM, Coulson EJ, Lutze-Mann LH (2005) A microarray study of post-mortem mRNA degradation in mouse brain tissue. Brain Res Mol Brain Res 138:164–177
Preece P, Cairns NJ (2003) Quantifying mRNA in postmortem human brain: influence of gender, age at death, postmortem interval, brain pH, agonal state and inter-lobe mRNA variance. Brain Res Mol Brain Res 118:60–71
Fitzpatrick R, Casey OM, Morris D et al (2002) Postmortem stability of RNA isolated from bovine reproductive tissues. Biochim Biophys Acta 1574:10–14
Kobayashi H, Sakimura K, Kuwano R et al (1990) Stability of messenger RNA in postmortem human brains and construction of human brain cDNA libraries. J Mol Neurosci 2:29–34
Reddy PH, McWeeney S, Park BS et al (2004) Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: upregulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet 13:1225–1240
Preece P, Virley DJ, Costandi M et al (2003) An optimistic view for quantifying mRNA in post-mortem human brain. Brain Res Mol Brain Res 116:7–16
Bauer M, Polzin S, Patzelt D (2003) Quantification of RNA degradation by semi-quantitative duplex and competitive RT-PCR: a possible indicator of the age of bloodstains? Forensic Sci Int 138:94–103
Bauer M, Gramlich I, Polzin S, Patzelt D (2003) Quantification of mRNA degradation as possible indicator of postmortem interval—a pilot study. Leg Med (Tokyo) 5:220–227
Bauer M, Patzelt D (2003) A method for simultaneous RNA and DNA isolation from dried blood and semen stains. Forensic Sci Int 136:76–78
Loddenkötter B, Becker K, Hohoff C, Brinkmann B, Bajanowski T (2005) Real-time quantitative PCR assay for the detection of Helicobacter pylori: no association with sudden infant death syndrome. Int J Legal Med 119:202–206
Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23–39
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Goidin D, Mamessier A, Staquet MJ, Schmitt D, Berthier-Vergnes O (2001) Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal Biochem 295:17–21
Aerts JL, Gonzales MI, Topalian SL (2004) Selection of appropriate control genes to assess expression of tumor antigens using real-time RT-PCR. Biotechniques 36:84–86, 88, 90–91
Phang TW, Shi CY, Chia JN, Ong CN (1994) Amplification of cDNA via RT-PCR using RNA extracted from postmortem tissues. J Forensic Sci 39:1275–1279
Mall G, Eckl M, Sinicina I, Peschel O, Hubig M (2005) Temperature-based death time estimation with only partially known environmental conditions. Int J Legal Med 119:185–194
Henssge C, Madea B (2004) Estimation of the time since death in the early post-mortem period. Forensic Sci Int 144:167–175
Takamiya M, Saigusa K, Kumagai R, Nakayashiki N, Aoki Y (2005) Studies on mRNA expression of tissue-type plasminogen activator in bruises for wound age estimation. Int J Legal Med 119:16–21
Hayashi T, Ishida Y, Kimura A, Takayasu T, Eisenmenger W, Kondo T (2004) Forensic application of VEGF expression to skin wound age determination. Int J Legal Med 118:320–325
Suay L, Salvador ML, Abesha E, Klein U (2005) Specific roles of 5′ RNA secondary structures in stabilizing transcripts in chloroplasts. Nucleic Acids Res 33:4754–4761
Ikematsu K, Tsuda R, Nakasono I (2006) Gene response of mouse skin to pressure injury in the neck region. Leg Med (Tokyo) 8:128–131
Liu J, Lewohl JM, Dodd PR, Randall PK, Harris RA, Mayfield RD (2004) Gene expression profiling of individual cases reveals consistent transcriptional changes in alcoholic human brain. J Neurochem 90:1050–1058
Acknowledgement
The authors thank Benedikt Vennemann and Markus Große Perdekamp for help with the sample collection and Manuela Liebers for helpful discussion. This work is part of the bachelor thesis of KM.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Sequences of the forward primers (in 5′-3′ orientation) for the amplification of different lengths of the GAPDH transcript. For all amplicons, the same reverse primer (5′-tgccctgtagaaattcgttg-3′) was used. The different forward primers were chosen to span at least one exon/exon boundary. E/E Exon/Exon boundary according to GenBank NM002046 (mRNA) and J04038 (genomic sequence) (DOC 29 kb)
Supplementary Fig. 1
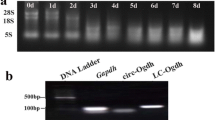
a Example of the real-time PCR results of the GAPDH assay using RNA extracted from muscle tissue of the individuals A–E (according to Table 2). b Example of a denaturing agarose gel electrophoresis using RNA extracted from skeletal muscle of individual A. The band representing 28s ribosomal RNA is stronger than the one representing 18s ribosomal RNA indicating a high quality of the extracted RNA (DOC 96 kb)
Supplementary Fig. 2
Example for the amplification of different lengths of the GAPDH transcripts for individual E. a PCR GAPDH_F1–F3. b PCR GAPDH_F4–F5 (DOC 305 kb)
Rights and permissions
About this article
Cite this article
Heinrich, M., Matt, K., Lutz-Bonengel, S. et al. Successful RNA extraction from various human postmortem tissues. Int J Legal Med 121, 136–142 (2007). https://doi.org/10.1007/s00414-006-0131-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-006-0131-9