
Abstract
Chromatin in the interphase nucleus is dynamic, decondensing where genes are activated and condensing where they are silenced. Local chromatin remodelling to a more open structure during gene activation is followed by changes in nucleosome distribution through the action of the transcriptional machinery. This leads to chromatin expansion and looping out of whole genomic regions. Such chromatin loops can extend beyond the chromosome territory. As several studies point to the location of transcription sites inside chromosome territories as well as at their periphery, extraterritorial loops cannot simply be a mechanism for making transcribed genes accessible to the transcriptional machinery and must occur for other reasons. The level of decondensation within an activated region varies greatly and probably depends on the density of activated genes and the number of engaged RNA polymerases. Genes that are silenced during development form a more closed chromatin structure. Specific histone modifications are correlated with gene activation and silencing, and silenced genes may become associated with heterochromatin protein 1 homologues or with polycomb group complexes. Several levels of chromatin packaging are found in the nucleus relating to the different functions of and performed by active genes; euchromatic and heterochromatic regions and the models explaining higher-order chromatin structure are still disputed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Unravelling higher-order chromatin structure
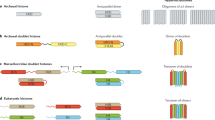
In eukaryotes, DNA is complexed with histones. One hundred and forty-six base pairs of DNA are wound in 1.75 turns around an octamer of the core histones H2A, H2B, H3 and H4 in the nucleosome core particle. The interaction of a linker histone (H1) with the DNA between two core nucleosomes (linker DNA) increases the number of base pairs to 165 corresponding to two turns (Bednar et al. 1998). The addition of linker histone therefore contributes to chromatin condensation (Horn and Peterson 2002). Short linker DNA also contributes to DNA compaction, whereas longer linker DNA has the opposite effect. Thus, the primary level of chromatin structure is represented by the 10-nm chromatin fibre or beads-on-a-string conformation of extended arrays of nucleosomes (Woodcock and Dimitrov 2001) (Fig. 1). Naked B-DNA has a length of 2.9 kb μm−1 and becomes about sevenfold compacted in a 10-nm fibre (Watson and Crick 1953; Goodrich and Tweedie 2002). Secondary chromatin structure is formed by nucleosome interactions, the most prominent of which is a 30-nm-diameter fibre with a compaction of 40- to 50-fold (Woodcock and Dimitrov 2001). The 30-nm fibre is thought to consist of a nucleosome helix, but its exact structure is still being debated (Dorigo et al. 2004). Still, higher levels of chromatin structure are formed by long-range interactions between the 30-nm fibres. A classical model of such higher-order structure is the chromonema fibre in which thinner fibres are folded to yield thicker ones with a diameter of 100 to 130 nm and about 500-fold compaction (Belmont and Bruce 1994). Other models propose radial 30-nm-fibre loops of various lengths connected to a central protein scaffold (Cremer and Cremer 2001). The nucleosome affinity, random chain model, dispenses with 30-nm fibres altogether and assumes random chains of nucleosomes exploring a given space (Müller et al. 2004). None of these models have been proven yet, and all are consistent with the observation that decondensed chromatin forms a series of adjacent beads and that active transcription is required for the maintenance of decondensed chromatin, an observation that has been made repeatedly in different organisms (Tsukamoto et al. 2000; Müller et al. 2001, 2004; Wegel et al. 2005).

Progressive chromatin condensation. The DNA helix with a diameter of 2 nm is wrapped around nucleosomes to yield the beads-on-a-string conformation of the 10-nm chromatin fibre. The latter condenses to a helical stucture, the 30-nm fibre, which can compact into chromonema fibres with a diameter of 100–130 nm
Histones are subject to a variety of post-translational modifications, principally in their conformationally flexible N-terminal tails. Modifications of specific residues include phosphorylation, ubiquitination, acetylation and mono-, di- or tri-methylation. The number of possible permutations of the various modifications is extremely large and has been proposed to constitute a ‘histone code’, an epigenetic mechanism for conferring differing degrees of transcribability on different regions of chromatin (Jenuwein and Allis 2001).
Chromosome territories and the distribution of transcription sites
Each chromosome occupies a distinct space, its own territory, in interphase nuclei (Cremer et al. 1982, 1988). Chromosome territories have been visualized using chromosome-specific in situ paints or by genomic in situ hybridisation (GISH), detecting alien chromosomes in addition lines (Hochstrasser et al. 1986; Abranches et al. 1998; Jin et al. 2000; Cremer and Cremer 2001). These studies have shown variability in shape and positioning of chromosome territories. In yeast, Drosophila and wheat, chromosomes span the width of the nucleus in rod-like structures with the two arms of each chromosome close together and centromeres and telomeres located at opposite poles in a Rabl configuration (Abranches et al. 1998). In contrast, mammalian chromosomes usually have a radial distribution where chromosomes are either peripherally or centrally positioned and have an irregular territory shape (Cremer and Cremer 2001). Selective labelling of individual chromosomes has suggested that chromosome territories are mostly separated into non-overlapping spaces (Visser and Aten 1999; Visser et al. 2000), although definitive proof of this will be hard to obtain.
Incorporation of bromo-UTP in run-on transcription, followed by immunofluorescence detection of the incorporated BrUTP, has shown that transcription occurs in many distinct small foci. In wheat, these transcription sites are uniformly distributed throughout the nucleoplasm, while the nucleoli contain a much higher concentration of transcription sites and are more intensely labelled by BrUTP (Abranches et al. 1998). No evidence has been found for a preferential localisation of transcription sites near the chromosome territorial boundaries or exclusion from the interior of chromosome territories. For mammalian nuclei, a study using BrUTP labelling of nascent transcripts in a cell line expressing green fluorescent protein (GFP) tagged histone H2B showed that, except for nucleoli and speckles, almost all nascent RNA co-localises with chromatin domains and that there is no preferential localisation in chromatin-depleted areas (Sadoni and Zink 2004). However, another study analysing bromo-UTP incorporation at the electron microscope level found newly synthesised RNA mainly at the periphery of condensed chromatin regions in perichromatin fibrils (Cmarko et al. 1999), which might be transcription sites on chromatin loops outside the chromosome territory.
Heterochromatin and euchromatin
Heterochromatin was originally cytologically defined as highly condensed chromatin (Heitz 1928). In Arabidopsis, for example, the heterochromatic knob on chromosome 4 shows about 350-fold condensation (1 Mb μm−1), whereas an adjacent euchromatic region has a condensation ratio of about 60-fold (180 kb μm−1) (Fransz et al. 2002). Heterochromatin is thought to consist of regular nucleosomal arrays, which impede access by nucleases and contain a high proportion of transcriptionally inactive repetitive sequences interspersed by relatively few genes (Elgin and Grewal 2003; Grewal and Moazed 2003). Heterochromatin has traditionally been subdivided into constitutive heterochromatin, chromatin that is always condensed, and facultative heterochromatin, which may decondense in some circumstances. Chromosomal regions around the centromeres and telomeres are examples of constitutive heterochromatin, whereas genes silenced from a certain point in development onwards can form facultative heterochromatin interspersed along chromosome arms. In organisms with large genomes, constitutively heterochromatic regions are also found along chromosome arms. Euchromatin is considered to be decondensed because of irregular nucleosome spacing, is relatively gene rich and is potentially transcriptionally active (Elgin and Grewal 2003). However, these differences are not always clear-cut as a recent analysis of the human genome showed that some pericentromeric regions are decondensed and that some euchromatic regions are condensed (Gilbert et al. 2004). Epigenetic marks of silent chromatin in higher eukaryotes are histone hypoacetylation, di- or trimethylation of lysine 9 at histone H3 (H3K9 di- or trimethylation) as well as cytosine methylation (Fischle et al. 2003; Grewal and Moazed 2003; Wu et al. 2005). Euchromatin is characterised by histone hyperacetylation and dimethylation of lysine 4 at histone H3 (H3K4 dimethylation) (Fischle et al. 2003).
Large-scale chromatin decondensation
Transcription-related chromatin decondensation was first observed when specific transcriptional activators were directed to multi-transgene loci labelled in vivo by GFP and its variants in mammalian cells (Tumbar et al. 1999; Tsukamoto et al. 2000). Although considerable chromatin decompaction was detected by in vivo labelling and confirmed by fluorescence in situ hybridisation, the loci in these studies were highly artificial because they used heterologous sequences and promoters. However, chromatin decondensation was also seen in the developmentally activated, murine HoxB gene cluster (Chambeyron and Bickmore 2004). In addition, the active genes in the HoxB cluster as well as the major histocompatibility complex (MHC) and other gene-rich regions on mammalian chromosomes were found in loops emanating from the chromosome territory (Volpi et al. 2000; Mahy et al. 2002; Chambeyron and Bickmore 2004). In all cases, the frequency with which a genomic region was detected on an external chromatin loop appeared to be related to the number of active genes in that region. In plants, few studies on transcription-related chromatin decondensation have been published so far. The Arabidopsis gene ddm1 encodes a SWI2/SNF2-like chromatin-remodelling factor. In the ddm1 mutant background, transcriptional gene silencing of a transgenic locus is released, the transgenes are transcribed and the locus is decondensed (Probst et al. 2003). In wheat, transgene loci containing developmentally regulated endogenous genes for the seed storage protein glutenin under the control of their own promoters decondense upon transcriptional activation during seed development (Wegel et al. 2005). Based on their size and shape, the decondensed transgene loci appeared to extend beyond the confines of the wheat chromosome territory.
Chromatin has been shown to be transcriptionally active at various degrees of compaction. A highly amplified heterochromatic transgene locus in a mammalian cell line spanning 90 Mb and containing multiple repeats of the lac operator decondensed from about 30,000-fold compaction to 1,000-fold after activation (Tumbar et al. 1999). A 375-kb region of the human MHC showed a packing order of about 100-fold before induction with interferon and about 60-fold after induction (Müller et al. 2004). A higher decondensation ratio than in the mammalian systems can be calculated for the transgene locus in wheat mentioned above (Wegel et al. 2005). This locus consists of about 20 transgene copies of 10 kb each. Fibre spreads of the locus suggested that there are short genomic regions interspersed with the repeated transgenes (EW and PJS, unpublished data). The entire locus, transgene copies and interspersed genomic sequences are probably less than 500 kb and the vast majority of the genes are transcriptionally active. Given the locus sizes visualized by FISH (Wegel et al. 2005), this would correspond to a compaction of about 100-fold before activation and of about 11-fold in its most decondensed form after activation. These values would mean that transcription takes place with the chromatin dispersed almost to the level of the 10-nm fibre. Possibly, the highest degree of decondensation has been found in the active 75S RNA genes in the Balbiani rings of dipterian chromosomes showing a DNA compaction ratio of 3.6, i.e. below the histone-coated 10-nm fibre (Daneholt et al. 1982). The last example illustrates the degree of decondensation in a highly transcribed gene with few nucleosomes left on the chromatin during transcription (Daneholt et al. 1982). Likewise, active rRNA genes each have many engaged RNA polymerases and are decondensed to similar compaction ratios (Gonzalez-Melendi et al. 2001). An explanation for this is that in order for the RNA polymerase to gain access to the template, 30-nm fibres have to uncoil locally followed by histone displacement where the polymerase moves through. How far a gene decondenses then depends on the number of engaged RNA polymerases at any given moment, and packing ratios at transcribed loci reflect the amount of chromatin that is transcriptionally activated in a sequence.
Effectors of chromatin decondensation during gene activation
There is good evidence that during transcriptional activation, chromatin expands in three stages: first, the initial factor (activator) gets access to the nucleosomal DNA; second, chromatin opens locally, mediated by an activator/coactivator; and third, the transcription machinery causes extensive chromatin opening (for extensive reviews see, Lemon and Tjian 2000; Li et al. 2004). Once gene-specific transcriptional activators occupy their binding sites on promoters or enhancers, local chromatin decondensation is mediated by the recruitment of two types of coactivators: an adenosine-5′-triphosphate (ATP) dependent, SWI/SNF-like chromatin-remodelling complex and a histone acetyltransferase (HAT) (Lemon and Tjian 2000; Horn and Peterson 2002; Li et al. 2004). Both HATs and SWI/SNF seem to disrupt higher-order folding of nucleosomal arrays, and SWI-SNF enzymes can also weaken the nucleosome-DNA interaction (Horn and Peterson 2002). The above-mentioned heterochromatic transgene locus spanning 90 Mb and containing multiple repeats of the lac operator decondensed within minutes upon induction with the transcriptional activator (Tumbar et al. 1999). Elevated levels of histone acetylation (acetylated H3K9) were observed early in the activation of the HoxB locus at both hoxb1 and hoxb9 several days before activation of the latter (Chambeyron and Bickmore 2004). Recruitment of a chromatin-remodelling enzyme and two HATs was shown during the induction of a tandem array of the mouse mammary tumour virus promoter (Müller et al. 2001). This study also showed a role for RNA polymerase II in producing and maintaining decondensed chromatin, because decondensation was blocked by two-transcription elongation inhibiting drugs, DRB and α-amanitin. Equally, the frequency of extraterritorial decondensed loops of human genomic regions was reduced when transcription elongation was inhibited by DRB or actinomycin D (Mahy et al. 2002). However, the recruitment of the transcriptional machinery seemed to suffice, and ongoing transcription was not necessary for decondensation of the highly amplified heterochromatic transgene locus mentioned above (Tumbar et al. 1999). Inasmuch as the transgene contruct used in this study contained a high number of activator binding sites, their decondensation upon activator binding may well have drowned out any visible effects of transcription-triggered decondensation. Two more studies have addressed the question of whether decondensation precedes gene expression or is the consequence of it, one in animals and one in plants (Janicki et al. 2004; Wegel et al. 2005). In both, nascent RNA was first detected before visible decondensation at the transgene loci. This may argue against the idea that local decondensation is a prerequisite for transcriptional initiation. However, in both cases, it seems that only a subset of genes in the arrays is activated initially and that large-scale decondensation visible by microscopy does not occur until later when a large proportion of the genes have become activated (Janicki et al. 2004).
Enhancers are cis-acting elements that increase transcription in an orientation- and distance-independent manner. They play a role in chromatin opening by relocating the target gene locus away from heterochromatin (Francastel et al. 1999; Ragoczy et al. 2003), by affecting histone modifications (Chua et al. 2003) or by initiating intergenic transcription (Li et al. 2004). It has been suggested that intergenic and locus control region (LCR) transcription play a role in maintaining an open chromatin structure and, through this mechanism, affect globin gene expression (Gribnau et al. 2000; Plant et al. 2001). The LCR of the β-globin locus acts as an enhancer, and the current model of its action is the formation of a loop bringing the distant LCR and the promoter into close proximity (De Laat and Grosveld 2003), which could actually be described as condensation rather than decondensation. This might also explain that the frequency of looping from the chromosome territory is increased before activation and is reduced during transcription of the locus (Ragoczy et al. 2003). Cis-acting elements may therefore open chromatin locally and at the same time produce a more closed higher-order structure through loop formation. A more detailed analysis of their involvement in gene activation may result in more instances where transcriptional activation does not equal visible chromatin decondensation because of this effect.
Effectors of chromatin condensation during gene-specific silencing
There is accumulating evidence that RNA interference (RNAi) is a pathway by which centromeric heterochromatin is formed in fission yeast, Drosophila, mammals and plants (Matzke and Birchler 2005). In this pathway, the repetitive sequences in pericentromeric regions generate transcripts that form double-stranded RNAs. These are processed into short-interfering (si) RNAs, which in turn trigger silencing of homologous sequences through H3K9 methylation and DNA methylation (Finnegan and Matzke 2003; Craig 2005; Matzke and Birchler 2005). Gene silencing during development is generally initiated by DNA sequence-specific transcription factors that act as transcriptional repressors and bind to gene promoters, recruit histone deacetylases and interact with DNA-methyltransferases and histone methyltransferases (Craig 2005). In some cases, silenced genes are moved into the vicinity of heterochromatin. One example is the brown locus in Drosophila (Dernburg et al. 1996). Another is the mouse terminal transferase gene (Dntt), which becomes silenced during thymocyte (immature lymphocyte) maturation. Silencing starts at the promoter with H3K9 deacetylation, loss of H3K4 methylation and methylation of H3K9, followed by bidirectional spreading of each event (Su et al. 2004). Coincidentally with deacetylation of histone 3, the gene is repositioned to pericentromeric heterochromatin (Su et al. 2004). One factor implicated in repositioning and permanent silencing of genes during lymphocyte development is the Ikaros DNA-binding protein. Ikaros has been shown to interact with chromatin-remodelling components, such as histone deacetylases, it co-localises with many inactive genes in lymphocytes and it can bind to specific sequences in the promoters of many lymphoid-specific genes as well as the repetitive DNA that surrounds mouse centromeres (Fisher and Merkenschlager 2002). It has been suggested that Ikaros might function as a transcriptional repressor and mediate the permanent inactivation of genes through recruitment to heterochromatin domains (Fisher and Merkenschlager 2002).
Several chromosomal proteins have been shown to mediate heterochromatin formation by binding to histones and condensing nucleosomal arrays. Heterochromatin protein 1 (HP1) in Drosophila and mammals is a structural component of silent chromatin at telomeres and centromeres. It was first discovered as a modifier of position effect variation (PEV), the variable expression of heterochromatic and euchromatic genes that have been relocated to the vicinity of a novel breakpoint between heterochromatin and euchromatin created by the relocation (Weiler and Wakimoto 1995). HP1 recognises H3 methylated at lysine 9 by the Drosophila histone methyltransferase SU(VAR)3-9 (Bannister et al. 2001). According to a model proposed by the same authors to explain subsequent heterochromatin spreading, HP1 binds to methylated H3K9 and recruits SU(VAR)3-9. SU(VAR)3-9 then methylates adjacent histones, which allows HP1 to spread linearly along the chromatin fibre. It has recently been shown for Drosophila and mammalian cells that HP1 tethered to a lac operator array causes silencing of downstream reporter genes and local chromatin condensation (Danzer and Wallrath 2004; Verschure et al. 2005). However, HP1 also seems to play a role as transcriptional activator (De Lucia et al. 2005 and references therein). HP1 and SU(VAR)3-9 have homologues in fission yeast, Swi6 and Clr4 (Schramke and Allshire 2003). HP1 also has an Arabidopsis homologue, TFL2 (LHP1), which is involved in, amongst others, the repression of several floral homoeotic genes but does not seem to be responsible for the assembly of constitutive pericentromeric heterochromatin (Gaudin et al. 2001; Kotake et al. 2003; Kim et al. 2004; Lindroth et al. 2004). Polycomb group (PcG) proteins are thought to form several distinct complexes that silence genes responsible for developmental regulation in Drosophila and mammals. They contain another protein with similarity to HP1, polycomb. Like HP1, it binds methylated histones and the histone methyltransferases responsible for their methylation (Craig 2005). It has recently been shown that core components of one of these complexes compact nucleosomal arrays in vitro and do not require histone tails for their action (Francis et al. 2004).
Models of higher-order chromatin structure in a functional context
It is possible that different models of higher-order chromatin structure simply describe different nuclear environments and particular stages in the regulation of genes. The nucleosome affinity, random chain model, might be correct for euchromatic regions with very low heterochromatin content and low compaction, where nucleosomal chains can freely explore nuclear space. Chromonema-type fibres might occur in heterochromatic regions through HP1 dimerization and the ability of HP1 to bring distant chromosomal sites into proximity (Li et al. 2003). The latter could also involve loop formation. As mentioned above for the β-globin locus, the formation of loops of less than 100 kb seems to play a role in bringing cis-regulatory elements into proximity with the genes they control (De Laat and Grosveld 2003; Kato and Sasaki 2005). Loops could also be formed through the Ikaros-mediated recruitment of silenced genes to heterochromatin. It has been shown that gene-rich euchromatic loops with a length of 0.2 to 2 Mbp emanate from a condensed chromocentre that comprises the few heterochromatic regions on chromosome 4, i.e. the pericentromeric regions and the nucleolus organising region (Fransz et al. 2002). This led van Driel and Fransz (2004) to suggest that interphase chromosomes are organised into transcriptionally active loops around heterochromatic centres. Inasmuch as transcription occurs throughout the chromosome territory, extraterritorial loops of active chromosomal regions might have little functional significance. They are only seen in a proportion of nuclei (Mahy et al. 2002) and could simply be a consequence of decondensation per se while the position of the loop with respect to the territory might not matter. The direction of loops might, however, be influenced by the number, position, availability and mode of assembly of transcription sites throughout the nucleus. If transcription sites turn out to be relatively stationary, preassembled and bound to the nuclear matrix, a protein scaffold present in the nucleus, then chromatin needs to be reeled in and passed through the polymerase (Cook 1999; Szentirmay and Sawadogo 2000; Bode et al. 2003). In this case, chromatin will have to be guided to transcription sites and decondensation will be directed. Matrix attachment regions (MARs) are AT-rich sequences that act as anchors to the nuclear matrix. MARs can either form selective and transient or permanent anchors and have been proposed among other functions to facilitate transcription by positioning adjacent genes in the vicinity of the transcriptional machinery (Bode et al. 2003; Heng et al. 2004). It might be the myriad of regulatory and often transient chromatin interactions within a chromosome in addition to MARs–matrix interactions that hold the territory together (Bode et al. 2003; Taddei et al. 2004) (Fig. 2).

Model of interactions that keep the chromosome territory together. The model depicts part of a chromosome arm starting with centromeric heterochromatin. (a) Centromeric heterochromatin made up of chromonema fibres and associated with heterochromatin protein 1. (b) Chromatin loops anchored to the protein structure of the nuclear matrix (grey) via matrix attachment regions (MARs; light blue). (c) Translocation of silenced genes to centromeric heterochromatin via the Ikaros protein (dark blue). (d) Interstitial heterochromatin associated with polycomb. (e) Highly-transcribed region with several transcription factories (green). Transcription factories are assumed to be attached to the nuclear matrix and transcribe to a large extent genes belonging to the same chromosome arm, dark purple gene belonging to a different chromosome (Osborne et al. 2004). (f) Active chromatin hub where positive cis-regulatory elements are in close proximity to the transcriptionally active gene while intervening inactive genes loop-out
Outlook
Clear differences between heterochromatin and euchromatin are disappearing. There is some evidence that HP1 plays a role in silencing in a euchromatic context, which is currently investigated (Danzer and Wallrath 2004). The importance of the RNAi machinery in silencing of euchromatic genes and heterochromatin will probably only increase. The nature and function of nuclear matrix anchors are still debated (Heng et al. 2004). Final proof of the importance of such anchors and of which enzyme complexes are bound to the matrix may give us a better understanding of functional and structural chromatin loops. Some progress has been made towards elucidating the structure of the 30-nm fibre, but the nature of higher-order chromatin folding is still debated and explained by several very different models without conclusive evidence for any of them. The discovery of histone-binding proteins, such as MENT and PcG complexes that contribute to chromatin condensation and in vitro studies of their mode of action, may be the clue to chromatin structure within the interphase nucleus (Springhetti et al. 2003; Francis et al. 2004).
References
Abranches R, Beven AF, Aragón-Alcaide L, Shaw PJ (1998) Transcription sites are not correlated with chromosome territories in wheat nuclei. J Cell Biol 143:5–12
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL (1998) Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci U S A 95:14173–14178
Belmont AS, Bruce K (1994) Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J Cell Biol 127:287–302
Bode J, Goetze S, Heng H, Krawetz SA, Benham C (2003) From DNA structure to gene expression: mediators of nuclear compartmentalization and dynamics. Chromosome Res 11:435–445
Chambeyron S, Bickmore WA (2004) Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev 18:1119–1130
Chua YL, Watson LA, Gray JC (2003) The transcriptional enhancer of the pea plastocyanin gene associates with the nuclear matrix and regulates gene expression through histone acetylation. Plant Cell 15:1468–1479
Cmarko D, Verschure PJ, Martin TE, Dahmus ME, Krause S, Fu XD, van Driel R, Fakan S (1999) Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol Biol Cell 10:211–223
Cook PR (1999) The organization of replication and transcription. Science 284:1790–1795
Craig JM (2005) Heterochromatin—many flavours, common themes. Bioessays 27:17–28
Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2:292–301
Cremer T, Cremer C, Schneider T, Baumann H, Hens L, Kirsch-Volders M (1982) Analysis of chromosome positions in the interphase nucleus of Chinese hamster cells by laser-UV-microirradiation experiments. Hum Genet 62:201–209
Cremer T, Lichter P, Borden J, Ward DC, Manuelidis L (1988) Detection of chromosome aberrations in metaphase and interphase tumor cells by in situ hybridization using chromosome-specific library probes. Hum Genet 80:235–246
Daneholt B, Andersson K, Bjorkroth B, Lamb MM (1982) Visualization of active 75 S RNA genes in the Balbiani rings of Chironomus tentans. Eur J Cell Biol 26:325–332
Danzer JR, Wallrath LL (2004) Mechanisms of HP1-mediated gene silencing in Drosophila. Development 131:3571–3580
De Laat W, Grosveld F (2003) Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11:447–459
De Lucia F, Ni JQ, Vaillant C, Sun FL (2005) HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res 33:2852–2858
Dernburg AF, Broman KW, Fung JC, Marshall WF, Philips J, Agard DA, Sedat JW (1996) Perturbation of nuclear architecture by long-distance chromosome interactions. Cell 85:745–759
Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ (2004) Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306:1571–1573
Elgin SC, Grewal SI (2003) Heterochromatin: silence is golden. Curr Biol 13:R895–R898
Finnegan EJ, Matzke MA (2003) The small RNA world. J Cell Sci 116:4689–4693
Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15:172–183
Fisher AG, Merkenschlager M (2002) Gene silencing, cell fate and nuclear organisation. Curr Opin Genet Dev 12:193–197
Francastel C, Walters MC, Groudine M, Martin DI (1999) A functional enhancer suppresses silencing of a transgene and prevents its localization close to centrometric heterochromatin. Cell 99:259–269
Francis NJ, Kingston RE, Woodcock CL (2004) Chromatin compaction by a polycomb group protein complex. Science 306:1574–1577
Fransz P, De Jong JH, Lysak M, Castiglione MR, Schubert I (2002) Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc Natl Acad Sci U S A 99:14584–14589
Gaudin V, Libault M, Pouteau S, Juul T, Zhao G, Lefebvre D, Grandjean O (2001) Mutations in LIKE HETEROCHROMATIN PROTEIN 1 affect flowering time and plant architecture in Arabidopsis. Development 128:4847–4858
Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA (2004) Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118:555–566
Gonzalez-Melendi P, Wells B, Beven AF, Shaw PJ (2001) Single ribosomal transcription units are linear, compacted Christmas trees in plant nucleoli. Plant J 27:223–233
Goodrich J, Tweedie S (2002) Remembrance of things past: chromatin remodeling in plant development. Annu Rev Cell Dev Biol 18:707–746
Grewal SI, Moazed D (2003) Heterochromatin and epigenetic control of gene expression. Science 301:798–802
Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P (2000) Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell 5:377–386
Heitz E (1928) Das Heterochromatin der Moose, vol 69. Jahrb Wiss Botanik, pp 762–818
Heng HH, Goetze S, Ye CJ, Liu G, Stevens JB, Bremer SW, Wykes SM, Bode J, Krawetz SA (2004) Chromatin loops are selectively anchored using scaffold/matrix-attachment regions. J Cell Sci 117:999–1008
Hochstrasser M, Mathog D, Gruenbaum Y, Saumweber H, Sedat JW (1986) Spatial organization of chromosomes in the salivary gland nuclei of Drosophila melanogaster. J Cell Biol 102:112–123
Horn PJ, Peterson CL (2002) Molecular biology. Chromatin higher order folding—wrapping up transcription. Science 297:1824–1827
Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, Spector DL (2004) From silencing to gene expression: real-time analysis in single cells. Cell 116:683–698
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Jin QW, Fuchs J, Loidl J (2000) Centromere clustering is a major determinant of yeast interphase nuclear organization. J Cell Sci 113(Pt 11):1903–1912
Kato Y, Sasaki H (2005) Imprinting and looping: epigenetic marks control interactions between regulatory elements. Bioessays 27:1–4
Kim JH, Durrett TP, Last RL, Jander G (2004) Characterization of the Arabidopsis TU8 glucosinolate mutation, an allele of TERMINAL FLOWER2. Plant Mol Biol 54:671–682
Kotake T, Takada S, Nakahigashi K, Ohto M, Goto K (2003) Arabidopsis TERMINAL FLOWER 2 gene encodes a heterochromatin protein 1 homolog and represses both FLOWERING LOCUS T to regulate flowering time and several floral homeotic genes. Plant Cell Physiol 44:555–564
Lemon B, Tjian R (2000) Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 14:2551–2569
Li Y, Danzer JR, Alvarez P, Belmont AS, Wallrath LL (2003) Effects of tethering HP1 to euchromatic regions of the Drosophila genome. Development 130:1817–1824
Li YJ, Fu XH, Liu DP, Liang CC (2004) Opening the chromatin for transcription. Int J Biochem Cell Biol 36:1411–1423
Lindroth AM, Shultis D, Jasencakova Z, Fuchs J, Johnson L, Schubert D, Patnaik D, Pradhan S, Goodrich J, Schubert I, Jenuwein T, Khorasanizadeh S, Jacobsen SE (2004) Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. Embo J 23:4286–4296
Mahy NL, Perry PE, Bickmore WA (2002) Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J Cell Biol 159:753–763
Matzke MA, Birchler JA (2005) RNAi-mediated pathways in the nucleus. Nat Rev Genet 6:24–35
Müller WG, Walker D, Hager GL, McNally JG (2001) Large-scale chromatin decondensation and recondensation regulated by transcription from a natural promoter. J Cell Biol 154:33–48
Müller WG, Rieder D, Kreth G, Cremer C, Trajanoski Z, McNally JG (2004) Generic features of tertiary chromatin structure as detected in natural chromosomes. Mol Cell Biol 24:9359–9370
Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36:1065–1071
Plant KE, Routledge SJ, Proudfoot NJ (2001) Intergenic transcription in the human beta-globin gene cluster. Mol Cell Biol 21:6507–6514
Probst AV, Fransz PF, Paszkowski J, Scheid OM (2003) Two means of transcriptional reactivation within heterochromatin. Plant J 33:743–749
Ragoczy T, Telling A, Sawado T, Groudine M, Kosak ST (2003) A genetic analysis of chromosome territory looping: diverse roles for distal regulatory elements. Chromosome Res 11:513–525
Sadoni N, Zink D (2004) Nascent RNA synthesis in the context of chromatin architecture. Chromosome Res 12:439–451
Schramke V, Allshire R (2003) Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science 301:1069–1074
Springhetti EM, Istomina NE, Whisstock JC, Nikitina T, Woodcock CL, Grigoryev SA (2003) Role of the M-loop and reactive center loop domains in the folding and bridging of nucleosome arrays by MENT. J Biol Chem 278:43384–43393
Su RC, Brown KE, Saaber S, Fisher AG, Merkenschlager M, Smale ST (2004) Dynamic assembly of silent chromatin during thymocyte maturation. Nat Genet 36:502–506
Szentirmay MN, Sawadogo M (2000) Spatial organization of RNA polymerase II transcription in the nucleus. Nucleic Acids Res 28:2019–2025
Taddei A, Hediger F, Neumann FR, Gasser SM (2004) The function of nuclear architecture: a genetic approach. Annu Rev Genet 38:305–345
Tsukamoto T, Hashiguchi N, Janicki SM, Tumbar T, Belmont AS, Spector DL (2000) Visualization of gene activity in living cells. Nat Cell Biol 2:871–878
Tumbar T, Sudlow G, Belmont AS (1999) Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J Cell Biol 145:1341–1354
van Driel R, Fransz P (2004) Nuclear architecture and genome functioning in plants and animals: what can we learn from both? Exp Cell Res 296:86–90
Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, van Driel R (2005) In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol 25:4552–4564
Visser AE, Aten JA (1999) Chromosomes as well as chromosomal subdomains constitute distinct units in interphase nuclei. J Cell Sci 112(Pt 19):3353–3360
Visser AE, Jaunin F, Fakan S, Aten JA (2000) High resolution analysis of interphase chromosome domains. J Cell Sci 113(Pt 14):2585–2593
Volpi EV, Chevret E, Jones T, Vatcheva R, Williamson J, Beck S, Campbell RD, Goldsworthy M, Powis SH, Ragoussis J, Trowsdale J, Sheer D (2000) Large-scale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei. J Cell Sci 113(Pt 9):1565–1576
Watson JD, Crick FHC (1953) The structure of DNA. Cold Spring Harb Symp Quant Biol 18:123–131
Wegel E, Vallejos RH, Christou P, Stoger E, Shaw P (2005) Large-scale chromatin decondensation induced in a developmentally activated transgene locus. J Cell Sci 118:1021–1031
Weiler KS, Wakimoto BT (1995) Heterochromatin and gene expression in Drosophila. Annu Rev Genet 29:577–605
Woodcock CL, Dimitrov S (2001) Higher-order structure of chromatin and chromosomes. Curr Opin Genet Dev 11:130–135
Wu R, Terry AV, Singh PB, Gilbert DM (2005) Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol Biol Cell 16:2872–2881
Acknowledgements
Eva Wegel and Peter Shaw were supported by funding from the UK Biotechnology and Biological Science Research Council (BBSRC). We thank Hans de Jong, Paul Fransz, Anne Osbourn and Silvia Costa for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E.A. Nigg
Rights and permissions
About this article
Cite this article
Wegel, E., Shaw, P. Gene activation and deactivation related changes in the three-dimensional structure of chromatin. Chromosoma 114, 331–337 (2005). https://doi.org/10.1007/s00412-005-0015-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-005-0015-7