
Abstract
Eukaryotic nuclear DNA is packaged into nucleosomes. During the past decade, genome-wide nucleosome mapping across species revealed the high degree of order in nucleosome positioning. There is a conserved stereotypical nucleosome organization around transcription start sites (TSSs) with a nucleosome-depleted region (NDR) upstream of the TSS and a TSS-aligned regular array of evenly spaced nucleosomes downstream over the gene body. As nucleosomes largely impede access to DNA and thereby provide an important level of genome regulation, it is of general interest to understand the mechanisms generating nucleosome positioning and especially the stereotypical NDR-array pattern. We focus here on the most advanced models, unicellular yeasts, and review the progress in mapping nucleosomes and which nucleosome positioning mechanisms are discussed. There are four mechanistic aspects: How are NDRs generated? How are individual nucleosomes positioned, especially those flanking the NDRs? How are nucleosomes evenly spaced leading to regular arrays? How are regular arrays aligned at TSSs? The main candidates for nucleosome positioning determinants are intrinsic DNA binding preferences of the histone octamer, specific DNA binding factors, nucleosome remodeling enzymes, transcription, and statistical positioning. We summarize the state of the art in an integrative model where nucleosomes are positioned by a combination of all these candidate determinants. We highlight the predominance of active mechanisms involving nucleosome remodeling enzymes which may be recruited by DNA binding factors and the transcription machinery. While this mechanistic framework emerged clearly during recent years, the involved factors and their mechanisms are still poorly understood and require future efforts combining in vivo and in vitro approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA packaging in the nucleus with histone and nonhistone proteins as well as RNAs is a hallmark of eukaryotic genome organization. The resulting structures are collectively called “chromatin” and differentially affect DNA accessibility. As all DNA-guided processes rely on factors binding DNA, chromatin modulation is an important level of genome regulation (Bell et al. 2011). Higher order chromatin structures are still ill defined (Maeshima et al. 2014), but the most basic level, the nucleosome core particle, is known at crystallographic resolution: ~147 base pairs (bp) of DNA are wrapped in ~1.65 left-handed turns around octamers containing two copies each of four histone types, H2A, H2B, H3, and H4 (Luger et al. 1997; Richmond and Davey 2003). This nucleosome core structure is well conserved and impedes DNA access for most factors. Core particles are connected by more accessible linker DNA with average linker length varying between species, or between different cell types within species (van Holde 1989). Core particle plus linker DNA are called “nucleosome,” but often this term is also used for the core particle. In addition to nucleosomes, there are regions of more extended accessibility (typically ~150–200 bp, but also much longer), originally called nuclease hypersensitive sites (Wu 1980), but now usually called nucleosome-free or nucleosome-depleted regions (NFRs or NDRs, respectively; Jiang and Pugh 2009b; Struhl and Segal 2013). Such NDRs are often linked to functional elements like promoters, enhancers, and replication origins (Almer and Hörz 1986; Bell et al. 2011; Berbenetz et al. 2010; Eaton et al. 2010; Elgin 1981). Since 2005 (Yuan et al. 2005), the distribution of nucleosomes and NDRs can be mapped genome-wide and shows a striking degree of well-ordered nucleosome positioning along eukaryotic genomes. This kicked off studies on the mechanisms responsible for such organization. As most regulation through chromatin homes in on the question if a particular stretch of DNA is accessible or occluded by nucleosomes, the question of nucleosome positioning mechanisms is fundamental for eukaryotic DNA biology. Here, we summarize the currently used methods, the observed patterns, and the discussed mechanisms as studied in yeasts, especially Saccharomyces cerevisiae. We also refer to other reviews (Cole et al. 2012b; Hughes and Rando 2014; Iyer 2012; Jansen and Verstrepen 2011; Jiang and Pugh 2009a, 2009b; Radman-Livaja and Rando 2010; Segal and Widom 2009b; Struhl and Segal 2013).
Definitions
“Nucleosome positioning” as used here and most commonly understood refers to the “translational position” of a nucleosome core particle along a DNA sequence, i.e., which bp is at the central dyad position of the core particle. If this is more or less the same with respect to the same genomic DNA sequence in a population of templates, for example in different cells, the nucleosome is considered more or less well positioned, or, respectively, less or more “fuzzy” (Fig. 1a).

Nucleosome positioning concepts. a Illustration of the terms defined in the section “Definitions.” b Different genes with the same nucleosome organization relative to a common reference point, e.g., the TSS. The stereotypical organization consists of an NDR just upstream of the TSS flanked by well-positioned nucleosomes (−1 and +1) and an array of evenly spaced nucleosomes in the direction of transcription (+2, +3, …). If the majority of genes show this stereotypical organization, the sum of the nucleosome positioning signals (dyad positions or occupancy) for each position along the genes and aligned at the TSSs gives a composite plot as in Fig. 3. c Variation of the organization shown in b such that NDR and flanking −1/+1 nucleosomes are unchanged and nucleosomes in dark blue still form regular arrays, but the arrays are not aligned at the TSS with the same register at different genes. Such organizations probably underlie the mutant composite patterns shown in Fig. 4b, c
“Rotational positioning” describes the orientation of a given base pair relative to the histone octamer, i.e., if it faces toward or away from the histone octamer. Given the ~10.5-bp periodicity of the DNA helix, which is a bit different within the nucleosome core (Prunell 1998), several translational positions of octamers that differ in frame shifts of ~10 bp multiples (10 × n) will show the same inside-out orientation of a given base pair. So nucleosomes may share the same rotational positioning (also called “rotational phasing”) despite quite different translational positions (Fig. 1a).
If consecutive nucleosomes on the same template have the same linker length, they are “regularly spaced” and one can derive the “nucleosomal repeat length” (NRL) or “spacing” (= dyad-to-dyad distance). In vivo spacing is hardly ever an exact property of individual nucleosomes but an aggregate feature of an overlay of many nucleosomal arrays. Regularly spaced nucleosomes can have a high degree of translational positioning (also called “translational phasing”; Fig. 1a, orange nucleosomes) but may also be offset between different templates (Fig. 1a, yellow nucleosomes). This offset may or may not lead to the same rotational positioning of overlapping nucleosomes on different templates.
“Nucleosome occupancy” describes the probability for a given base pair to be part of any nucleosome core and amounts to a 147-bp sliding window of translational positioning. For a given position (e.g., peak position), it corresponds to the percentage of a template/cell population with a nucleosome here (partially represented by peak height, see “Comparison of different methods and the problem of nucleosome occupancy”). For more detailed definitions, see Kaplan et al. (2010).
Note that nucleosome maps usually may have two different formats (Zhang and Pugh 2011): either “nucleosome occupancy,” i.e., the signal at each bp is contributed by any nucleosome covering this bp, or “nucleosome dyad occupancy/density,” i.e., only base pairs at nucleosome dyads contribute. Consequently, the former plots show less distinct peaks than the latter (see also Fig. 2 and Table 1).

Single locus comparisons of different nucleosome mapping methods. a, b Browser shots of nucleosome mapping data at the S. cerevisiae loci indicated at the bottom of the panels. References and method comparison as in Table 1. Boxed region in a shows an example for stereotypical NDR-array pattern and in b for a nonstereotypical promoter nucleosome organization. The asterisk marks “hypersensitive site 2” in the PHO5 promoter (Almer et al. 1986). This PHO5 promoter NDR is much further upstream than the NDRs of the stereotypical organization
Methods: genome-wide nucleosome mapping
Nucleosome mapping monitors the biologically relevant chromatin feature of differential DNA accessibility by using either enzymes, like nucleases or methylases, or chemicals, like hydroxyl radicals, to distinguish nucleosomal from nonnucleosomal DNA. Most common is mapping by micrococcal nuclease (MNase), which preferentially cuts in linkers and NDRs (Axel 1975). MNase digestion can be carefully titrated to yield mostly mononucleosomal DNA fragments. The continuous protection of ~140–150 bp from MNase is unique to nucleosomes and has been used as an operational definition for a canonical nucleosome since the discovery of the nucleosome (Kornberg and Lorch 1999). So mononucleosome-sized DNA length after MNase digestion is usually a sufficient criterion for the identification of DNA that was nucleosomal, i.e., the histone octamer DNA footprint. Such mononucleosomal DNA is now readily analyzed on the genome level (Table 2).
MNase-chip (-array)
The first genome-scale nucleosome mapping was based on high-resolution tiling arrays (Yuan et al. 2005). Mononucleosomal DNA after MNase digestion of whole genome chromatin is gel extracted, labeled, and hybridized to microarrays with a resolution of <20 bp. Continuous hybridization across probes spanning a mononucleosome-sized region corresponds to a well-positioned nucleosome. Hybridization signals are usually plotted along the genome giving nucleosome occupancy rather than translational positions (Zhang and Pugh 2011). An advantage of this approach is that PCR is not necessary, thereby avoiding amplification biases. Further, custom arrays allow the analysis of just a selection of a genome, which reduces costs particularly for larger, e.g., human, genomes. Nonetheless, arrays become more and more expensive relative to deep sequencing and are less flexible as they detect only what they are designed for. A special caveat for Affymetrix tiling arrays with 25 bp probes is the requirement for additional fragmentation of mononucleosomal DNA prior to hybridization. Hybridization of full-length mononucleosomal DNA leads to artifactually shifted nucleosome occupancy (see Pointner et al. 2012 and Fig. S18a in Whitehouse et al. 2007).
MNase-seq
The very same mononucleosomal DNA as for MNase-chip can be analyzed by high-throughput sequencing. Depending on the sequencing platform, the entire mononucleosomal fragment is sequenced, or only its ends, either both ends together (paired end), or just one end (single end). The former two modes readily allow the determination of nucleosome dyads, while the latter requires either bona fide read start extension by 73 bp, assuming uniform fragment lengths of 147 bp, or computational analysis of average fragment length by cross correlation of forward and reverse reads. In any case, MNase-seq provides nucleosome dyad positions more readily and at higher resolution than MNase-chip. For example, nucleosomal arrays upstream of the transcription start site (TSS) in Schizosaccharomyces pombe are hardly detected by MNase-chip but apparent by MNase-seq (compare S. pombe wild-type pattern in Fig. 4a vs. c; Mojardin et al. 2013). Sequencing library preparation as well as sequencing itself involves PCR. This allows analyzing small quantities of starting material but may also introduce biases.
MNase-ChIP-seq
The Pugh lab refined MNase-seq by a chromatin immunoprecipitation (ChIP) step after MNase digestion to mononucleosomes, e.g., using an anti-H3-C-terminus antibody (Albert et al. 2007; Mavrich et al. 2008a). This provides a second selection criterion, in addition to MNase protection, to ensure monitoring of nucleosomes. ChIP is done with cross-linking, usually by formaldehyde prior to cell harvesting, and the washing steps are so stringent that all noncross-linked material is lost. Such MNase-ChIP-seq is probably closest to a snapshot of the true in vivo situation. Of note, cross-linking is hardly ever complete and both cross-linked and noncross-linked nucleosomes yield the characteristic mononucleosomal DNA fragments upon MNase digestion. Therefore, cross-linking without immunoprecipitation does not guard against mapping artifactual noncross-linked nucleosomes that become repositioned or even assembled during chromatin preparation. Nonetheless, for wt S. cerevisiae, a comparison between MNase-seq without cross-linking and MNase-ChIP-seq showed not much difference (Kaplan et al. 2009; Zhang et al. 2011b) making cross-linking not urgently necessary if working with yeasts. Besides these biological issues, immunoprecipitation allows easier downstream sample handling as all steps are done on beads sparing repeated DNA collection. Finally, the combination of nucleosome selection by MNase with immunoprecipitation using virtually any antibody allows monitoring factor binding to nucleosomes (Koerber et al. 2009; Yen et al. 2012).
Hydroxyl-radical-seq
MNase-based nucleosome mapping was criticized because of sequence biases of MNase digestion (Chung et al. 2010; Locke et al. 2010; McGhee and Felsenfeld 1983). Indeed, MNase cuts at dA:dT base pairs with higher probability than at dG:dC base pairs (Cockell et al. 1983; Dingwall et al. 1981; Hörz and Altenburger 1981) such that higher MNase activities may even cleave canonical nucleosomes at intranucleosomal dA:dT-rich regions (Caserta et al. 2009; Cockell et al. 1983). Further, MNase-digested free DNA may produce similar patterns as chromatin (Chung et al. 2010; Locke et al. 2010). Since long, it is known that MNase indirect end labeling requires MNase-digested free DNA as control in order to distinguish if banding patterns are caused by chromatin or by DNA sequence bias (Svaren et al. 1995). Nonetheless, the MNase sequence bias at the very low MNase activities used for free DNA digestion is mitigated relative to the nucleosome-imposed bias at the much higher activities used for chromatin digestion. A free DNA control is therefore not necessarily informative, and pattern similarities between free DNA and chromatin rather reflect the sequence bias of nucleosome positioning than that of MNase digestion. Recently, chromatin digestions with MNase versus a caspase-activated DNase, which has different sequence bias and is even less prone to intranucleosomal cuts, produced quite similar cleavage patterns arguing for a negligible MNase bias (Allan et al. 2012).
The long-standing controversy about MNase bias in nucleosome mapping was recently resolved by taking an early MNase-independent high-resolution method (Flaus et al. 1996) to the genome level: hydroxyl-radical-seq (Brogaard et al. 2012a). Here, histone H4 is mutated to contain a unique cysteine residue (H4S47C) close to the DNA backbone at the nucleosome dyad. After covalent coupling of copper-chelating phenanthroline to this cysteine, addition of copper ions generates short-lived and therefore locally well-defined hydroxyl radicals that cleave the DNA backbone at defined distances relative to the dyad. Sequencing of the resulting DNA fragments directly maps dyad positions with single-base pair resolution. Importantly, the resulting nucleosome positions correlate very well with those obtained by MNase-based methods thereby validating the MNase approach (Brogaard et al. 2012a; Moyle-Heyrman et al. 2013). Nonetheless, the resolution of this chemical mapping is considerably higher and enables to delineate more precisely and more accurately DNA sequence effects. For example, this technique uncovered the high propensity of dA:dT base pairs at nucleosome borders in earlier maps as an MNase artifact (Brogaard et al. 2012a).
MNase-exoIII-seq
The need for the H4S47C mutation somewhat limits the applicability of hydroxyl-radical-seq. Another way to reduce the MNase bias at nucleosome borders is combining MNase digestion with trimming by exonuclease III (exo III; Nikitina et al. 2013). This technique was applied on the genome level (Rodriguez et al. 2014; Whitehouse et al. 2007), but the extent to which it increases accuracy, e.g., in comparison with MNase-seq and hydroxyl-radical-seq, was not analyzed so far.
FAIRE
An also MNase-independent but reverse approach is formaldehyde-assisted isolation of regulatory elements (FAIRE) (Nagy et al. 2003) as it does not monitor nucleosomes but NDRs, especially to screen for and map regulatory elements. FAIRE is based on the differential distribution of protein-bound and free DNA, particularly nucleosomal versus nucleosome-free DNA, during phenol-chloroform extraction after cross-linking and sonication. Cross-linking is adjusted such that mainly histones are coupled to DNA. The fragments recovered from the aqueous phase are therefore mainly NDRs and analyzed by microarrays or deep sequencing. The resolution is limited by the resolution of sonication, but there is the clear advantage of directly showing the DNA regions that are lost in the nucleosome mapping approaches.
Particle spectrum analysis
Even though continuous protection of ~140–150 bp from MNase digestion is typical only for nucleosomes, other DNA binding factors/complexes also protect against MNase digestion. To obtain those MNase footprints, not only the mononucleosomal DNA but also shorter and longer fragments are prepared and mapped by deep sequencing (Henikoff et al. 2011; Kent et al. 2011). Fragments of ~25–50 bp are primarily located in NDRs and probably correspond to transcription factors (TFs), whereas somewhat larger subnucleosomal fragments may correspond to partially unwrapped nucleosomes and may be bound to remodelers like the “remodels the structure of chromatin” (RSC) complex (Floer et al. 2010; Henikoff et al. 2011). However, only nucleosomes may be confidently identified by the length of MNase-protected DNA, whereas for the identification of other factors, additional immunoprecipitation or mutant analysis is necessary. Such particle spectrum analyses are likely to provide an ever richer picture of the chromatin landscape.
Comparison of different methods and the problem of nucleosome occupancy
S. cerevisiae genome nucleosome maps obtained by different techniques correlate rather well (Fig. 2). Inconsistencies at particular loci may be resolved by classical single locus techniques, like DNaseI/MNase indirect end labeling (Svaren et al. 1995).
Recent systematic analyses (Ozonov and van Nimwegen 2013; Quintales et al. 2014; Rizzo et al. 2011) as well as Fig. 2 show that nucleosome occupancy (peak height/trough depth) varies considerably between data sets. This is due to different MNase digestion degrees and hybridization or deep sequencing biases (Stein et al. 2010; Zhang et al. 2009). Especially digestion degrees affect occupancy levels, e.g., for promoter NDRs and +1 nucleosomes (DeGennaro et al. 2013; Flores et al. 2014; Givens et al. 2011; Rizzo et al. 2012; Weiner et al. 2010). All MNase-based mapping operates in a limited rather than exhaustive digestion regime as exhaustive digestion would digest even nucleosomal DNA. So the probability for MNase cuts scales both with the accessibility as imposed by chromatin structure and with the chosen MNase activity. Occupancy directly reflects this composite probability. As their name implies, hypersensitive sites, i.e., long linkers or NDRs, are cut with the highest probability due to their open chromatin structure, but the extent of cutting, i.e., the resulting trough depth in nucleosome occupancy plots, will also depend on the digestion degree. Further, nucleosome mapping depends on MNase introducing two double strand breaks, i.e., at both sides of the nucleosome, in order to generate a mononucleosomal fragment for further analysis. This coupled probability for a double cut is higher if a nucleosome is flanked by a long linker or hypersensitive site, in contrast to nucleosomes in closely packed arrays flanked by short linkers only. So especially the +1 nucleosome occupancy will be high already at low MNase digestion degree, while the occupancy levels of array nucleosomes come up mainly at higher digestion (Weiner et al. 2010). In addition, nucleosomes with internal dA/dT-rich sequences may be cleaved more often intranucleosomally by increasing MNase activity than others leading to their preferentially decreased occupancy (Caserta et al. 2009; Cockell et al. 1983).
In order to account for different MNase digestion degrees, these may be matched by analyzing fragment distributions in gels (Rizzo et al. 2012) or can be assessed in paired end or 454-type sequencing data via the distribution of fragment lengths (Cole et al. 2011a). Using the exact same digestion degree for different samples is challenging. The alternative, nucleosome occupancy normalization for digestion degrees, is not published so far and may not even work sufficiently. MNase digestibility bias varies for each individual nucleosome according to flanking linker lengths, i.e., the probability of MNase cutting at each side, and the sequence-dependent bias for intranucleosomal cleavage. So individual nucleosome occupancy levels are unlikely to scale uniformly with digestion degree. Collectively, we caution that MNase-based nucleosome maps are sufficiently accurate in terms of nucleosome positions (peak/trough positions), but not very reliable with regard to occupancy levels. Global occupancy differences, e.g., in aged versus young yeast cells, are only detected using spike-in controls (Hu et al. 2014). Nonetheless, for comparison of MNase accessibility at different genomic regions within one sample, a normalization for nucleosome occupancy as determined by MNase-independent anti-histone ChIP was proposed (NCAM method; Rodriguez et al. 2014).
Finally, the bioinformatics analysis of the nucleosome mapping data is not standardized yet. There is a sizeable collection of different algorithms for peak calling, smoothing, etc. that may affect the conclusions and have to be tried for each application in question (nonexhaustive reference list: Becker et al. 2013; Chen et al. 2013; Flores and Orozco 2011; Kuan et al. 2009; Lee et al. 2007; Nellore et al. 2012; Quintales et al. 2014; Shivaswamy et al. 2008; Tirosh 2012; Weiner et al. 2010; Yuan et al. 2005).
Maps: genome-wide nucleosome organizations
There are many genome-wide nucleosome maps for S. cerevisiae available including many mutants and different biological conditions as well as maps for at least 19 other yeasts (Table 3).
Stereotypical nucleosome organization around TSSs
Wild-type log phase cells show a stereotypical nucleosome organization around TSSs for most genes: an “NDR-array” pattern (Figs. 1b and 3). An NDR is located just upstream of the TSS placing the TSS on average 12 bp within the +1 nucleosome 5′ borderFootnote 1 (Lee et al. 2007; Mavrich et al. 2008a). Well-positioned +1/−1 nucleosomes flank the TSS and arrays of regularly spaced nucleosomes start here, most prominently downstream into the gene. The degree of translational positioning is most pronounced for the +1/+2 nucleosomes and gradually decreases along the array. Even well-positioned nucleosomes rarely occupy a unique position, but rather show Gaussian distributions (“position clusters”; Cole et al. 2012b) around average midpoints with alternative positions offset by multiples of ~10 bp, i.e., rotationally phased (Albert et al. 2007; Cole et al. 2011a; Jiang and Pugh 2009a). Only the single nucleosomes that contain the histone H3 variant Cse4 and occupy the point centromeres of S. cerevisiae are uniquely positioned (Cole et al. 2011b). The upstream arrays starting from the −1 nucleosomes are usually much less pronounced than the genic arrays, unless bidirectional promoters share a common NDR such that both the upstream and downstream arrays will be genic. In S. pombe, upstream arrays are even less prominent (Fig. 4a; Givens et al. 2012; Lantermann et al. 2010). The NDR of the stereotypical NDR-array organization in S. cerevisiae has an average size of 150 bp (Jiang and Pugh 2009a); is enriched for TF binding sites (Lee et al. 2007; Ozonov and van Nimwegen 2013), TATA boxes, or TATA-like elements (Basehoar et al. 2004; Rhee and Pugh 2012); and provides an “open door policy” for assembly of the pre-initiation complex (PIC) (Morse 2007). Both the +1 and −1 nucleosomes in S. cerevisiae (Albert et al. 2007) but only the +1 nucleosomes in S. pombe (Buchanan et al. 2009) are enriched for the histone variant H2A.Z and show the highest histone turnover rates (Dion et al. 2007; Jamai et al. 2007; Rufiange et al. 2007). The average NRL in arrays varies among yeast species from 5 to 7 bp in S. pombe and Aspergillus nidulans (Givens et al. 2012; Lantermann et al. 2010; Moyle-Heyrman et al. 2013; Nishida et al. 2013) to 18 bp in S. cerevisiae (Jiang and Pugh 2009a; Lee et al. 2007; Mavrich et al. 2008a; Thomas and Furber 1976) and up to 30 bp in Kluyveromyces lactis (Tsankov et al. 2010).


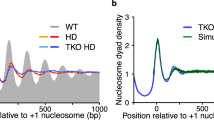
S. pombe shows tighter nucleosome spacing in genic arrays than S. cerevisiae and lack of ISWI- and/or CHD1-type remodeling ATPases impairs array alignment in both yeasts. Comparison of patterns as in Fig. 3 for a S. cerevisiae versus S. pombe (Tsankov et al. 2010, 2011), b S. cerevisiae wild type versus Δisw1 Δisw2 Δchd1 mutant lacking all ISWI- and CHD1-type remodeler ATPase subunits (Gkikopoulos et al. 2011a), and c S. pombe wild type versus Δhrp1 Δhrp3 mutant lacking two of three CHD1-type remodeler ATPases (Pointner et al. 2012)
Nonstereotypical promoter nucleosome organizations
TSS-aligned composite plots of nucleosome occupancy or dyad position (Fig. 3) combine two features that are not necessarily linked. A narrow, rather than a wide, peak for a +1 nucleosome signifies two things. First, such a nucleosome occupies the same position at the same gene compared among different cells (well positioned vs. fuzzy, Fig. 1a). Second, it has the same position at different genes relative to their respective TSS (same translational phasing register relative to a common alignment point; Fig. 1b). Conversely, broader peaks in composite plots may correspond to fuzzy nucleosomes, or to nucleosomes that do not share a common register relative to the alignment point, or both. Genic nucleosomes in S. cerevisiae indeed become fuzzier further downstream (Zhang et al. 2011b). On the other hand, there are also genes with particularly well-positioned nucleosomes around TSSs, e.g., at the PHO5 promoter (Fig. 2b; Almer et al. 1986), but their TSS-aligned nucleosome occupancy plot does not yield a regular pattern together with other genes as their positions lack a common register relative to their TSSs (Lee et al. 2007; Zhang et al. 2011b). These genes have nonstereotypical and very diverse nucleosome patterns. For example, the NDR may be further upstream or not present at all. Each such gene is essentially a case of its own.
Relationship between promoter nucleosome organization and transcription
The two broad types of stereotypical versus nonstereotypical promoter nucleosome organizationFootnote 2 correlate with transcriptional features (Cairns 2009; Hughes and Rando 2014; Tirosh and Barkai 2008). Promoters with stereotypical NDR-array pattern tend to drive constitutive expression (“growth” or housekeeping genes), lack classical TATA boxes but show TATA-like elements (Rhee and Pugh 2012), depend on TFIID (Basehoar et al. 2004), have TF binding sites mostly in or at the NDR (Lee et al. 2007), and show rather steady transcription rates (Raser and O’Shea 2005) and low turnover rates for histones (Dion et al. 2007) and the TATA box binding protein (van Werven et al. 2009). In contrast, promoters with nonstereotypical nucleosome organizations often belong to inducible genes with a wide dynamic range that respond to environmental cues (“stress” genes), e.g., the PHO promoters that are regulated by phosphate levels (Almer et al. 1986; Lam et al. 2008). Their expression is less steady but “bursty” (Brown et al. 2013; Raser and O’Shea 2004). They often contain TATA boxes and depend on chromatin cofactors, like the SWI/SNF complex, to overcome their repressive promoter nucleosome organization where, for example, TF binding sites (UAS elements) are covered by nucleosomes in the noninduced state (Elfving et al. 2014; Lam et al. 2008; Venter et al. 1994). Therefore, nucleosome positioning relative to UAS elements can determine the threshold of promoter induction, i.e., if high or low affinity UAS are intra- or internucleosomal. Further, the extent of nucleosome remodeling can regulate induction strength, i.e., to what extent initially intranucleosomal UAS become accessible (Lam et al. 2008). Even though such chromatin-regulated promoters are the minority in yeast, they are excellent models to study gene regulation through tuning chromatin states. The stereotypical S. cerevisiae NDR-array pattern as well as the dichotomy between “growth” and “stress” genes was also observed in 12 other yeasts (Tsankov et al. 2010; Tsui et al. 2011), but with some differences in absolute terms. For example, the average 5′NDR width varies between 109 and 155 bp, and the average linker length between 7 and 30 bp (Lantermann et al. 2010; Tsankov et al. 2011; Tsankov et al. 2010). Even in larger eukaryotes, the basic distinction between stereotypical NDR-array and nonstereotypical promoter chromatin architectures still holds. Human cells show the NDR-array pattern predominantly at active genes and genes with paused RNA polymerase but not at inactive genes (Schones et al. 2008; Valouev et al. 2011).
Single locus studies, for example with inducible genes like PHO5 (Almer et al. 1986; Korber and Barbaric 2014), heat shock genes (Zhao et al. 2005), or CUP1 (Shen et al. 2001), shaped the view that nucleosome positioning changes dramatically upon gene activation or repression such that nucleosome occupancy and/or positioning is diminished at promoters and over genes in the activated state. However, this tendency is not as apparent on the genome level as one might have expected. Global expression changes of many genes, for example by shift to different carbon source (Kaplan et al. 2009; Zawadzki et al. 2009), drug treatment (Cole et al. 2011a), meiosis (Soriano et al. 2013; Zhang et al. 2011a), and oxidative (Huebert et al. 2012) or heat shock stress (Shivaswamy et al. 2008), left the overall nucleosome patterns in most cases surprisingly unchanged. Genes with changed expression levels display all kinds of changes in nucleosome positioning. For example, upregulation could correspond to unchanged, increased, or decreased promoter nucleosome occupancy.Footnote 3 Especially the “open door policy” of the stereotypical NDR-array pattern seems to provide a rather stable chromatin organization that is poised for transcription, but the actual transcription rate is regulated by the recruitment/activity of transactivators (Zawadzki et al. 2009) or antisense transcription (Zhang et al. 2011a). Conversely, genes with changed nucleosome positioning often do not show much expression changes (at least regarding sense transcripts), particularly in chromatin cofactor mutants (Gkikopoulos et al. 2011a; Tolkunov et al. 2011). Also evolutionary comparisons between yeasts show that expression levels of orthologous genes are more conserved than their respective nucleosome organizations (Nishida 2012; Tsui et al. 2011). Collectively, the predictive power of nucleosome organization for expression level, and vice versa, is weak. This does not discount a role for nucleosome organization in gene regulation as there are many examples for such a role (this review and Korber and Barbaric 2014), and there still is a correlation of lower promoter nucleosome occupancy, wider NDRs, and disrupted intragenic arrays with elevated expression (Cui et al. 2012; Kristell et al. 2010; Zawadzki et al. 2009), most prominently for the inducible promoters with nonstereotypical nucleosome patterns. Nonetheless, there is no straightforward relationship or “code” between nucleosome pattern and transcriptional output.
Mechanisms: what determines nucleosome positions in vivo?
We focus on the mechanism for generating the stereotypical NDR-array pattern as this will explain the vast majority of positioned nucleosomes in yeasts. This mechanism can be subdivided into four mechanisms generating the individual features of the NDR-array pattern, in some temporal order or in parallel:
-
1.
NDR formation
-
2.
Positioning of the +1 nucleosome
-
3.
Generation of the nucleosomal arrays (= equalizing linker length)
-
4.
Array alignment to the NDR/TSS/PIC/+1 nucleosome
As the relative distances between NDR, TSS, and +1 nucleosome are rather uniform within the NDR-array organizations, each can be used for descriptive purposes and TSS or +1 nucleosome alignments are most common for bioinformatical representations. However, it matters mechanistically what the molecular correlate of the actual alignment point is. These four mechanisms in the end amount to the observed translational positions relative to the DNA sequence. So there should be something that “reads” the sequence and translates sequence information into nucleosome positions. The following factors are discussed to contribute to nucleosome positioning and may read the DNA sequence:
-
(a)
Histone-DNA interactions
-
(b)
Nonhistone DNA binding factors, e.g., general regulatory factors (GRFs) or TFs
-
(c)
Nucleosome remodeling enzymes (“remodelers”)
-
(d)
DNA-templated processes, e.g., transcription
Remodelers are ATPases that assemble, slide, and disassemble nucleosomes and change their composition, e.g., by exchanging canonical for variant histones (Becker and Workman 2013; Clapier and Cairns 2009). In a nucleosome-centric view, DNA and histones are cis- and everything else transfactors for nucleosome positioning (Radman-Livaja and Rando 2010). Regarding the extent to which DNA sequence is involved, there is a continuum between the two extremes that either each individual nucleosome position is determined by reading the DNA sequence in or immediately around the nucleosome, or that only few positions are sequence-determined while the majority of nucleosomes are positioned relative to such “barriers” (Kornberg and Stryer 1988) by some sequence-independent propagation mechanism. As an example of the first alternative, sequence-driven positioning of many (>50 %) individual nucleosomes was prominently suggested as a “genomic code for nucleosome positioning” (Kaplan et al. 2009, 2010; Segal et al. 2006), where DNA sequence preferences of histone octamer binding define nucleosome positions. The second alternative, a combination of barriers with sequence-independent positioning, was considered early on as the “statistical positioning” mechanism (Kornberg and Stryer 1988; Mobius and Gerland 2010). Here, nucleosomes are modeled as hard noninteracting spheres that move freely along the DNA until they meet one of few barrier elements, which are determined by some DNA sequence feature, e.g., a factor binding site. Solely due to statistical distribution, nucleosomes align at the barriers in regularly spaced arrays, similar to those observed in vivo, without contribution of the underlying DNA sequence. In this model, only very few nucleosome/barrier positions are directly sequence determined.
We first review these two alternatives and why neither of them suffices to explain in vivo nucleosome positioning. Second, we discuss that both still contribute importantly to a mainly trans-factor, especially remodeler-driven mechanism.
Sequence preferences of the histone octamer (“intrinsic” nucleosome positioning)
The histone octamer binds 147 bp of DNA mainly through many ionic and hydrogen bonds between histones and DNA (Davey and Richmond 2002; Luger et al. 1997) and can be viewed as a DNA binding factor with a particularly long footprint (Struhl and Segal 2013). This cumulative binding energy compensates the energetically unfavorable tight bending of DNA around the octamer such that all DNA sequences may be incorporated into a nucleosome. Nonetheless, some DNA sequences are more resistant to this bending than others so that there are sequence preferences for the histone octamer. A particularly well-known example are poly(dA:dT) stretches that are less prone to become reconstituted into nucleosomes by salt gradient dialysis (SGD) in vitro (Kaplan et al. 2009; Zhang et al. 2009, 2011b). The biophysical basis of this is not really understood (Segal and Widom 2009a). Such poly(dA:dT) elements are strongly enriched in S. cerevisiae promoter NDRs (Lee et al. 2007; Yuan et al. 2005), and the introduction of such elements can generate NDRs and influence expression levels in vivo (Raveh-Sadka et al. 2012; Small et al. 2014; Zhang and Reese 2007). This is a particularly fine example for transcription regulation through nucleosome organization. Also poly(dG:dC) has similar properties (Sekinger et al. 2005; Tsankov et al. 2011) such that poly(dA:dT) and poly(dG:dC) tend to be nucleosome free in several yeast species (Tsankov et al. 2011). This seems to be an intrinsic or cis mechanism to generate NDRs in vivo.
However, the intrinsic histone octamer sequence preferences under physiological conditions are not known as there is no technique that could determine these. Therefore, the correlation between intrinsic binding preferences as measured under nonphysiological reconstitution conditions and in vivo nucleosome positioning need not argue that there are the same intrinsic preferences in vivo and/or that they determine positioning. There could still be other factors, e.g., remodeling enzymes that read the DNA sequence with similar sequence preferences for nucleosome positioning. We consider this question not resolved yet, but acknowledge the common assumption in the field that some DNA sequences, like poly(dA:dT) elements, cause NDRs due to their intrinsic biophysical properties. Even if this is a true mechanism in vivo, it is certainly not necessary or universal. There are many NDRs without obvious intrinsic nucleosome excluding sequences, for example, S. pombe promoter NDRs are not poly(dA:dT) enriched (Lantermann et al. 2010), and NDR generation through poly(dA:dT) at promoters (Tsankov et al. 2010, 2011) or replication origins (Xu et al. 2012) is not evolutionarily conserved. Even more, the recent high-resolution hydroxyl-radical-seq map for fission yeast confirmed that dA/dT-rich sequences are not particularly nucleosome depleted here but showed instead their enrichment at nucleosome dyads (Moyle-Heyrman et al. 2013). This is just opposite to the intrinsic nucleosome positioning rules as derived from biophysical reconstitutions and suggests that nucleosome positioning DNA sequence rules as derived from in vivo nucleosome maps reflect the preferences of trans-factors in combination or not with the intrinsic preferences of the histone octamer. Very much to the point, the transfer of large genomic fragments (yeast artificial chromosomes, YACs) between yeast species clearly showed that the host cell environment mostly dictates nucleosome positioning along the heterologous DNA sequence (Hughes et al. 2012). NDRs were generated in the acceptor yeast species at positions where no NDR was present in the donor species and also where no poly(dA:dT) elements were present. Much earlier, long stretches of S. pombe DNA were introduced in mouse chromosomes and adopted the NRL of the host mouse cell (McManus et al. 1994). Clearly, DNA sequence rules for nucleosome positioning are species specific and not as universal as one would expect from a purely biophysical mechanism based on the interaction of DNA and the highly conserved histone core.Footnote 4 This is probably the reason for the limited success of biophysics-based algorithms predicting nucleosome positions from DNA sequence only (overview in Liu et al. 2013).
Nonetheless, there seems to be one universal and probably biophysical feature of nucleosome positioning: dinucleotide periodicity. Bending of DNA around the histone octamer necessitates widening and compression of the helix. The position of certain dinucleotides, like dA:dA, dT:dA, and dT:dT in minor grooves facing toward and dG:dC in minor grooves facing away from the histone octamer, favors these distortions (Drew and Travers 1985). Indeed, all eukaryotic nucleosome maps, and especially the high-resolution hydroxyl-radical-seq maps for S. cerevisiae and S. pombe (Brogaard et al. 2012a; Moyle-Heyrman et al. 2013), show a pronounced ~10-bp periodicity of such dinucleotides (with ~5 bp offset between dA/dT and dG/dC dinucleotides) in overlays of all nucleosome sequences. This periodicity is even more pronounced in SGD reconstituted nucleosomes than in vivo and seems to be encoded to a certain degree in eukaryotic genomes, but not in Escherichia coli DNA (Zhang et al. 2009). While this periodicity is usually not sufficient to direct translational positioning—maybe with the exception of some sequences underlying strongly positioned +/−1 nucleosomes (Cui et al. 2012; Mavrich et al. 2008a)—it likely drives the rotational positioning in nucleosome “position clusters” (Albert et al. 2007; Cole et al. 2012b; Jiang and Pugh 2009b).
Statistical positioning
The beauty of the statistical positioning model (Kornberg and Stryer 1988; Mobius and Gerland 2010) lies in the evolutionary freedom of genomes to evolve their DNA sequence without many constraints regarding the encoding of nucleosome positioning. Only relatively few regions, the barriers, have to be encoded. Further, it appeals by combining simplicity with explanatory power. Nonetheless, some of the assumptions are not experimentally validated. Most importantly, statistical positioning predicts that the NRL of barrier-aligned nucleosomal arrays is solely dictated by the nucleosome density. Fewer nucleosomes would have wider spacing and vice versa. However, this is not true if the histone concentration is reduced in in vitro reconstitutions (Zhang et al. 2011b), or in vivo if nucleosome density is lower because of mutations (compromised function of the transcription elongation coupled histone chaperone complex FACT, i.e., nhp6a/b mutant in S. cerevisiae (Celona et al. 2011) or pob3 mutant in S. pombe (Hennig et al. 2012)), or upon histone expression shut off (Gossett and Lieb 2012; van Bakel et al. 2013), or in aged S. cerevisiae mother cells (Hu et al. 2014). In these six cases, nucleosome density was substantially lower, even down to ~50 % of the wild-type situation, but global nucleosome spacing remained largely unchanged. This strongly argues for something like an “active packing” (Zhang et al. 2011b) or “clamping” mechanism that keeps nucleosomes together despite low nucleosome density. Indeed, just including into the statistical positioning model a hypothetical factor that binds two nucleosomes at the same time and sets their relative distance is already sufficient to keep spacing constant (Mobius et al. 2013). Structural studies of the budding yeast ISW1a complex suggested that the Ioc3 subunit may act as a “protein ruler” by bridging two nucleosomes and maybe setting their relative spacing (Yamada et al. 2011). This exciting possibility needs to be validated in functional assays, but it cannot be the only answer as an isw1 mutant retains regular arrays (Gkikopoulos et al. 2011a; Tirosh et al. 2010). The linker histone H1 was implied in determining the NRL (Oberg et al. 2012). However, yeasts do not have canonical H1, and the lack of the closest homologue in S. cerevisiae, Hho1, which is indeed localized to the linkers (Bryant et al. 2012), does not affect global spacing. Finally, statistical positioning treats nucleosomes as hard noninteracting spheres. However, nucleosomes are dynamic particles where histone octamer composition can be altered (Clapier and Cairns 2009; Mizuguchi et al. 2004) and where DNA undergoes “breathing” motions (Polach and Widom 1995) and nucleosomes can even invade each other, maybe pushed into each other by remodelers (Engeholm et al. 2009). Hydroxyl-radical-seq directly monitors dyad-to-dyad distances of neighboring nucleosomes on the same template and revealed for S. cerevisiae that “crowded nucleosomes” closer than 147 bp apart are quite common (almost 40 %; Chereji and Morozov 2014) arguing for many noncanonical nucleosomal particles, maybe including H3-H4 tetrasomes, across the genome. Indeed, replacing hard with soft nucleosome cores of variable footprint size allows the statistical positioning model to better explain different nucleosome spacing across 12 yeast species (Mobius et al. 2013).
Summarizing both preceding sections, intrinsic DNA sequence preferences of the histone octamer cannot account for a sufficient and/or universal nucleosome positioning mechanism, but appear to form NDRs in some and underlie rotational positioning in most species. The statistical positioning mechanism in its original form does not reflect the in vivo mechanism as it cannot explain constant nucleosome spacing despite varied nucleosome density and does not account for nucleosome breathing. Nonetheless, if additional trans-factors are introduced such that linkers are not only equalized but held constant, the basic statistical positioning concept of a DNA sequence-independent propagation mechanism that regularly aligns nucleosomes at barriers is still very appealing and was recently nicely demonstrated in S. cerevisiae (Ganguli et al. 2014). RSC remodeler depletion narrowed NDRs and shifted +1 nucleosomes upstream. As predicted by a barrier-aligned propagation mechanism and consistent with constant spacing, downstream array nucleosomes shifted accordingly. Furthermore, depending on whether the distance between two barriers, such as between NDRs of two divergent genes, is or is not an integer multiple of the NRL, the array forming mechanisms originating from either barrier are either reinforced or annulled, leading to regular or fuzzy arrays between the barriers, respectively.
Trans-factor-driven nucleosome positioning
The predominant role of trans-factors in nucleosome positioning is not only suggested by the inadequacies of mere intrinsic positioning and clearly required for the implementation of a modified statistical positioning mechanism, but also suggested by mutant studies in vivo and directly demonstrated by in vitro reconstitution.
Role of trans-factors indicated by mutant studies in vivo
Many candidate mutations were tested for their effects on genome-wide nucleosome positioning, especially in S. cerevisiae (Table 3): mutations affecting transcription or TFs; histone variants/modifiers/chaperones; remodelers; GRFs, such as Abf1, Reb1, Rap1, and Cbf1 in S. cerevisiae and Sap1 in S. pombe (Tsankov et al. 2011); and others. Besides some locus-specific effects, the overall stereotypical NDR-array patterns were only mildly affected if at all. Not surprisingly, the most severe effects were caused by global impairment of histone deposition, either by shut down of histone expression or depletion of the essential FACT or Spt6 histone chaperone/deposition activities (Celona et al. 2011; Gossett and Lieb 2012; Hennig et al. 2012; Hu et al. 2014; van Bakel et al. 2013). Other single mutations with substantial effects were those depleting GRFs, e.g., Reb1 or Abf1, or RSC (Badis et al. 2008; Ganapathi et al. 2011; Hartley and Madhani 2009; Parnell et al. 2008; van Bakel et al. 2013), which both increased nucleosome occupancy in promoter NDRs. Less disturbing was transcription shutdown (van Bakel et al. 2013; Weiner et al. 2010) and single deletions of genes encoding remodelers such as Isw2, Chd1, or Isw1, which all led to some rearrangements, e.g., upstream shifts of genic arrays including the +1 nucleosome (Tirosh et al. 2010; van Bakel et al. 2013; Whitehouse et al. 2007; Yen et al. 2012). Only the combined deletion of ISW1 and CHD1 in S. cerevisiae (Gkikopoulos et al. 2011a) or of the CHD1-type remodeler encoding genes hrp1 and hrp3 in S. pombe (Hennig et al. 2012; Pointner et al. 2012; Shim et al. 2012) also affected the global NDR-array patterns rather drastically. In these mutants, the NDRs and +1 nucleosomes are still comparable to those of the wild-type, but the array regularity over the genes largely disappeared in TSS-aligned composite plots (Fig. 4b, c). Importantly, this was not mainly due to a general lack of array regularity as these mutants still showed substantial, though less extensive, array regularity in MNase ladder assays of bulk chromatin (Pointner et al. 2012). Rather, the genic nucleosome arrays were probably differently aligned relative to the TSS from one gene to the next (Fig. 1b vs. c) and maybe also from one template to the next of the same gene, thereby disturbing the regularity in TSS-aligned composite plots. Therefore, equalizing linker length within arrays (“spacing”) and array alignment may be mechanistically separated (Pointner et al. 2012). In both yeasts, remodelers of the ISWI or CHD1 type were redundantly involved as only double mutants showed the full effect, and there seems to be mechanistic flexibility as S. cerevisiae uses a combination of ISWI- and CHD1- but S. pombe only CHD1-type remodelers. Given the far evolutionary divergence between both yeasts and the different set of factors involved, it is quite striking how similar the respective mutant nucleosomal patterns are (Fig. 4b, c). This points to an evolutionarily highly conserved function of linking nucleosomal arrays to the TSS that can be implemented by different factors.
Of note, the NDR/+1 nucleosome organization was substantially and globally disturbed only if the function of essential genes was compromised. Viable mutants showed only small effects at a minority of genes here, e.g., the isw2 mutant (Whitehouse et al. 2007), or large effects only downstream in the genes, e.g., the isw1 chd1 double mutant (Gkikopoulos et al. 2011a). This argues that a proper global NDR/+1 nucleosome organization is essentially linked to viability and underscores the biological importance of proper nucleosome positioning. But also the array alignment and regularity further downstream over the gene body is functional as their disturbance leads to increased cryptic or antisense transcription both in S. cerevisiae (Smolle et al. 2012) and in S. pombe (Hennig et al. 2012; Pointner et al. 2012; Shim et al. 2012).
Collectively, these mutant studies suggest the following. First, specific DNA binding proteins like GRFs and TFs are involved in promoter NDR formation where their binding sites are enriched (Badis et al. 2008; Hartley and Madhani 2009; Lee et al. 2007; Ozonov and van Nimwegen 2013; Rhee and Pugh 2011; Tsankov et al. 2010, 2011; van Bakel et al. 2013). Engineering a Reb1 site into a coding region caused NDR formation, but only in connection with a poly(dA:dT) element (Raisner et al. 2005). In contrast, deletion of the Reb1 sites at the ILV1 or GAL1-10 and/or of the poly(dA:dT) elements at the ILV1 or PHO5 promoter did not substantially change nucleosome positions (Angermayr and Bandlow 1997; Fascher et al. 1993; Reagan and Majors 1998). At the CLN2 promoter NDR, only the combined deletion of several GRF and TF sites increased nucleosome occupancy and repressed CLN2 (Bai et al. 2011). Deletion of two TFs for meiotic genes in S. pombe affected only few NDRs with the respective binding sites (Soriano et al. 2013). About half of the nucleosome occupancy changes upon induction/repression of Msn2/4-regulated promoters depended on Msn2/4 (Elfving et al. 2014). Therefore, multiple, often redundant factors contribute to the displacement of nucleosomes at NDRs in vivo. Which mechanism is used at a particular gene is evolutionarily flexible. The 5′NDR of homologous genes may involve poly(dA:dT) elements in one, a combination of poly(dA:dT) and GRFs in another and GRFs only in yet another yeast species (Tsankov et al. 2010). It is unclear if GRFs and TFs generate NDRs by mere binding competition with the histone octamer or if they mainly recruit remodelers that actually displace nucleosomes. Importantly, manipulating GRFs/TFs or their binding sites usually affects transcription levels. Are changes in NDR nucleosome occupancy cause or consequence of changes in transcription (see also “Role of transcription”)?
Second, remodelers are involved in generating all aspects of the NDR-array pattern. RSC seems to be required in particular for NDR generation and ISWI- and CHD1-type remodelers for array formation and linking arrays to the NDRs. In vitro assays demonstrated “spacing activity”, i.e., turning irregular into regular nucleosomal arrays, for ISWI-, and CHD1-, but not for SWI/SNF-/RSC- or SWR-type remodelers (Clapier and Cairns 2009). These assays explain how regularity is achieved in the sense of equalizing linker length (“length sensor model”; Yang et al. 2006), but not how arrays are aligned at the TSS or how constant spacing is set regardless of nucleosome density. Further, the ISWI, Chd1, RSC, and also the INO80 remodelers seem to push and pull at the +1/−1 nucleosomes whose position is of course intimately linked to NDR generation. Apparently, the mechanisms for generating NDR position and width can be separated. Poly(dA:dT) generates properly positioned NDRs on heterologous YAC sequences in S. cerevisiae, but not necessarily with the proper width, i.e., proper positions of +1/−1 nucleosomes (Hughes et al. 2012), which likely depend on host cell-specific remodelers.
Third, it is difficult to decide from in vivo results if remodelers have a direct or indirect, specific or generic, sufficient or necessary role in nucleosome positioning. Mutations in remodeler encoding or other genes may affect expression of some other factors and therefore have indirect effects. Even though RSC as well as the remodelers Isw1, Chd1, INO80, and SWR1 bind NDRs (Koerber et al. 2009; Yen et al. 2012; Yen et al. 2013; Zentner et al. 2013), at least for some of these, the NDR may be just a preferred binding/cross-linking site due to the available long stretch of free DNA, or a kind of repository, but they may actually function elsewhere, e.g., in coding regions. Regarding specificity, remodelers have been seen as mere “nucleosome lubricators” that enable nucleosomes to sample alternative positions within physiological time scales, but that do not carry positioning information, i.e., do not affect the decision for one over another position. This view is akin to the principles of molecular chaperones (Walter and Buchner 2002). These assist protein folding by speeding up sampling conformations and by preventing irreversible aggregation but they do not determine or participate in the native 3D structure, which is solely intrinsically determined by the amino acid sequence. With regard to nucleosome positioning, this view has been called the “dynamic equilibrium model of nucleosome positioning” (Segal and Widom 2009b). Here, integrating the concentrations and binding affinities of all DNA binding factors, like the histone octamers and GRFs, determines nucleosome positions (= positioning information), while remodelers just keep nucleosomes mobile by lowering the activation energies for nucleosome repositioning. It is difficult to distinguish in vivo if remodelers only mobilize nucleosomes or if they co-determine nucleosome positions.
Direct and specific roles of trans-factors, especially remodelers, indicated by reconstitution in vitro
To overcome these limitations and to study nucleosome positioning mechanisms biochemically, our group established an in vitro reconstitution system that recapitulates very in vivo-like nucleosome positioning for S. cerevisiae (Hertel et al. 2005; Korber and Horz 2004; Krietenstein et al. 2012; Wippo and Korber 2012; Zhang et al. 2011b) and allows dissecting if a factor’s role is direct or indirect, necessary and/or sufficient, and specific or unspecific. Plasmids carrying yeast genome inserts, even a whole genome plasmid library, are reconstituted into nucleosomes by SGD. This will generate NDRs at proper positions (maybe not with proper width) over poly(dA:dT) elements and some individual in vivo-like nucleosome positions, but not the canonical NDR-array patterns (Kaplan et al. 2009; Zhang et al. 2009, 2011b). The degree of similarity to the in vivo situation, including the basic NDR-array pattern at most genes, is dramatically increased, although still not perfect, upon incubation of the SGD chromatin with an S. cerevisiae whole cell extract. Importantly, such nucleosome repositioning is strictly ATP dependent and does not include transcription. This strongly argues for direct and predominant effects of trans-factors, especially for the ATP-dependent remodelers. For some single loci, like PHO8, we showed using purified RSC and RSC-depleted extracts, that RSC has a direct, necessary, but not sufficient role in the formation of proper promoter NDRs (Wippo et al. 2011). Importantly, RSC depletion was only compensated by the addition of purified RSC but not by purified SWI/SNF or ISW2, even though all three remodelers were added with similar activities and all of them slide nucleosomes in vitro. This argues against an unspecific “lubricating” but strongly for a specific positioning contribution of RSC. We advocate that remodelers (co-)determine nucleosome positioning on their own. It is conceivable that remodelers modify the DNA sequence preferences of histone octamers, e.g., by direct octamer contacts or distortion and ATP input. In addition, remodelers may mediate the contribution of other, e.g., specific DNA binding factors. This need not mean that they remain bound to a nucleosome in order to maintain positioning. Once remodelers positioned a nucleosome, it will stay there also in the absence of remodelers as nucleosome positions are mostly kinetically frozen on DNA under physiological conditions (Korolev et al. 2007), even in intrinsically unfavorable positions. Therefore, nucleosomes are not necessarily at their thermodynamic equilibrium position in vivo, but are actively placed by remodelers (Cole et al. 2012b; Korber 2012; Wippo et al. 2011). This is supported by single loci assays for the remodeler RSC but remains to be shown directly on the genome level for this and other remodelers and trans-factors.
Specific remodeler recruitment
A specific role for remodelers in nucleosome positioning calls for specific remodeler recruitment. Indeed, remodelers show type-specific distributions as to which nucleosomes they can be cross-linked and detected by immunoprecipitation (MNase-anti-remodeler-ChIP-seq; Yen et al. 2012). For example, ISW1a, Isw2, and the INO80 Arp5 subunit preferentially bind the +1 nucleosome; RSC and SWI/SNF complexes bind more broadly nucleosomes around the NDR, ISW1b rather the +2 and downstream nucleosomes; and the Ino80 ATPase is found everywhere. This distribution specificity is intriguing and somewhat fits to the nucleosome repositioning trends in the respective remodeler mutants (“Role of trans-factors indicated by mutant studies in vivo”). Nonetheless, such data probably do not reveal the full picture as some remodelers, e.g., Chd1, were not scored and as the selection for MNase-resistant mononucleosomes prior to immunoprecipitation biases against noncanonical nucleosomes that may be sites of remodeler action.
There are several demonstrated mechanisms for remodeler recruitment. The INO80 and SWR complexes preferentially bind long (>60 bp for SWR; Ranjan et al. 2013) stretches of free DNA and are therefore enriched at NDRs (Fig. 5a; Ranjan et al. 2013; Yen et al. 2013) where they participate in the rapid histone exchange and incorporation of H2A.Z typical for the +1/−1 nucleosomes (Papamichos-Chronakis et al. 2011; Watanabe et al. 2013). RSC may target itself to NDRs through the DNA binding specificity of its subunit Rsc3 (Fig. 5b; Badis et al. 2008), although mutation of the Rsc3 site at the CLN2 promoter did not decrease the anti-RSC-ChIP signal (Bai et al. 2011). GRFs may target remodelers to NDRs (Fig. 5c), but direct evidence is lacking. Remodeler recruitment may even extend from TF-specified primary to ectopic sites via DNA looping (Fig. 5d; Yadon et al. 2013). Several remodeling complexes carry subunits with domains recognizing histone modifications (Clapier and Cairns 2009), like the acetyl-lysine binding bromodomains of the RSC and SWI/SNF complexes (Fig. 5e) or the PWWP domain of Ioc4 in the ISW1b complex binding H3K36me (Fig. 5f; Smolle et al. 2012). The latter is associated with transcribed regions. Maybe also the RNA polymerase holoenzyme directly recruits remodelers, e.g., Chd1 via the CTD (Fig. 5f; also awaiting direct demonstration). This way transcription would be a remodeler recruiting process.

Remodeler recruitment modes. Remodelers may a be recruited by long free DNA stretches; b carry sequence-specific DNA binding domains/subunits; c be directly recruited through specific DNA binding factors, e.g., maybe by GRFs; d be directly and indirectly recruited through specific DNA binding factors and DNA looping mediated by TFIIB; e bind histone modifications; or f be co-transcriptionally recruited via transcription coupled histone modifications or via the RNA polymerase II CTD
Role of transcription
Due to the intimate links between nucleosome organization and transcription, their mutual causal relationships amount to chicken-and-egg questions. Recent high-resolution ChIP-exo mapping of the PIC showed a strong correlation of TFIID-associated factors (TAFs) with the +1 nucleosome (Rhee and Pugh 2012). Maybe the +1 nucleosome slows down RNA polymerase scanning and thereby participates in TSS selection (Jiang and Pugh 2009b). This may explain the canonical TSS position just within the +1 nucleosome 5′ flank. Conversely, PIC formation may contribute to positioning of the +1 nucleosome (Struhl and Segal 2013; Zhang et al. 2009). During zebrafish development, nucleosomes are repositioned around TSSs before these TSSs are activated (Haberle et al. 2014). Due to this in vivo result and our own in vitro reconstitution of the basic NDR-array pattern without transcription (see “Direct and specific roles of trans-factors, especially remodelers, indicated by reconstitution in vitro”), we tend toward the view that the +1 nucleosome is prior to PIC formation. Nonetheless, there may be a continuum between the two alternatives and also truly circular causalities on a case-by-case basis. Such potential circularity is hard to resolve in most in vivo experiments.
As the regularity and extent of nucleosomal arrays correlates with the direction and extent of transcription, also for de novo generated transcription units in YAC transfer experiments (Hughes et al. 2012), it was suggested (Hughes et al. 2012; Struhl and Segal 2013; Zhang et al. 2009) that not only transcription initiation may be involved in generating NDRs and positioning +1 nucleosomes but also that transcription elongation mediates regular spacingFootnote 5 and array formation. Again, our reconstitutions of the basic NDR-array pattern without transcription makes such an essential role of transcription in array formation unlikely. Nonetheless, reconstituted arrays were not as extensive as in vivo, the +1 nucleosome position was shifted slightly downstream and the regular spacing a bit too wide. Interestingly, inactivation of RNA polymerase II in S. cerevisiae also led to a downstream shift of the arrays over coding regions (Weiner et al. 2010). The authors proposed a kind of “conveyor belt plus chopper” mechanism, i.e., polymerase passage constantly moves nucleosomes toward the NDR where the nucleosomes are then evicted. This idea is also based on following the fate of ancestral histones after several cell generations in S. cerevisiae. The covalently labeled ancestral histones became enriched in the 5′ region of long, lowly transcribed genes arguing for an upstream movement of nucleosomes by transcription (Radman-Livaja et al. 2011).
All this argues that +1 nucleosome positioning and array generation is somehow linked to transcription and that transcription has at least a fine-tuning role. We suppose that ultimately the remodelers position the nucleosomes while transcription mainly provides their specific recruitment rather than some specific positioning mechanism itself. However, this question is still open.
Integrative model
We suggest a model for nucleosome positioning mechanisms that integrates the contributions of intrinsic positioning, trans-factors, and statistical positioning (Fig. 6). Nucleosomes are kept out of NDRs (Fig. 6a) either by nucleosome excluding sequences (e.g., poly(dA:dT)) and/or by trans-factors like remodelers (e.g., RSC) and GRFs (e.g., Reb1) or TFs (e.g., Msn2/4). The exact NDR borders are determined by positioning the +1/−1 nucleosomes (Fig. 6b), mainly through remodelers (e.g., Isw2) with or without trans-factor recruitment (e.g., by GRFs), maybe also through intrinsic sequence contributions and the PIC. Something around the NDR/TSS/PIC?/+1 nucleosome serves as a barrierFootnote 6 in the sense of the original statistical positioning mechanism. However, the alignment of regularly spaced nucleosomes is an active process that keeps linker length constant regardless of nucleosome density and is mediated by remodelers (Fig. 6c). These may either act on individual nucleosomes from their recruitment site at the barrier and/or function as dinucleosome clamps that propagate setting the NRL from the barrier onwards. Especially for the latter, it may be crucial that transcription and associated histone marks recruit remodelers over gene bodies to yield sufficient local concentrations also further downstream of the TSS (Fig. 5f). This may explain why array extent scales with the direction and extent of transcription. Finally, intrinsic sequence features (e.g., dinucleotide periodicities) fine tune nucleosomes with regard to rotational positioning. In this sense, we adopt Geeta Narlikar’s “Chromatin remodelers act globally, sequence positions nucleosomes locally” (Partensky and Narlikar 2009).

Integrative model of mechanisms leading to the stereotypical NDR-array organization. a NDRs may be formed by intrinsic histone octamer sequences preferences, e.g., nucleosome repelling poly(dA:dT), or by remodelers with or without recruitment by specific DNA binding factors, e.g., GRFs, or a combination of all these. b Nucleosomes flanking the NDR are positioned by remodelers with or without recruitment by specific DNA binding factors, e.g., GRFs, and maybe a role for the PIC and nucleosome positioning sequences. c Something linked to the NDR/TSS/PIC?/+1 nucleosome serves as barrier. Regular nucleosomal arrays are formed and aligned at barriers by an active remodeler driven mechanism. Remodelers either act from their recruitment sites at barriers (left) and/or clamp nucleosomes together (right), maybe while co-transcriptionally recruited (Fig. 5f)
Our integrative model is similar to the “unified model for nucleosome positioning” proposed by Hughes et al. (2012) and Struhl and Segal (2013), but we emphasize more the role of remodelers versus the role of transcription. Both views may be reconciled if transcription is a recruitment mechanism for remodelers, but are substantially different if transcription has a genuine function in nucleosome positioning.
We highlight that all aspects of the model involve remodeling enzymes and propose, based on their specific roles, that they truly contribute positioning information and not just kinetic lubrication for nucleosomes. This view is similar to the idea of a “remodeler code” (Rippe et al. 2007) and consistent with sequence-dependent kinetics and steady-state outcome of remodeling reactions in vitro (Rippe et al. 2007; van Vugt et al. 2009). Such a thermodynamic contribution of remodeling ATPases implies that nucleosome positioning never reaches equilibrium but at most a steady state as it constantly requires net consumption of ATP and that nucleosomes need not reside at intrinsically most favored positions, but can be actively placed and maintained anywhere in the genome.
Open questions
The integrative model for nucleosome positioning mechanisms provides a fruitful framework, but our understanding of which factor implements which aspect in which way is still rather sparse. For example, we do not understand yet how remodelers are recruited and how their specific mechanisms are tailored toward nucleosome eviction, deposition, positioning, and spacing. We do not know the molecular correlate of the barriers nor how the +1 nucleosome position is determined nor how spacing is set in absolute termsFootnote 7 nor how GRFs function nor how transcription participates nor if histone chaperones have nucleosome positioning information or which other factors may still be involved. During the past 10 years, nucleosome positioning was reanimated to an exciting and vibrant field that promises to answer how the most basic level of chromatin structure is generated.
Notes
While the average +1 nucleosome position relative to the TSS is conserved across yeasts, it differs in other eukaryotes. For example, the Drosophila TSSs are often upstream of the +1 nucleosome within the NDR (Mavrich et al. 2008b).
Alternative nomenclatures: canonical versus noncanonical (Jiang and Pugh 2009b), open versus covered (Cairns 2009), depleted versus occupied proximal nucleosome (DPN vs. OPN; Tirosh and Barkai 2008), and much earlier in the context of Drosophila promoters: preset versus remodeling promoters (Lu et al. 1994).
The comparison of nucleosome occupancy between different data sets may be problematic (see “Comparison of different methods and the problem of nucleosome occupancy”).
There is the formal possibility of species-specific intrinsic sequence preferences. However, we think this unlikely given the high conservation of the histone octamer and physiological biophysical conditions.
While yeast cells show constant spacing throughout the genome, human cells vary the NRL inversely with transcription rate (Valouev et al. 2011).
It is quite striking that one of the oldest observations in the chromatin field, regular nucleosome spacing seen in MNase ladders, is still not explained mechanistically.
References
Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446:572–576
Allan J, Fraser RM, Owen-Hughes T, Keszenman-Pereyra D (2012) Micrococcal nuclease does not substantially bias nucleosome mapping. J Mol Biol 417:152–164
Almer A, Hörz W (1986) Nuclease hypersensitive regions with adjacent positioned nucleosomes mark the gene boundaries of the PHO5/PHO3 locus in yeast. EMBO J 5:2681–2687
Almer A, Rudolph H, Hinnen A, Hörz W (1986) Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J 5:2689–2696
Angermayr M, Bandlow W (1997) The general regulatory factor Reb1p controls basal, but not Gal4p-mediated, transcription of the GCY1 gene in yeast. Mol Gen Genet 256:682–689
Axel R (1975) Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry 14:2921–2925
Badis G et al (2008) A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32:878–887
Bai L, Ondracka A, Cross FR (2011) Multiple sequence-specific factors generate the nucleosome-depleted region on CLN2 promoter. Mol Cell 42:465–476
Basehoar AD, Zanton SJ, Pugh BF (2004) Identification and distinct regulation of yeast TATA box-containing genes. Cell 116:699–709
Becker J, Yau C, Hancock JM, Holmes CC (2013) NucleoFinder: a statistical approach for the detection of nucleosome positions. Bioinformatics (Oxford, England) 29:711–716. doi:10.1093/bioinformatics/bts719
Becker PB, Workman JL (2013) Nucleosome remodeling and epigenetics. Cold Spring Harbor perspectives in biology 5 doi:10.1101/cshperspect.a017905
Bell O, Tiwari VK, Thoma NH, Schubeler D (2011) Determinants and dynamics of genome accessibility. Nat Rev Genet 12:554–564
Berbenetz NM, Nislow C, Brown GW (2010) Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet 6:e1001092
Brogaard K, Xi L, Wang JP, Widom J (2012a) A map of nucleosome positions in yeast at base-pair resolution. Nature 486:496–501. doi:10.1038/nature11142
Brogaard KR, Xi L, Wang JP, Widom J (2012b) A chemical approach to mapping nucleosomes at base pair resolution in yeast. Methods Enzymol 513:315–334. doi:10.1016/b978-0-12-391938-0.00014-8
Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H (2013) Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol 11:e1001621. doi:10.1371/journal.pbio.1001621
Bryant JM, Govin J, Zhang L, Donahue G, Pugh BF, Berger SL (2012) The linker histone plays a dual role during gametogenesis in Saccharomyces cerevisiae. Mol Cell Biol 32:2771–2783
Buchanan L et al (2009) The Schizosaccharomyces pombe JmjC-protein, Msc1, prevents H2A.Z localization in centromeric and subtelomeric chromatin domains. PLoS Genet 5:e1000726
Cairns BR (2009) The logic of chromatin architecture and remodelling at promoters. Nature 461:193–198
Caserta M, Agricola E, Churcher M, Hiriart E, Verdone L, Di Mauro E, Travers A (2009) A translational signature for nucleosome positioning in vivo. Nucleic Acids Res 37:5309–5321
Celona B et al (2011) Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol 9:e1001086
Chen K et al (2013) DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23:341–351. doi:10.1101/gr.142067.112
Chereji RV, Morozov AV (2014) Ubiquitous nucleosome crowding in the yeast genome. Proc Natl Acad Sci U S A 111:5236–5241. doi:10.1073/pnas.1321001111
Chung HR et al (2010) The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS ONE 5:e15754
Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273–304
Cockell M, Rhodes D, Klug A (1983) Location of the primary sites of micrococcal nuclease cleavage on the nucleosome core. J Mol Biol 170:423–446
Cole HA, Howard BH, Clark DJ (2011a) Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res 39:9521–9535. doi:10.1093/nar/gkr643
Cole HA, Howard BH, Clark DJ (2011b) The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc Natl Acad Sci U S A 108:12687–12692. doi:10.1073/pnas.1104978108
Cole HA, Howard BH, Clark DJ (2012a) Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods Enzymol 513:145–168
Cole HA, Nagarajavel V, Clark DJ (2012b) Perfect and imperfect nucleosome positioning in yeast. Biochim Biophys Acta 1819:639–643. doi:10.1016/j.bbagrm.2012.01.008
Cui F, Cole HA, Clark DJ, Zhurkin VB (2012) Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res 40:10753–10764. doi:10.1093/nar/gks870
Davey CA, Richmond TJ (2002) DNA-dependent divalent cation binding in the nucleosome core particle. Proc Natl Acad Sci U S A 99:11169–11174
de Castro E, Soriano I, Marin L, Serrano R, Quintales L, Antequera F (2012) Nucleosomal organization of replication origins and meiotic recombination hotspots in fission yeast. EMBO J 31:124–137
DeGennaro CM et al (2013) Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol Cell Biol 33:4779–4792. doi:10.1128/mcb. 01068-13
Dingwall C, Lomonossoff GP, Laskey RA (1981) High sequence specificity of micrococcal nuclease. Nucleic Acids Res 9:2659–2673
Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ (2007) Dynamics of replication-independent histone turnover in budding yeast. Science 315:1405–1408
Drew HR, Travers AA (1985) DNA bending and its relation to nucleosome positioning. J Mol Biol 186:773–790
Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM (2010) Conserved nucleosome positioning defines replication origins. Genes Dev 24:748–753
Elfving N, Chereji RV, Bharatula V, Bjorklund S, Morozov AV, Broach JR (2014) A dynamic interplay of nucleosome and Msn2 binding regulates kinetics of gene activation and repression following stress. Nucleic Acids Res 42:5468–5482. doi:10.1093/nar/gku176
Elgin SC (1981) DNAase I-hypersensitive sites of chromatin. Cell 27:413–415
Engeholm M, de Jager M, Flaus A, Brenk R, van Noort J, Owen-Hughes T (2009) Nucleosomes can invade DNA territories occupied by their neighbors. Nat Struct Mol Biol 16:151–158
Fascher KD, Schmitz J, Hörz W (1993) Structural and functional requirements for the chromatin transition at the PHO5 promoter in Saccharomyces cerevisiae upon PHO5 activation. J Mol Biol 231:658–667
Field Y et al (2008) Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol 4:e1000216
Flaus A, Luger K, Tan S, Richmond TJ (1996) Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci U S A 93:1370–1375
Floer M et al (2010) A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141:407–418
Flores O, Deniz O, Soler-Lopez M, Orozco M (2014) Fuzziness and noise in nucleosomal architecture. Nucleic Acids Res. doi:10.1093/nar/gku165
Flores O, Orozco M (2011) nucleR: a package for non-parametric nucleosome positioning. Bioinformatics (Oxford, England) 27:2149–2150 doi:10.1093/bioinformatics/btr345
Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, Morse RH (2011) Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res 39:2032–2044. doi:10.1093/nar/gkq1161
Ganguli D, Chereji RV, Iben JR, Cole HA, Clark DJ (2014) RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res. doi:10.1101/gr.177014.114
Giresi PG, Lieb JD (2009) Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 48:233–239. doi:10.1016/j.ymeth.2009.03.003
Givens RM et al (2012) Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res 40:7176–7189
Givens RM, Mesner LD, Hamlin JL, Buck MJ, Huberman JA (2011) Integrity of chromatin and replicating DNA in nuclei released from fission yeast by semi-automated grinding in liquid nitrogen. BMC Res Notes 4:499
Gkikopoulos T et al (2011a) A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 333:1758–1760
Gkikopoulos T et al (2011b) The SWI/SNF complex acts to constrain distribution of the centromeric histone variant Cse4. EMBO J 30:1919–1927
Gossett AJ, Lieb JD (2012) In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet 8:e1002771
Haberle V et al (2014) Two independent transcription initiation codes overlap on vertebrate core promoters. Nature 507:381–385. doi:10.1038/nature12974
Hartley PD, Madhani HD (2009) Mechanisms that specify promoter nucleosome location and identity. Cell 137:445–458
Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S (2011) Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A 108:18318–18323
Hennig BP, Bendrin K, Zhou Y, Fischer T (2012) Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep 13:997–1003
Hertel CB, Längst G, Hörz W, Korber P (2005) Nucleosome stability at the yeast PHO5 and PHO8 promoters correlates with differential cofactor requirements for chromatin opening. Mol Cell Biol 25:10755–10767
Hörz W, Altenburger W (1981) Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res 9:2643–2658
Hu Z et al (2014) Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev 28:396–408. doi:10.1101/gad.233221.113
Huebert DJ, Kuan PF, Keles S, Gasch AP (2012) Dynamic changes in nucleosome occupancy are not predictive of gene expression dynamics but are linked to transcription and chromatin regulators. Mol Cell Biol 32:1645–1653. doi:10.1128/mcb.06170-11
Hughes AL, Jin Y, Rando OJ, Struhl K (2012) A functional evolutionary approach to identify determinants of nucleosome positioning: a unifying model for establishing the genome-wide. Pattern Mol Cell 48:5–15
Hughes AL, Rando OJ (2014) Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys 43:41–63. doi:10.1146/annurev-biophys-051013-023114
Iyer VR (2012) Nucleosome positioning: bringing order to the eukaryotic genome. Trends Cell Biol 22:250–256
Jamai A, Imoberdorf RM, Strubin M (2007) Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell 25:345–355
Jansen A, Verstrepen KJ (2011) Nucleosome positioning in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 75:301–320. doi:10.1128/mmbr.00046-10
Jiang C, Pugh BF (2009a) A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 10:R109
Jiang C, Pugh BF (2009b) Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10:161–172
Kaplan N, Hughes TR, Lieb JD, Widom J, Segal E (2010) Contribution of histone sequence preferences to nucleosome organization: proposed definitions and methodology. Genome Biol 11:140–152
Kaplan N et al (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458:362–366
Kent NA, Adams S, Moorhouse A, Paszkiewicz K (2011) Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res 39:e26
Koerber RT, Rhee HS, Jiang C, Pugh BF (2009) Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol Cell 35:889–902. doi:10.1016/j.molcel.2009.09.011
Korber P (2012) Active nucleosome positioning beyond intrinsic biophysics is revealed by in vitro reconstitution. Biochem Soc Trans 40:377–382
Korber P, Barbaric S (2014) The yeast PHO5 promoter: from single locus to systems biology of a paradigm for gene regulation through chromatin. Nucleic Acids Res 42:10888–10902. doi:10.1093/nar/gku784
Korber P, Horz W (2004) In vitro assembly of the characteristic chromatin organization at the yeast PHO5 promoter by a replication-independent extract system. J Biol Chem 279:35113–35120. doi:10.1074/jbc.M405446200
Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98:285–294
Kornberg RD, Stryer L (1988) Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res 16:6677–6690
Korolev N, Vorontsova OV, Nordenskiold L (2007) Physicochemical analysis of electrostatic foundation for DNA-protein interactions in chromatin transformations. Prog Biophys Mol Biol 95:23–49. doi:10.1016/j.pbiomolbio.2006.11.003
Krietenstein N, Wippo CJ, Lieleg C, Korber P (2012) Genome-wide in vitro reconstitution of yeast chromatin with in vivo-like nucleosome positioning. Methods Enzymol 513:205–232
Kristell C, Orzechowski Westholm J, Olsson I, Ronne H, Komorowski J, Bjerling P (2010) Nitrogen depletion in the fission yeast Schizosaccharomyces pombe causes nucleosome loss in both promoters and coding regions of activated genes. Genome Res 20:361–371. doi:10.1101/gr.098558.109
Kuan PF, Huebert D, Gasch A, Keles S (2009) A non-homogeneous hidden-state model on first order differences for automatic detection of nucleosome positions. Stat App Genet Mol Biol 8: Article29 doi:10.2202/1544-6115.1454
Lam FH, Steger DJ, O’Shea EK (2008) Chromatin decouples promoter threshold from dynamic range. Nature 453:246–250
Lantermann A, Stralfors A, Fagerstrom-Billai F, Korber P, Ekwall K (2009) Genome-wide mapping of nucleosome positions in Schizosaccharomyces pombe. Methods 48:218–225. doi:10.1016/j.ymeth.2009.02.004
Lantermann AB, Straub T, Stralfors A, Yuan GC, Ekwall K, Korber P (2010) Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 17:251–257
Lee JS et al (2012) Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev 26:914–919
Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39:1235–1244
Liu H, Zhang R, Xiong W, Guan J, Zhuang Z, Zhou S (2013) A comparative evaluation on prediction methods of nucleosome positioning. Brief Bioinform. doi:10.1093/bib/bbt062
Locke G, Tolkunov D, Moqtaderi Z, Struhl K, Morozov AV (2010) High-throughput sequencing reveals a simple model of nucleosome energetics. Proc Natl Acad Sci U S A 107:20998–21003. doi:10.1073/pnas.1003838107
Lu Q, Wallrath LL, Elgin SCR (1994) Nucleosome positioning and gene regulation. J Cell Biochem 55:83–92
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Maeshima K, Imai R, Tamura S, Nozaki T (2014) Chromatin as dynamic 10-nm fibers. Chromosoma 123:225–237. doi:10.1007/s00412-014-0460-2
Mavrich TN et al (2008a) A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18:1073–1083
Mavrich TN et al (2008b) Nucleosome organization in the Drosophila genome. Nature 453:358–362
McGhee JD, Felsenfeld G (1983) Another potential artifact in the study of nucleosome phasing by chromatin digestion with micrococcal nuclease. Cell 32:1205–1215
McManus J et al (1994) Unusual chromosome structure of fission yeast DNA in mouse cells. J Cell Sci 107(Pt 3):469–486
Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C (2004) ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303:343–348
Mobius W, Gerland U (2010) Quantitative test of the barrier nucleosome model for statistical positioning of nucleosomes up- and downstream of transcription start sites. PLoS Comput Biol 6:e1000891
Mobius W, Osberg B, Tsankov AM, Rando OJ, Gerland U (2013) Toward a unified physical model of nucleosome patterns flanking transcription start sites. Proc Natl Acad Sci U S A 110:5719–5724
Mojardin L, Vazquez E, Antequera F (2013) Specification of DNA replication origins and genomic base composition in fission yeasts. J Mol Biol 425(23):4706–4713
Morse RH (2007) Transcription factor access to promoter elements. J Cell Biochem 102:560–570
Moyle-Heyrman G et al (2013) Chemical map of Schizosaccharomyces pombe reveals species-specific features in nucleosome positioning. Proc Natl Acad Sci U S A 110:20158–20163. doi:10.1073/pnas.1315809110
Nagy PL, Cleary ML, Brown PO, Lieb JD (2003) Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci U S A 100:6364–6369. doi:10.1073/pnas.1131966100
Nellore A, Bobkov K, Howe E, Pankov A, Diaz A, Song JS (2012) NSeq: a multithreaded Java application for finding positioned nucleosomes from sequencing data. Front Genet 3:320. doi:10.3389/fgene.2012.00320
Nikitina T, Wang D, Gomberg M, Grigoryev SA, Zhurkin VB (2013) Combined micrococcal nuclease and exonuclease III digestion reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J Mol Biol 425:1946–1960. doi:10.1016/j.jmb.2013.02.026
Nishida H (2012) Conservation of nucleosome positions in duplicated and orthologous gene pairs. Sci World J 2012:298174. doi:10.1100/2012/298174
Nishida H, Katayama T, Suzuki Y, Kondo S, Horiuchi H (2013) Base composition and nucleosome density in exonic and intronic regions in genes of the filamentous ascomycetes Aspergillus nidulans and Aspergillus oryzae. Gene 525:5–10. doi:10.1016/j.gene.2013.04.077
Nishida H, Kondo S, Matsumoto T, Suzuki Y, Yoshikawa H, Taylor TD, Sugiyama J (2012) Characteristics of nucleosomes and linker DNA regions on the genome of the basidiomycete Mixia osmundae revealed by mono- and dinucleosome mapping. Open Biol 2:120043. doi:10.1098/rsob.120043
Oberg C, Izzo A, Schneider R, Wrange O, Belikov S (2012) Linker histone subtypes differ in their effect on nucleosomal spacing in vivo. J Mol Biol 419:183–197. doi:10.1016/j.jmb.2012.03.007
Ozonov EA, van Nimwegen E (2013) Nucleosome free regions in yeast promoters result from competitive binding of transcription factors that interact with chromatin modifiers. PLoS Comput Biol 9:e1003181
Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL (2011) Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144:200–213
Parnell TJ, Huff JT, Cairns BR (2008) RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J 27:100–110
Partensky PD, Narlikar GJ (2009) Chromatin remodelers act globally, sequence positions nucleosomes locally. J Mol Biol 391:12–25
Platt JL, Kent NA, Harwood AJ, Kimmel AR (2013) Analysis of chromatin organization by deep sequencing technologies. Methods Mol Biol (Clifton, NJ) 983:173–183. doi:10.1007/978-1-62703-302-2_9
Pointner J et al (2012) CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J 31:4388–4403
Polach KJ, Widom J (1995) Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254:130–149
Prunell A (1998) A topological approach to nucleosome structure and dynamics: the linking number paradox and other issues. Biophys J 74:2531–2544. doi:10.1016/s0006-3495(98)77961-5
Quintales L, Vazquez E, Antequera F (2014) Comparative analysis of methods for genome-wide nucleosome cartography. Brief Bioinform. doi:10.1093/bib/bbu037
Radman-Livaja M, Rando OJ (2010) Nucleosome positioning: how is it established, and why does it matter? Dev Biol 339:258–266
Radman-Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F (2011) Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol 9:e1001075
Raisner RM et al (2005) Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123:233–248
Rando OJ (2010) Genome-wide mapping of nucleosomes in yeast. Methods Enzymol 470:105–118. doi:10.1016/s0076-6879(10)70005-7
Ranjan A et al (2013) Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154:1232–1245. doi:10.1016/j.cell.2013.08.005
Raser JM, O’Shea EK (2004) Control of stochasticity in eukaryotic gene expression. Science 304:1811–1814. doi:10.1126/science.1098641
Raser JM, O’Shea EK (2005) Noise in gene expression: origins, consequences, and control. Science 309:2010–2013. doi:10.1126/science.1105891
Raveh-Sadka T et al (2012) Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44:743–750
Reagan MS, Majors JE (1998) The chromatin structure of the GAL1 promoter forms independently of Reb1p in Saccharomyces cerevisiae. Mol Gen Genet 259:142–149
Rhee HS, Pugh BF (2011) Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147:1408–1419
Rhee HS, Pugh BF (2012) Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483:295–301. doi:10.1038/nature10799
Richmond TJ, Davey CA (2003) The structure of DNA in the nucleosome core. Nature 423:145–150
Rippe K, Schrader A, Riede P, Strohner R, Lehmann E, Längst G (2007) DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc Natl Acad Sci U S A 104:15635–15640
Rizzo JM, Bard JE, Buck MJ (2012) Standardized collection of MNase-seq experiments enables unbiased dataset comparisons. BMC Mol Biol 13:15. doi:10.1186/1471-2199-13-15
Rizzo JM, Mieczkowski PA, Buck MJ (2011) Tup1 stabilizes promoter nucleosome positioning and occupancy at transcriptionally plastic genes. Nucleic Acids Res 39:8803–8819. doi:10.1093/nar/gkr557
Rodriguez J, McKnight JN, Tsukiyama T (2014) Genome-wide analysis of nucleosome positions, occupancy, and accessibility in yeast: nucleosome mapping, high-resolution histone ChIP, and NCAM. Current protocols in molecular biology / edited by Frederick M Ausubel [et al.] 108:21.28.21-21.28.16 doi:10.1002/0471142727.mb2128s108
Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A (2007) Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 27:393–405
Schones DE et al (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132:887–898
Segal E et al (2006) A genomic code for nucleosome positioning. Nature 442:772–778
Segal E, Widom J (2009a) Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol 19:65–71
Segal E, Widom J (2009b) What controls nucleosome positions? Trends Genet 25:335–343
Sekinger EA, Moqtaderi Z, Struhl K (2005) Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell 18:735–748
Shen CH, Leblanc BP, Alfieri JA, Clark DJ (2001) Remodeling of yeast CUP1 chromatin involves activator-dependent repositioning of nucleosomes over the entire gene and flanking sequences. Mol Cell Biol 21:534–547. doi:10.1128/mcb. 21.2.534-547.2001
Shim YS et al (2012) Hrp3 controls nucleosome positioning to suppress non-coding transcription in eu- and heterochromatin. EMBO J 31:4375–4387. doi:10.1038/emboj.2012.267
Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR (2008) Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol 6:e65
Simon JM, Giresi PG, Davis IJ, Lieb JD (2013) A detailed protocol for formaldehyde-assisted isolation of regulatory elements (FAIRE). Current protocols in molecular biology / edited by Frederick M Ausubel [et al.] Chapter 21: Unit21.26 doi:10.1002/0471142727.mb2126s102
Small EC, Xi L, Wang JP, Widom J, Licht JD (2014) Single-cell nucleosome mapping reveals the molecular basis of gene expression heterogeneity. Proc Natl Acad Sci U S A 111:E2462–E2471. doi:10.1073/pnas.1400517111
Smolle M et al (2012) Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol 19:884–892
Soriano I, Quintales L, Antequera F (2013) Clustered regulatory elements at nucleosome-depleted regions punctuate a constant nucleosomal landscape in Schizosaccharomyces pombe. BMC Genomics 14:813. doi:10.1186/1471-2164-14-813
Stein A, Takasuka TE, Collings CK (2010) Are nucleosome positions in vivo primarily determined by histone-DNA sequence preferences? Nucleic Acids Res 38:709–719. doi:10.1093/nar/gkp1043
Struhl K, Segal E (2013) Determinants of nucleosome positioning. Nat Struct Mol Biol 20:267–273
Svaren J, Venter U, Hörz W (1995) In vivo analysis of nucleosome structure and transcription factor binding in Saccharomyces cerevisiae. Methods in Mol Genet 6:153–167
Thomas JO, Furber V (1976) Yeast chromatin structure. FEBS Lett 66:274–280
Tirosh I (2012) Computational analysis of nucleosome positioning. Methods Mol Biol (Clifton, NJ) 833:443–449. doi:10.1007/978-1-61779-477-3_27
Tirosh I, Barkai N (2008) Two strategies for gene regulation by promoter nucleosomes. Genome Res 18:1084–1091
Tirosh I, Sigal N, Barkai N (2010) Widespread remodeling of mid-coding sequence nucleosomes by Isw1. Genome Biol 11:R49
Tolkunov D, Zawadzki KA, Singer C, Elfving N, Morozov AV, Broach JR (2011) Chromatin remodelers clear nucleosomes from intrinsically unfavorable sites to establish nucleosome-depleted regions at promoters. Mol Biol Cell 22:2106–2118
Tsankov A, Yanagisawa Y, Rhind N, Regev A, Rando OJ (2011) Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res 21:1851–1862
Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ (2010) The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol 8:e1000414
Tsui K, Dubuis S, Gebbia M, Morse RH, Barkai N, Tirosh I, Nislow C (2011) Evolution of nucleosome occupancy: conservation of global properties and divergence of gene-specific patterns. Mol Cell Biol 31:4348–4355
Tsui K, Durbic T, Gebbia M, Nislow C (2012) Genomic approaches for determining nucleosome occupancy in yeast. Methods Mol Biol (Clifton, NJ) 833:389–411. doi:10.1007/978-1-61779-477-3_23
Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A, Sidow A (2011) Determinants of nucleosome organization in primary human cells. Nature 474:516–520
van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C (2013) A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9:e1003479
van Holde KE (1989) Chromatin. Springer, New York
van Vugt JJ, de Jager M, Murawska M, Brehm A, van Noort J, Logie C (2009) Multiple aspects of ATP-dependent nucleosome translocation by RSC and Mi-2 are directed by the underlying DNA sequence. PLoS ONE 4:e6345
van Werven FJ, van Teeffelen HA, Holstege FC, Timmers HT (2009) Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol 16:1043–1048
Venter U, Svaren J, Schmitz J, Schmid A, Hörz W (1994) A nucleosome precludes binding of the transcription factor Pho4 in vivo to a critical target site in the PHO5 promoter. EMBO J 13:4848–4855
Wal M, Pugh BF (2012) Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol 513:233–250. doi:10.1016/B978-0-12-391938-0.00010-0
Walter S, Buchner J (2002) Molecular chaperones—cellular machines for protein folding. Angewandte Chemie (International ed in English) 41:1098–1113
Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL (2013) A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 340:195–199. doi:10.1126/science.1229758
Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N (2010) High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20:90–100
Whitehouse I, Rando OJ, Delrow J, Tsukiyama T (2007) Chromatin remodelling at promoters suppresses antisense transcription. Nature 450:1031–1035
Wippo CJ, Israel L, Watanabe S, Hochheimer A, Peterson CL, Korber P (2011) The RSC chromatin remodelling enzyme has a unique role in directing the accurate positioning of nucleosomes. EMBO J 30:1277–1288
Wippo CJ, Korber P (2012) In vitro reconstitution of in vivo-like nucleosome positioning on yeast DNA. Methods Mol Biol 833:271–287
Wu C (1980) The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 286:854–860
Xu J et al (2012) Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol 13:R27
Yadon AN, Singh BN, Hampsey M, Tsukiyama T (2013) DNA looping facilitates targeting of a chromatin remodeling enzyme. Mol Cell 50:93–103. doi:10.1016/j.molcel.2013.02.005
Yamada K et al (2011) Structure and mechanism of the chromatin remodelling factor ISW1a. Nature 472:448–453
Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ (2006) The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol 13:1078–1083
Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF (2012) Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149:1461–1473
Yen K, Vinayachandran V, Pugh BF (2013) SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154:1246–1256
Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ (2005) Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309:626–630
Zawadzki KA, Morozov AV, Broach JR (2009) Chromatin-dependent transcription factor accessibility rather than nucleosome remodeling predominates during global transcriptional restructuring in Saccharomyces cerevisiae. Mol Biol Cell 20:3503–3513. doi:10.1091/mbc.E09-02-0111
Zentner GE, Tsukiyama T, Henikoff S (2013) ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet 9:e1003317
Zhang H, Reese JC (2007) Exposing the core promoter is sufficient to activate transcription and alter coactivator requirement at RNR3. Proc Natl Acad Sci U S A 104:8833–8838
Zhang L, Ma H, Pugh BF (2011a) Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res 21:875–884
Zhang Y et al (2009) Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16:847–852
Zhang Z, Pugh BF (2011) High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144:175–186
Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF (2011b) A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332:977–980
Zhao J, Herrera-Diaz J, Gross DS (2005) Domain-wide displacement of histones by activated heat shock factor occurs independently of Swi/Snf and is not correlated with RNA polymerase II density. Mol Cell Biol 25:8985–8999
Acknowledgments
We apologize to all colleagues whose work we could not cite because of space restrictions. This work was funded by the German Research Community (DFG) through the Collaborative Research Cluster SFB1064 and by the Bavarian State Ministry of Education and Culture, Science and the Arts through the Bavarian Research Network for Molecular Biosystems (BioSysNet).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lieleg, C., Krietenstein, N., Walker, M. et al. Nucleosome positioning in yeasts: methods, maps, and mechanisms. Chromosoma 124, 131–151 (2015). https://doi.org/10.1007/s00412-014-0501-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-014-0501-x