
Abstract
The incorporation of histone variants into chromatin plays an important role for the establishment of particular chromatin states. Six human histone H3 variants are known to date, not counting CenH3 variants: H3.1, H3.2, H3.3 and the testis-specific H3.1t as well as the recently described variants H3.X and H3.Y. We report the discovery of H3.5, a novel non-CenH3 histone H3 variant. H3.5 is encoded on human chromosome 12p11.21 and probably evolved in a common ancestor of all recent great apes (Hominidae) as a consequence of H3F3B gene duplication by retrotransposition. H3.5 mRNA is specifically expressed in seminiferous tubules of human testis. Interestingly, H3.5 has two exact copies of ARKST motifs adjacent to lysine-9 or lysine-27, and lysine-79 is replaced by asparagine. In the Hek293 cell line, ectopically expressed H3.5 is assembled into chromatin and targeted by PTM. H3.5 preferentially colocalizes with euchromatin, and it is associated with actively transcribed genes and can replace an essential function of RNAi-depleted H3.3 in cell growth.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first level of chromatin compaction is the 10-nm fibre, in which the DNA is wrapped around protein octamers composed of the four core histone types H2A, H2B, H3 and H4, thus forming nucleosomal arrays (Woodcock and Ghosh 2010; Olins and Olins 2003; Luger et al. 1997). This 10-nm fibre becomes further compacted by the interaction with linker histone H1 and other proteins. Posttranslational modifications (PTMs) at the N-termini of all histones can alter the degree of chromatin compaction (Kouzarides 2007). In vivo observations suggest that transcriptionally silent genes occur mostly within domains of condensed heterochromatin, whereas in many cases actively transcribed genes are found within domains of less condensed chromatin. Histone modifications occur in a dynamic fashion. Through altering the degree of chromatin compaction, PTMs of histone tails create chromatin structures favourable either for activation or repression of genes, depending on the genomic context and the combination of modifications at a given site. However, epigenetic information is not only conveyed through numerous combinations of PTMs. The assembly of histone variants into chromatin is another important level of chromatin structure regulation. The number of H3 variants differs among species (Postberg et al. 2010). Since decades, it has been known that four main non-CenH3 variants occur in mammals. H3.3 is expressed throughout the cell cycle. In contrast, H3.1 and H3.2 are expressed only during the S-phase. Another variant, which is apparently restricted to testes, is H3.1t (occasionally referred to as H3.4). Whereas H3.3 and H3.2 probably occur in all animals, H3.1 and H3.1t seem to be restricted to mammals. Very recently, two additional genes specific to Catarrhines encoding the H3 variants H3.X and H3.Y, which were formerly annotated as pseudogenes in GeneBank, have been recognized to be protein-coding (Wiedemann et al. 2010). The selective incorporation of histone variants into chromatin largely depends on the function of histone chaperones. For example, it has been shown that discrete amino acids in the “chaperone recognition site” are important for the replication-independent incorporation of H3.3 into chromatin by HIRA, whereas CAF-1 is specific for the replication-dependent histone variant H3.1 (Hake and Allis 2006; Park and Luger 2008). CENP-A is a divergent form of the H3 family, which is specifically present at centromeres (Black and Bassett 2008). Defined PTM patterns are apparently established depending on a given histone H3 variant, e.g. it has been observed that H3.3, when compared with H3.1, is enriched in histone modifications considered as representatives of a transcriptionally competent or “open” chromatin state. The observation that different H3 variants establish different PTM patterns led to the assumption that a selective incorporation of histone variants into chromatin acts as a determinant for the establishment of particular histone modification patterns (Henikoff and Ahmad 2005; Loyola and Almouzni 2007). A keynote of this concept is the general view that the assembly of histone H3 variants into chromatin can impart structural differences to nucleosomal arrays or chromatin domains before PTMs are introduced. This could lead to an “indexing” of the genome into functionally separate domains. Thus, histone variants could contribute not only to the regulation of gene expression, but also to the establishment of “epigenetic memory”—the inheritance of expression and silencing patterns from one generation to the next. It has been proposed that histone variants positioned in this way in a non-random fashion are then targeted by PTMs. The authors of this “H3 barcode hypothesis” envision that particularly mammals have evolved an additional way of regulating their genetic information over many cell generations (Hake and Allis 2006). Although it is well-accepted that histone H3 variants play a major role for the establishment of particular chromatin states, the extent how H3 variants are involved into these events is largely unexplored to date. Here we report the discovery of H3.5, a seventh non-CenH3 variant of histone H3. Due to its specific amino acid composition, H3.5 excludes selective PTM suggesting a potential role in discriminative predetermination of chromatin structure.
Results and discussion
A unique histone H3 variant is encoded on human chromosome 12p11.21
First evidence for the existence of a yet uncharacterized histone H3 variant encoded in the human genome was given by the results of tBlastn searches for less conserved H3 genes or pseudogenes. The H3F3C gene (gene ID: 440093) is located on the small arm of chromosome 12 at 12p11.21 (Fig. 1a). Fluorescence in situ hybridization (FISH) using a fluorescein isothiocyanate (FITC)-labelled bacterial artificial chromosome (BAC) probe containing the H3F3C locus in combination with a human whole chromosome 12 paint confirmed the chromosome 12 localization in spread metaphase chromosomes (Fig. 1b) or in chromosome 12 territories (CT #12) in interphase nuclei (Fig. 1c) of human embryonic kidney (Hek)293 cells. In single light optical sections of CT #12, it became obvious that the H3F3C locus was localized adjacent to but not within condensed centromeric heterochromatin (Fig. 1d).

H3.5 is encoded on chromosome 12p11.21. a Chromosome #12 cartoon. The localization of 12p11.21 is marked with a red asterisk. b Spread metaphase chromosomes of Hek293 cells. DNA was stained with DAPI. Chromosome #12 is coloured in red, the H3F3C locus is green to yellow. c Projection of chromosome #12 territories in interphase nuclei of Hek293 cells. Chromosome #12 is coloured in red, the H3F3C locus is green. d Single light optical section (section #9 out of 30) of the same nucleus as shown in (c) (×1.5 magnification). The position of condensed centromeric heterochromatin is indicated (cen). Chromosome #12 is coloured in red, the H3F3C locus is green. e Protein alignment of human H3 variants and H3.5 homologs in hominids. Identical residues are shaded in grey
H3F3C encodes a unique histone H3 variant (GenBank accession no. HQ873957). In continuation of the histone H3 nomenclature, we decided to name the novel variant H3.5. The H3.5 protein sequence (134 amino acids) identity in comparison to the most closely related human H3 variant, H3.3 is ∼96% (129/135 residues) (Fig. 1e). Differences to all other human H3 variants are the presence of two exact copies of the ARKST motif adjacent to lysine-9 or lysine-27, respectively, a cysteine (C33) at the basis of the N-terminus, a degenerated motif adjacent to lysine-36 (lack of lysine-36 or lysine-37, depending on the method of counting) and the presence of an asparagine instead of lysine-79. It is remarkable that the two last mentioned sites, H3K36 as well as H3K79, are frequently found to be targets of lysine methylation in H3 variants associated with actively transcribed euchromatin (Kouzarides 2007). Since at least H3K79 is not present in H3.5, this site cannot be targeted by methylation PTM. It is not known whether the same could be true for H3K36 due to the degenerated adjacent motif. H3 methylated at lysine-9 or lysine-27 in the context of A7R8K9S10T11 or A25R26K27S28A29 motifs, respectively, frequently propagates binding of heterochromatin binding protein 1 (HP1)-like proteins via their chromodomains, often resulting in repressive chromatin states. Interestingly, slight differences in the motif compositions lead to preferential binding of HP1 to H3K9me3, respectively, or the polycomb protein to H3K27me3 (Fischle et al. 2003). It may be speculated that the affinity of H3.5K27me for HP1-like proteins, if this modification exists in vivo, could be altered through the presence of two exact copies of the ARKST motif.
H3.5 is specific to hominids and is a pseudogene in chimpanzee and bonobo
By similarity searches, we found homologous H3F3C genes on chromosome 12 of the Neanderthal genome (chromosome 12: 31,835,960-31,836,367; http://neandertal.ensemblgenomes.org/index.html), the chimpanzee (Pan troglodytes; Gene ID: 736298) and orangutan (Pongo abelii; Gene ID: 100452761), but not in non-hominid primates such as Macaca mulata (Supporting Data 1). Noteworthy, due to the lack of sequence data, we cannot exclude the existence of H3F3C homologs in lower hominoid taxa, such as gibbons (Hylobatidae). Common features of all homologous H3.5 protein sequences from orangutan to human were duplex ARKST motifs and the presence of asparagine instead of H3K79, whereas the lack of H3K36 or H3K37 was unique to modern human and Neanderthal, which shared identical H3F3C sequences.
Glycine-33 is encoded by H3.3 encoding genes of all great apes and orangutan H3F3C. Instead in H3F3C of modern human and Neanderthal, cysteine-33 is encoded by a TGC triplet. Interestingly, the chimpanzee H3F3C gene clearly seemed to be a pseudogene due to a C to A transversion of the third nucleotide of triplet 33 resulting in a STOP codon (Fig. 1e). To strengthen this finding, we sequenced a fragment of H3F3C from genomic DNA obtained from ejaculate of one bonobo (Pan paniscus) individual (GenBank accession no. HQ873956). Bonobos are closely related to chimpanzee. Our data showed that the bonobo H3F3C possessed an identical STOP codon (Fig. 1e; Supporting Data 1). Thus at least in the Pan genus H3F3C seemed to be a pseudogene. Moreover, we sequenced a H3F3C fragment using genomic DNA obtained from saliva samples of two female gorilla (Gorilla gorilla) individuals (GenBank accession nos. HQ873953, HQ873954, HQ873955). Similarly, like in modern human and Neanderthal, the gorilla H3.5 protein sequence contained a TGC-encoded cysteine-33 strongly suggesting that C33 was ancestral in H3F3C of all African great apes prior to the appearance of a STOP codon in the Pan lineage. Interestingly, in one gorilla individual we sequenced two alleles. Allele A was interrupted by a STOP codon within the α3-helix of the histone fold following isoleucine-124, whereas allele B encoded a full-length H3 homolog. A STOP codon within allele A would possibly lead to a loss-of-function, since the α3-helix is known to be involved in histone heterodimerization during nucleosome assembly (Luger et al. 1997). Only allele A was found in a second gorilla individual.
H3.5 mRNA is specifically expressed in seminiferous tubules of testicular tissue
To exclude that human H3F3C is a pseudogene despite its continuous protein-coding sequence, we undertook expression analyses. First evidence that human H3F3C is expressed in testicular tissue was obtained from database searches for full-length complementary deoxyribonucleic acid (cDNA) clones connected with the NIH Mammalian Gene Collection (MGC) program (Strausberg et al. 2002). To confirm and to extend these data, we decided to monitor the H3.5 mRNA expression in various human primary tissues or cell lines. In initial polymerase chain reaction (PCR)/cloning/sequencing experiments, we found evidence for H3.5 messenger ribonucleic acid (mRNA) expression in human normal testicular cDNA obtained from seminiferous tubules taken from patients that received orchiectomy due to prostate cancer. Follow-up experiments using cDNA from testicular and other tissues showed that primer specificity was insufficient to faithfully distinguish the H3.3/H3F3B mRNA from H3.5 mRNA, even if virtually H3.5-specific primers were used (P1+: 5′-GCA CCC CCT CTA CCT GCG GGG TGA AGC-3′; P1−: 5′-GAC GAC TGC GCT CTG AAA CCT CAG GTC AG-3′) (Fig. 2a). Our interpretation of this problem was that in most tissues the H3.5 mRNA occurred in much lower copy number than H3.3/H3F3B mRNA, thus propagating unspecific primer binding. However, if H3.3/H3F3B cDNA was digested with SmaI endonuclease prior to touchdown PCR under stringent annealing conditions, we specifically identified H3.5 mRNA (∼89% of all sequences determined) in human testicular tissues by direct PCR product sequencing or sequencing of PCR products cloned into the pGEM-T vector (Fig. 2a). Subsequently, we performed quantitative real-time PCR (qRT-PCR) analyses under optimized touchdown PCR conditions using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-actin expression as a calibrators. These analyses showed that the relative rate of H3.5 expression is more than 1,000-fold higher in testicular tissue than in other tissues (Fig. 2a, right handed). Sequencing analyses of PCR products occasionally revealed H3.5 mRNA in leucocytes and primary hepatocytes (each ∼25% of sequences determined)—besides H3.3/H3F3B mRNA, whereas exclusively H3.3/H3F3B mRNA was detected in bronchial epithelial tissue, mucosa epithelium, colon, ileum, brain, ovary, Hek293 cells and HeLa cells (Fig. 2a). To support this finding, we performed RNA FISH on human testicular tissue sections using an H3.5-specific oligonucleotide probe and stringent post-hybridization washing conditions (Fig. 2b, c, d). We observed annular signals within the seminiferous tubules as well as signals colocalizing with Leydig cells (Fig. 2b, d), whereby these results should be interpreted with care with regard to potential slight H3.3 mRNA cross-reactivity. In contrast, no signals exceeding threshold level could be detected in non-testicular tissue sections such as colon or ileum (data not shown). We concluded that H3.5 mRNA is highly enriched in seminiferous tubules of testicular tissue. In non-testicular tissues and some cell lines, the H3.5 mRNA copy number appeared to be very low in comparison with H3.3/H3F3B mRNA. Therefore, in support of database information mentioned above, our data strongly suggested that human H3F3C is not a pseudogene. H3.5 mRNA is specifically expressed in seminiferous tubules of human testes.

H3.5 mRNA is expressed in seminiferous tubules testicular tissue. a Alignment of the H3.5/H3F3C cDNA and (false-positive) H3.3/H3F3B amplicons. Primer positions P1+ and P1− (reverse complement is shown) are indicated. In sequenced PCR products, H3.5 cDNA and H3.3/H3F3B cDNA could be identified by two discrete residues (yellow). The position of a SmaI site in H3.3/H3F3B is shown in red. Two exemplary chromatograms of positive H3.5 cDNA sequences from testicular samples are shown below. H3.5 cDNA was mainly identified in testis, but also in ejaculate, leucocytes and liver. No evidence for H3.5 cDNA was found in other tissues or cell lines (compare table). Results of qRT-PCR demonstrated that relative H3.5 expression is more than 1,000-fold higher in human testes than in non-testicular tissues. In the diagram, the mean value of testicular H3.5 expression was set as 100%. Values of other tissues refer to the testicular value. b RNA FISH using a H3.5 RNA-specific probe revealed annular signals in seminiferous tubules of human testicular sections (“hot” colour in false colour illustration). Signals are also observed in Leydig cells. c Phase contrast illustration of the same section shown in (b). d Confocal single light optical section of a seminiferous tubule showing that the annular signals shown in (b) consist of numerous focal signals (green). Nuclei were visualized by DNA counterstaining with DAPI (blue)
The occurrence of the H3F3C gene possibly resulted from H3F3B gene duplication by retrotransposition and evolved with slightly elevated substitution rate
To elucidate the evolutionary history of H3F3C, we performed phylogenetic analyses using the coding sequences of numerous mammalian histone H3 variants. As described above, the human H3.5 protein most closely resembles H3.3, which in hominids is encoded by two genes on different chromosomes: human H3F3A is on chromosome 1q41, whereas H3F3B is on chromosome 17q25. The phylogenetic tree in Fig. 3a suggests that H3F3C most probably derived from H3F3B. H3F3C genes from modern human, Neanderthal, bonobo, chimpanzee, gorilla and orangutan form a well-supported clade, whereas two rhesus monkey H3 variants, which probably also derived from H3F3B exhibit none of the H3.5 character states, such as asparagine instead of H3K79 or the duplex ARKST motifs. We concluded that these variants evolved through events independent from the H3F3B gene duplication, which eventually resulted in H3F3C occurrence.

A phylogenetic analysis reveals the evolutionary history of H3F3C in hominids. a H3F3C-like genes form a well-supported clade in close neighborhood to H3F3B genes. H3F3C variants (red asterisks) were found in hominids only. Human histone H3 variant genes are coloured in dark blue. Murine H3F3A was used to root the tree, since it is well-known that a H3.3-like variant was ancestral to all animal H3 variants (Postberg et al. 2010). b The cartoon illustrates the large stretch along the H3F3C gene (light green: 5′-UTR or 3′-UTR; dark green: protein coding sequence) that exhibits homology to H3.3/H3F3B mRNA (light blue: 5′-UTR or 3′-UTR; dark blue: protein coding sequence). This high homology region extends the mRNA coding sequence at both sides (see also Supporting Data 2)
Besides some indels, transitions or transversions a large portion of the 5′-UTR of the human H3.3/H3F3B mRNA (NCBI reference sequence: NM_005324.3) is mirrored in the H3F3C gene (∼92% sequence identity). Along the H3F3C gene, a high homology region with the H3.3/H3F3B mRNA extends the H3.5 mRNA coding sequence (1.057 nt) at both sides and stretches over approximately 1.700 nucleotides (Fig. 3b; Supporting Data 2). These observations gave support to the idea that H3F3B gene duplication resulting in H3F3C may have occurred mechanistically via retrotransposition.
Since we assumed that selective pressure on duplicated H3 variant genes could deviate from their ancestral counterparts, we calculated substitution rates for the coding regions of various H3 variants using the distance matrix calculated for phylogenetic analyses and divergence times for Catarrhines as mentioned in (Zalmout et al. 2010). Interestingly, the substitution rate for H3F3B in human, chimpanzee and orangutan is relatively low (0,12–0,20%/Myr), whereby we calculated slightly elevated rates for H3F3C (0,15–0,29%/Myr). For comparison, we determined substitution rates of 0,27–0,54%/Myr for the fast evolving SEMG2 gene in Catarrhines and 0,03–0,10%/Myr for the conserved GAPDH gene in mammals. We conclude that the substitution rate of hominid H3F3C is in the range of moderately evolving genes. Remarkably, the substitution rate in both recently characterized human H3 variants, H3.X and H3.Y, is high with 0,98–1,18%/Myr or 1,00–1,21%/Myr, respectively.
Taken together, our data make a scenario very plausible that H3F3C evolved by gene duplication in an ancestor common to all hominids by gene duplication of H3F3B from chromosome 17 to chromosome 12 as a consequence of retrotransposition. Since then, H3F3C evolved with a moderate substitution rate.
Ectopically expressed GST-H3.5 is assembled into chromatin in mammalian cell lines
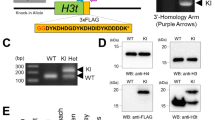
We next made an attempt to characterize the biological relevance of H3.5. Therefore, a glutathione S-transferase (GST)-H3.5 fusion construct was cloned into the pDEST27 expression vector (Invitrogen). For transgenic protein expression, we transfected pDEST27-GST-H3.5 into human cell lines such as HepG2 or Hek293. The occurrence of the ∼41 kDa GST-H3.5 protein was evaluated by Western analyses using whole cell extracts (Fig. 4a, lane 1). Subsequently, we monitored the cellular localization of GST-H3.5 by confocal laser scanning microscopy. These analyses showed that GST-H3.5 was localized to the nucleus. The nuclear distribution of GST-H3.5 was reminiscent of typical chromatin visualized with DNA-selective dyes in both Hek293 cells (Fig. 4b) or HepG2 cells (data not shown). To demonstrate that GST-H3.5 is assembled into chromatin, we performed further Western analyses using nuclear extracts (Fig. 4a, lane 2), or soluble chromatin fractions containing mononucleosomes obtained by Micrococcus nuclease (MNase) digest (Fig. 4a, lane 3). Since GST-H3.5 could be detected in all these fractions, we concluded that it most probably was assembled into chromatin. Immunofluorescence microscopy moreover suggested that GST-H3.5 was segregated to daughter cell nuclei during mitosis, since we observed that GST-H3.5 nicely co-localized with all condensed chromosomes of prospective daughter nuclei during metaphase (Fig. 5a).

GST-H3.5 exhibits nuclear localization and is most probably assembled into chromatin. a Western blot detection of GST-tagged H3.5 in Hek293 cells (Lane 1: whole cell extract; lane 2: nuclear extract; lane 3: soluble chromatin fraction digested with Micrococcus nuclease (MNase)). Protein samples in all lanes correspond to approximately 0.4 × 10^6 Hek293 cells. b Immunofluorescence microscopic image of two out of three Hek293 cells expressing GST-H3.5 (green). H3.5 was exclusively detected in nuclei

GST-H3.5 preferentially colocalizes with euchromatin. The nuclear distribution of GST-H3.5 (green to yellow) was compared with several chromatin markers (red), such as H3K4me3 (a), H3K9ac (b), H3K9me3 (c) or HP1alpha (d). e Semi-quantitative amplification of DNA pulled-down in association with GST-H3.5. Primer pairs used for amplification were (1) human GAPDH promoter, −1.0 kb; (2) human GAPDH promoter, TATA-box; (3) human GAPDH promoter; (4) human LMNA promoter; (5) human TSH2B. Top: input DNA was used as template. Bottom: co-precipitated DNA was used as template. f Quantitative real-time PCR analyses of ChIP experiments. For control chromatin from Hek293 cells expressing GST only was incubated with anti-GST antibodies and analysed as described above. The resulting “percent of input” value (<0.5%) was taken as zero. Columns display “percent of input DNA co-precipitated with GST-H3.5”. g Antibodies targeted to several PTMs react with a ∼41 kDa protein band in Western analyses when MNase digested soluble chromatin was separated with SDS PAGE and blotted onto a PVDF membrane. No signals could be detected using antibodies targeted to H3K36me3 or H3K79me3
GST-H3.5 is preferentially associated with euchromatin and is targeted by various PTM
To study the nuclear localization of GST-H3.5 in detail, we performed co-localization studies with various PTMs or proteins characteristic for permissive (H3K4me3, H3K9ac) or repressive chromatin states (H3K9me3, HP1α). These analyses showed that the nuclear distribution of GST-H3.5 largely overlapped with H3K4me3 (Fig. 5a) or H3K9ac (Fig. 5b). In contrast, GST-H3.5 was excluded from H3K9me3 (Fig. 5c) or HP1α (Fig. 5d) domains. Semi-quantitative chromatin immunoprecipitation (ChIP) analyses (Fig. 5e) as well as combined ChIP and qRT-PCR analyses (Fig. 5f) using mouse anti-GST monoclonal antibodies for immunoprecipitation of ectopically expressed H3.5 support this observation. We observed that sequence motifs of the GAPDH promoter and to some extent also a sequence within the LMNA promoter, which both correspond to regions of actively transcribed chromatin, were enriched in the pulled-down chromatin fraction, whereas TSH2B sequence fragments, corresponding to transcriptionally repressed chromatin, could at best weakly be amplified. These data suggested that GST-H3.5 was enriched in the fraction of actively transcribed euchromatin. To test whether GST-H3.5 assembled into chromatin was targeted by PTM, we performed Western analyses using a MNase-digested chromatin fraction enriched in mononucleosomes. We detected several bands at a size corresponding to the GST-H3.5 fusion protein using antibodies specific to H3K4me3, H3K9ac or H3K9me3 suggesting that GST-H3.5 was targeted by PTM. Remarkably, although our results indicated that GST-H3.5 was preferentially associated with euchromatin, it appeared that it also could be tri-methylated at lysine-9, a repressive chromatin marker. In contrast, we could not detect H3K36me3 or, as it could be expected, H3K79me3.
H3.3, as mentioned above, shows tendency to associate with “open” or transcriptionally competent chromatin. Our observations suggested that H3.5 behaved in a very similar manner. To test whether ectopically expressed H3.5 can replace H3.3 to some extent, we simultaneously knocked down H3.3 expressed by the H3F3A and H3F3B genes, respectively, in Hek293 cells by ribonucleic acid interference. Over several days post transfection of small interfering ribonucleic acid (siRNA), we observed decreased doubling times in H3.3 knockdown cells, whereas doubling times remained relatively constant in cells co-transfected with H3F3A/H3F3B siRNAs and the GST-H3.5 expression vector (Supporting Data 3A) as determined using a Neubauer counting chamber. For further support, we incubated cells 12 h with 10 μM 5-bromo-2-deoxyuridine (BrdU), which was incorporated into nascent DNA 72 h post siRNA transfection. BrdU-labelled DNA was visualized using mouse monoclonal anti-BrdU antibodies (Sigma-Aldrich; clone BU-33, cat. #B2531), showing that siRNA-treated cells exhibited less frequent and apparently less synchronous staining (Supporting Data 3B), whereas most cells co-transfected with H3F3A/H3F3B siRNAs and the GST-H3.5 expression vector frequently showed BrdU incorporation. These data suggested that ectopically expressed H3.5 transiently could replace an essential function of H3.3 in cellular growth. Except amino acid 89, which is isoleucine-89 in H3.3 but valine-89 in H3.5, the putative chaperone recognition sites (residues 86–99) of both H3 variants are identical. Using ConSurf (http://consurf.tau.ac.il/), we performed a structural simulation of H3.5 within the nucleosome using the 1AOI X-ray crystal structure of the nucleosome core particle by Luger and co-workers as a model, which is deposited in the RCSB Protein Data Bank (http://www.pdb.org/pdb/explore/explore.do?pdbId=1AOI). This simulation suggested that H3.5V89 is somewhat buried within the globular domain (Supporting Data 4). It may be speculated that H3.3 and H3.5 could be assembled into nucleosomes by the same molecular machinery.
Conclusions
H3.5 is an evolutionary novelty, as it most probably evolved by H3F3B gene duplication through retrotransposition in a common ancestor of all recent great apes (Hominids). H3.5 is specifically expressed in the seminiferous tubules of human testes. Ectopically expressed H3.5 is assembled into chromatin and is preferentially associated with euchromatin. H3.5 can replace an essential function of H3.3 in cell growth in H3F3A/H3F3B-depleted cells. Moreover, H3.5 is targeted by PTM, whereas some modifications observed in other histone H3 variants are excluded by discrete amino acid substitutions.
Methods
Animals or human specimen preparation and cell culture
Patient’s material was obtained in agreement with the June 1964 Declaration of Helsinki (Title: “Ethical Principles for Medical Research Involving Human Subjects”). In our experiments, we made use of routinely sampled human tissues. All experiments were performed without reference to individual patient’s records. Following surgery, all sampled material was immediately transferred into RNAlater (Qiagen). Bonobo genomic DNA was isolated from a sperm specimen obtained from the male bonobo Lusambo by masturbation in the Wuppertal Zoo. Gorilla genomic DNA was isolated from saliva specimen obtained from two female individuals (Rosalinde and Ukiwa, Wuppertal Zoo). Human HeLa cells were grown in Dulbecco’s complete minimum essential medium (MEM) containing 10% foetal calf serum (FCS). Human Hek293 cells were grown in Dulbecco’s complete MEM supplemented with 15% FCS. We used ectopically expressed tagged H3.5 for experiments in cell lines. Therefore, the H3.5 coding sequence (GeneBank: NM_001013699) was cloned into the GST-encoding pDEST27 mammalian expression vector by LR clonase reaction. Cells were transfected with pDEST27-GST-H3.5 using Fugene 6 transfection reagent (Roche) upon manufacturers’ recommendation. Prior to experiments, cells were grown 36–48 h.
Antibodies
Primary antibodies used in this study were: (1) mouse anti-GST (Cell Signaling Technologies), (2) rabbit anti-H3K4me3 (Diagenode), (3) rabbit anti-H3K9ac (Diagenode), (4) rabbit anti-H3K9me3 (Diagenode), (5) rabbit anti-H3K36me3 (Abcam), (6) rabbit anti-H3K79me3 (Abcam) and (7) rabbit anti-HP1alpha (Active Motif). Secondary antibodies were: (1) rabbit anti-DIG Cy3 (Jackson ImmunoResearch), (2) goat anti-mouse Alexa Fluor 488 (Invitrogen) and (3) goat anti-rabbit Alexa Fluor 555 (Invitrogen).
Mononucleosome preparation, ChIP assays and quantitative real-time PCR analyses
The 5 × 107 to 5 × 108 cells were grown to semi-confluency and then harvested with 2 ml trypsin at 37°C. Subsequently, 5-ml cell culture medium was added. Cells were spinned down at 4,000 g for 5 min at 4°C. The pellet was resuspended in buffer 1 [0.3 M sucrose in 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM ethylene glycol tetraacetic acid (EGTA), 15 mM Tris-HCl (pH 7.5), 0.5 mM dithiothreitol (DTT), 0.1 mM phenylmethanesulfonylfluoride (PMSF), 3.6 ng/mL aprotinin], then 2 ml ice-cold buffer 3 was added [0.3 M sucrose in 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM Tris-HCl (pH 7.5), 0.5 mM DTT, 0.1 mM PMSF, 3.6 ng/mL aprotinin, 0,4% (v/v) Tergitol/Nonidet P40 substituent], mixed gently and incubated on ice for 10 min. In a 15-ml reaction vial, 8 ml of a sucrose cushion (1.6 M sucrose in 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM Tris-HCl (pH 7.5), 0.5 mM DTT, 0.1 mM PMSF, 3.6 ng/mL aprotinin) were carefully layered each with 2 ml of the cell suspension and subsequently centrifuged at 10,000×g for 20 min at 4°C. The supernatant was carefully removed with a Pasteur pipette. Then, the pellet fraction was resuspended in 350 μl MNase shearing buffer (Active Motif), and an aliquot was microscopically checked for the presence of morphologically intact cell nuclei. In some experiments, this fraction was used for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) and subsequent Western analyses. For enrichment of mononucleosomes, 17 μl MNase cocktail (200 U/ml) (Active Motif) was added to pre-warmed nuclei and incubated for 5 min. The reaction was stopped by addition of 7 μl 0.5 M ethylenediaminetetraacetic acid (EDTA) and placed on ice for 10 min. Subsequently, the suspension was centrifuged at 18,000×g for 10 min at 4°C. The supernatant contained the soluble mononucleosome chromatin fraction. The chromatin quality was controlled on an agarose gel, 10 μl of the chromatin fraction was saved as input. In some experiments, this fraction was used for SDS PAGE and subsequent Western analyses.
For ChIP-sheared chromatin corresponding to 25 μg DNA was incubated in a rotator for 16 h at 4°C in a total volume of 200 μl with 20 μl ChIP incubation buffer [50 mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.1 mM PMSF, 5 mM EDTA] and 2 μl of mouse monoclonal anti-GST (26H1) antibody (Cell Signaling Technology, cat. #2624). Subsequently, 25 μl protein G magnetic beads (Active Motif) were added and incubated for 4 h at 4°C rotation. Protein G magnetic beads were separated using a magnetic rack and washed repeatedly. To elute, immunocomplexes were incubated with elution buffer [1% sodium dodecyl sulfate (SDS), 10 mM EDTA, 50 mM Tris-HCl, (pH 8.1)] for 30 min at 65°C on a shaker. Eluted immunocomplexes were treated with proteinase K. DNA was purified using phenol-chloroform extraction and ethanol precipitation.
Quantitative real-time PCR analyses were performed using a Rotorgene 6000 instrument (Qiagen). For PCR reactions, Maxima SYBR Green qPCR Master Mix (Fermentas) containing Hot Start Taq DNA polymerase and SYBR Green fluorescent dye for real-time detection of double-stranded DNA was used as recommended by the manufacturer. To generate standard curves for amplicons, input DNA of known concentration was serially diluted and analyzed in duplicates by qRT-PCR. Using individual standard curves for each amplicon, the relative amounts of specifically immunoprecipitated DNA analyzed by qRT-PCR in duplicates could be estimated as “percent of input”. The following primer pairs optimized for qRT-PCR (Diagenode) analysis were used as follows: (1) human GAPDH promoter, −1.0 kb (cat. #pp-1003); (2) human GAPDH promoter, TATA-box (cat. #pp-1001); (3) human GAPDH promoter (cat. #pp-1044); (4) human LMNA promoter (cat. #pp-1011); and (5) human TSH2B (cat. #pp-1041).
Primers and PCR and qRT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen) upon manufacturer’s recommendations. Subsequently, cDNA was synthesized from 1 μg RNA using the Qiagen QuantiTect Reverse Transcription kit. Prior to H3.5 cDNA, amplification samples were digested with SmaI for 2 h at 37°C to reduce unspecific amplification of H3.3/H3F3B cDNA (Fig. 2a). We performed touchdown PCR under stringent conditions using H3.5-specific primers (P1+: 5′-GCA CCC CCT CTA CCT GCG GGG TGA AGC-3′; P1−: 5′-GAC GAC TGC GCT CTG AAA CCT CAG GTC AG-3′): (1) initial denaturation was done for 15 min at 95°C, followed by 10 touchdown cycles (denaturation: 95°C for 30 s; annealing: 84°C for 30 s [to decrease 1°C per cycle]; elongation: 72°C for 12 s). (2) Subsequently, 25 cycles were performed (95°C for 30 s; 72°C for 40 s), then a final elongation at 72°C for 10 min was allowed. Quantitative real-time PCR analyses were performed using a Rotorgene 6000 instrument (Qiagen). GAPDH and β-actin expression were used as housekeeping genes for calibration. For PCR reactions, Maxima SYBR Green qPCR Master Mix (Fermentas) containing Hot Start Taq DNA polymerase and SYBR Green fluorescent dye for real-time detection of double-stranded DNA was used as recommended by the manufacturer. To generate standard curves for amplicons, input DNA of known concentration was serially diluted and analyzed in duplicates by qRT-PCR. For control, PCR products were directly sequenced each with P1+ or P1− as sequencing primers, or alternatively cloned into pGEM-T easy (Promega) and then, after plasmid purification, sequenced using the T7 forward or M13 reverse primers.
SDS PAGE and Western analyses
Cells were lysed, and subsequently protein fractions were resuspended in loading buffer (Laemmli 1970), heated for 10 min at 100°C, and separated on 15% SDS PAGE. Proteins were then transferred onto a nylon membrane and probed with specific antibodies. Detection was done using the Qdot Western blotting system (Invitrogen).
Fluorescence in situ hybridization
Probes for FISH or RNA FISH were labelled with digoxigenin-dUTP (chromosome #12 paint; H3.5 mRNA probe: 5′-CCC TCT ACC TGC GGG GTG AAG CCT CAT CGC TAC AGG CCT G-3′) or FITC-dUTP (BAC probe H3F3C locus). Prior to hybridization probes and Cot-1 human competitor DNA (Roche) were precipitated with ethanol, air dried and subsequently dissolved in hybridization mixture (50% formamide, 10% dextran sulfate, 2xSSC).
Metaphase chromosomes were spread as described earlier (Stehle et al. 2007). For some experiments, nuclei were permeabilised with 0.5% Triton X-100-phosphate buffered saline (PBS) for 20 min, followed by incubation with 0.1 M HCl for 5 min at room temperature. Preparations were then equilibrated in 50% formamide in 2xSSC for 7 days at 4°C. For DNA denaturation, nuclei or metaphase chromosomes, respectively, were treated with 70% formamide in 2xSSC pH 7.4 (1xSSC is 0.15 M NaCl and 0.015 M Na citrate) for 3 min at 72°C. Nuclei were then briefly washed in ice-cold 70% ethanol followed by 100% ethanol and finally in 2xSSC for 5 min. The probe was denatured simultaneously in a boiling water bath for 10 min, briefly chilled on ice and subsequently loaded onto a coverslip with immobilized and fixed cells/metaphase spreads. Hybridization was performed overnight at 42°C followed by post-hybridization washes in 2xSSC at 42°C and 0.1xSSC at 64°C. Blocking was done in 3% bovine serum albumin (BSA), 0.1% Triton X-100, PBS (blocking solution) for 20 min at room temperature. The rabbit anti-DIG Cy3 antibody was diluted in blocking solution. Incubation was performed for 1.5 h at 37°C.
RNA interference
Expression of H3.3 (H3F3A/H3F3B genes) was silenced using a combination of siRNAs (each 15 pmol/mL): (1) H3F3A sense: AAA UUG CUC AGG ACU UUA Att/H3F3A antisense: UUA AAG UCC UGA GCA AUU Uct; and (2) H3F3B sense: AGG UAC UAG AGU UGA UAA Att/H3F3B antisense: UUU AUC AAC UCU AGU ACC Uta). Transfection was performed using GeneSilencer siRNA Transfection Reagent (Amsbio) upon manufacturer’s recommendations. Transfection rates of 50–60% were achieved as determined by co-transfection of the pmaxGFP expression vector (Lonza) followed by microscopic evaluation.
Immunofluorescence
Cells were grown on coverslips, fixed in 2% paraformaldehyde and then washed twice with phosphate buffered saline. Cells were permeabilised 5 min in PBS/0.5% Triton X-100 and then washed three times with PBS. Prior to antibody incubation, cells were blocked in PBS/0.1% Triton X-100 containing 4% BSA (blocking buffer). Antibodies were diluted in blocking buffer upon manufacturer’s recommendations. Subsequently, immunostaining with specific antibodies was performed for 1.5 h at 37°C. Between applications of primary and secondary antibodies, nuclei were washed in PBS for 20 min.
Confocal laser scanning microscopy
Cells were analyzed by confocal laser scanning microscopy (CLSM). Acquisition of serial sections was done with a Zeiss LSM 5 Pascal confocal laser scanning microscope equipped with a water objective lens (Plan-Neofluar 25/0.8, or in some cases C-Apochromat 63/1.2). Fluorochromes were visualized with an argon laser with excitation wavelengths of 488 nm for Alexa Fluor 488 and 543 nm for Alexa Fluor 555 or Cy3. Fluorochrome images were scanned sequentially generating 8-bit grayscale images. Image resolution was 512 × 512 pixels with variable pixel size depending on the selected zoom factor. The axial distance between light optical serial sections was 300 nm. To obtain an improved signal-to-noise ratio, each section image was averaged from four successive scans. The 8-bit grayscale single channel images were overlaid to an RGB image assigning a false color to each channel and then assembled into tables using open source software ImageJ (Rasband, W.S., ImageJ, National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997–2004.) and Adobe Photoshop CS5 software.
Alignments and phylogenetic analyses
Aligments were performed using ClustalW included in molecular evolutionary genetics analysis (MEGA) 4.1 (Tamura et al. 2007) and subsequently manually refined. Phylogenetic tree calculations were conducted using MEGA 4.1 software. Hereby, the evolutionary relationships H3 variant genes were inferred using the neighbor-joining method (Saitou & Nei 1987). The bootstrap consensus tree was inferred from 5,000 replicates (Felsenstein 1985).
References
Black BE, Bassett EA (2008) The histone variant CENP-A and centromere specification. Curr Opin Cell Biol 20:91–100
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17:1870–1881
Hake SB, Allis CD (2006) Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc Natl Acad Sci USA 103:6428–6435
Henikoff S, Ahmad K (2005) Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol 21:133–153
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Loyola A, Almouzni G (2007) Marking histone H3 variants: how, when and why? Trends Biochem Sci 32:425–433
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260
Olins DE, Olins AL (2003) Chromatin history: our view from the bridge. Nat Rev Mol Cell Biol 4:809–814
Park YJ, Luger K (2008) Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol 18:282–289
Postberg J, Forcob S, Chang WJ, Lipps HJ (2010) The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol Biol 10:259
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Stehle IM, Postberg J, Rupprecht S, Cremer T, Jackson DA, Lipps HJ (2007) Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol 8:33
Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF et al (2002) Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA 99:16899–16903
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Wiedemann SM, Mildner SN, Bonisch C, Israel L, Maiser A, Matheisl S, Straub T, Merkl R, Leonhardt H, Kremmer E et al (2010) Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J Cell Biol 190:777–791
CL,Woodcock, RP Ghosh (2010) Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol 2:10.1101/cshperspect.a000596
Zalmout IS, Sanders WJ, Maclatchy LM, Gunnell GF, Al-Mufarreh YA, Ali MA, Nasser AA, Al-Masari AM, Al-Sobhi SA, Nadhra AO et al (2010) New Oligocene primate from Saudi Arabia and the divergence of apes and old world monkeys. Nature 466:360–364
Acknowledgements
Whole chromosome #12 paint was kindly donated by Dr. Marion Cremer, LMU Munich. Bonobo sperm as well as gorilla saliva samples were kindly provided by Dr. Arne Lawrenz, Wuppertal Zoo. Testicular tissue was sampled and kindly provided by Ulrich Gertenbach and Stephan Roth, HELIOS Klinikum Wuppertal, Urology. This work was supported by the HELIOS Research Center.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Nigg
Electronic supplementary material
Below is the link to the electronic supplementary material.

Supporting Data 1
{kind=link}
Alignment of the protein coding sequences of H3 variant genes used in this study. (JPEG 16451 kb)

Supporting Data 2
{kind=link}
Alignment of the H3F3C gene with the mRNA sequences of H3.5 as well as H3.3/H3F3B. (JPEG 2514 kb)

Supporting Data 3
{kind=link}
Ectopically expressed H3.5 can replace H3.3 to some extent. a Comparison of doubling times in cells co-transfected with H3F3A/H3F3B siRNAs and the GST-H3.5 expression vector and cells transfected with H3F3A/H3F3B siRNAs only at four successive days. b Nascent BrdU-labelled DNA (green) in cells transfected with H3F3A/H3F3B siRNAs. DAPI (blue) was used for DNA counterstaining. c. Nascent DNA (green) in cells co-transfected with H3F3A/H3F3B siRNAs and the GST-H3.5 expression vector. DAPI (blue) was used for DNA counterstaining. (JPEG 476 kb)

Supporting Data 4
{kind=link}
Structural simulation of H3.5 within the nucleosome with emphasis on the chaperone recognition domain. Selected residues are labelled and colour-shaded. (JPEG 809 kb)
Rights and permissions
About this article
Cite this article
Schenk, R., Jenke, A., Zilbauer, M. et al. H3.5 is a novel hominid-specific histone H3 variant that is specifically expressed in the seminiferous tubules of human testes. Chromosoma 120, 275–285 (2011). https://doi.org/10.1007/s00412-011-0310-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-011-0310-4