
Abstract
Most eukaryotic centromeres contain long arrays of tandem repeats, with unit lengths of 150–300 bp. We searched for such repeats in the functional centromeres of the model legume Medicago truncatula (Medicago) accession Jemalong A17. To this end three repeats, MtR1, MtR2 and MtR3, were identified in 20 Mb of a low-pass, whole genome sequencing data set generated by a random shotgun approach. The nucleotide sequence composition, genomic organization and abundance of these repeats were characterized. Fluorescent in situ hybridization of these repeats on chromosomes at meiosis I showed that only the MtR3 repeat, encompassing stretches of 450 kb to more than 1.0 Mb, is located in the functional portion of all eight centromeres. MtR1 and MtR2 occupy distinct regions in pericentromeric heterochromatin. We also studied the presence and distribution of MtRs in Medicago accession R108-1, a genotype with a genome that is 20% smaller than that of Jemalong A17. We determined that while MtR3 is also centromeric on all pachytene bivalents in R108-1, MtR1 and MtR2 are not present in the R108 genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Centromeres are the domains of chromosomes that are responsible for sister chromatid cohesion, kinetochore assembly and spindle attachment, and are essential for proper chromosome segregation during mitosis and meiosis. In most eukaryotes centromeres are embedded within long arrays of tandem DNA repeats (Csink and Henikoff 1998; Heslop-Harrison 2000; Lamb et al. 2004). The lengths of the repeat units show strikingly little variation, likely determined by the nucleosome unit length (150–180 bp) or its multiple (for review see Henikoff et al. 2001). Examples of such repeats are the 171 bp satellite in primates (Wevrick and Willard 1989), the 180 bp pAL1 repeat in Arabidopsis thaliana (Arabidopsis) (Martinez-Zapater et al. 1986; Maluszynska and Heslop-Harrison 1991) and the 155 bp CentO repeat in rice (Oryza sativa) (Cheng et al. 2002). These repeats form long arrays with a head-to-tail orientation and will be referred to here as satellite repeats, irrespective of their nucleotide composition.
The centromeric satellite repeats of different species display little or no sequence homology and even ecotype-specific sequence variants have been reported, as in the case of Arabidopsis (Hall et al. 2003). In contrast to the rapidly evolving centromeric DNA sequences, proteins associated with centromeric sequences can be highly conserved. One example is the centromere-specific histone H3, CENH3, specifically associated with the functional centromere in yeast, animals and plants (see for review: Jiang et al. 2003; Houben and Schubert 2003). But even in the case of CENH3 there is evidence of increased rapid evolution, while other centromere-associated proteins appear to be specific to certain organisms (Henikoff et al. 2001). The occurrence of satellite repeats is not restricted to centromeric regions, as they can also be located in pericentromeric heterochromatin and in non-centromeric heterochromatic regions such as heterochromatic knobs in maize (see for review Heslop-Harrison et al. 2003; Hall et al. 2004).
The legume Medicago truncatula (Medicago) has been selected as a model plant due to its close phylogenetic relationships to numerous crop legume species (Choi et al. 2004) and its well-characterized symbiosis with Rhizobium bacteria. It has a relatively small genome of 560–580 Mb per 1C and eight chromosome pairs (Agarwal and Gupta 1983; Blondon et al. 1994). Chromosome morphology is most distinct in 4′,6-diamidino-2-phenylindole (DAPI)-stained pachytene chromosomes, revealing blocks of heterochromatin in the pericentromeric and nucleolar organizer region, in contrast to the mostly euchromatic chromosome arms. We have identified the MtR1 satellite repeat by end sequencing of randomly selected bacterial artificial chromosome clones, and mapped it to six sites of pericentromeric heterochromatin (Kulikova et al. 2001). We pursued our search for other satellite repeats that are part of the functional centromere itself and the flanking heterochromatic areas. This was done by exploiting sequence data from a low-pass random shotgun survey of the Medicago accession Jemalong A17, revealing two additional satellite repeats, MtR2 and MtR3. The organization of these repeats was analyzed and their chromosomal positions established by fluorescent in situ hybridization (FISH) on pachytene and metaphase chromosomes. As repeats may contribute considerably to the variation in genome size between related taxa, as described for the rice indica and japonica cultivar groups (Ohmido et al. 2000), we compared the distribution of MtRs in Jemalong A17 with that of Medicago accession R108-1, a genotype that possesses a genome 20% smaller than that of Jemalong A17 (Blondon et al. 1994; Hoffmann et al. 1997).
Materials and methods
Plant material
Young leaves of the Medicago accessions Jemalong A17 and R108-1 were used for DNA isolation (Rogers and Bendish 1988) and Southern blot analysis. Young flower buds were used to prepare cell spreads containing pollen mother cells at meiosis (Ross et al. 1996; Kulikova et al. 2001).
DNA sequences and analysis
The procedures for random shotgun cloning, fluorescent-based DNA sequencing and subsequent analysis were described in Pan et al. (1994), Bodenteich et al. (1993) and Sambrook et al. (1989). Briefly, 50 μg portions of leaf genomic DNA of Medicago line A17 were randomly sheared for preparing blunt-end fragments. After T4 DNA polymerase, T4 kinase treatment and gel purification, fragments in the 1–3 kb range were ligated into SmaI-cut, bacterial alkaline phosphatase-treated pUC18 (Pharmacia), and Escherichia coli strain XL1BlueMRF’ (Stratagene) was transformed by electroporation. A random library of approximately 12,000 colonies from each transformation was picked to 384-well plates, grown in Terrific Broth (TB) medium supplemented with 100 μg of ampicillin for 14 h at 37°C with shaking at 250 rpm, and the sequencing templates were isolated by means of the alkaline lysate protocol.
Sequencing reactions were performed as previously described (Chissoe et al. 1995) using either the Applied Biosystems Big Dye 3.0 or Amersham ET terminator sequencing reaction mixes with the M13 universal forward and reverse primers. The reactions were incubated for 60 cycles in a Perkin-Elmer Cetus DNA Thermocycler 9600 at cycle conditions recommended by the manufacturer. Unincorporated dye terminators were removed by ethanol precipitation at 20°C, and after the fluorescent-labeled nested fragment sets had been dissolved in double-distilled water they were resolved by electrophoresis on an ABI 3700 capillary DNA sequencer. After base calling with the ABI Analysis Software, the analyzed data were transferred to a Sun Workstation Cluster and reanalyzed using Phred and assembled using Phrap (Ewing et al. 1998; Ewing and Green 1998). Overlapping sequences and contigs were analyzed using Consed (Gordon et al. 1998).
Southern and dot-blot analysis
Digested genomic DNA (3 μg/lane) was separated on a 1% agarose gel, blotted onto Hybond N+ membranes (Amersham Pharmacia Biotech) and hybridized with the 32P-labeled DNA probes. The probes were made by the polymerase chain reaction (PCR) with primers specific to each satellite repeat using plasmid DNA as template. MtR1 had been subcloned in pGEM-T (Promega) previously (Kulikova et al. 2001). A 283 bp fragment of MtR2 was amplified by PCR with two oligonucleotide primers 5′-tgactagtcaacttaggaac-3′ and 5′-tgagaaattcaagttactgc-3′, while the 169 bp MtR3 fragment was amplified with the primers 5′-tgttcattctagttatgtaatac-3′ and 5′-tatgagacatttagcaagaatc-3′. The PCR fragments were subcloned in pGEM-T. Southern and dot-blot hybridizations were carried out in phosphate–SDS buffer at 65°C for 16 h followed by washing with 2×SSC, 0.1% SDS (10 min), 1×SSC, 0.1% SDS (20 min) and 0.2×SSC, 0.1% SDS (20 min). The membranes were exposed in PhosphoImager cassettes and signals were visualized using a Storm PhosphoImager (Molecular Dynamics). Dilution series (100, 50, 10, 5, 1 and 0.1 ng) of genomic DNA and dilution series (5, 2.5, 1, 0.5, 0.25, 0.1 and 0.01 ng) of plasmid DNA containing a repeat sequence were spotted on a membrane and hybridized with the corresponding repeat. The hybridization signals were quantified with Image Analysis Software (Image Quant TL; Amersham Biosciences). Comparison of the radioactivity bound within the linear range of response was used to estimate the relative amount of the repeats per haploid genome.
Fluorescent in situ hybridization
The FISH protocol used was described in Kulikova et al. (2001). The MtR1, MtR2 and MtR3 probes were labeled by PCR with either digoxigenin-11-dUTP or biotin-16-dUTP (Roche), and clone pCT4.2, containing 5S rDNA (Campell et al. 1992), was labeled by nick translation according to the manufacturer’s protocol (Roche). Biotin-labeled probes were detected by initial binding with Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories), followed by amplification with biotinylated anti-streptavidin (Vector Laboratories, USA) and a second treatment with streptavidin-Cy3. Digoxigenin-labeled probes were detected with sheep anti-digoxigenin-fluorescein (Roche) and amplified with rabbit anti-sheep-FITC (fluorescein isothiocyanate; Jackson Immuno-Research Laboratories). Chromosomes were counterstained with 5 μg/ml DAPI in Vectashield antifade solution (Vector Laboratories). Slides were examined under a Zeiss Axioplan 2 Photomicroscope equipped with epifluorescence illumination, and filter sets for DAPI, FITC and Cy3 fluorescence. Selected images were captured by a Photometrics Sensys 1305×1024 pixel CCD camera and processed with Genus Image Analysis Workstation software (Applied Imaging Corporation, Newcastle upon Tyne, UK). Chromosome lengths and fluorescence signals were measured with Image ProPlus 5.0 (Media Cybernetics, Silver Springs, Md.).
Results
Identification of Medicago satellite sequences
In total 23,544 sequence reads were generated from randomly picked clones, encompassing 20.4 Mb or ∼4% of the Medicago genome. Micropeats software (Parsons 1995) was used to identify three different tandem repeats with unit lengths varying from 166–183 bp. One of these repeats is similar to the previously described repeat MtR1 (166 bp) (Kulikova et al. 2001). Two new repeats, named MtR2 and MtR3, were found that have motif lengths of 183 and 166 bp, respectively. Sequence data from individual reads revealed that MtR1, MtR2 and MtR3 are tandem repeats with a typical head-to-tail orientation. Within each satellite family, several polymorphic variants were found. Therefore we determined a consensus sequence for each of the three repeat elements (Fig. 1). Initial repeat units were used to locate all identical and degenerate units in the complete data set using standard BLAST (Altschul et al. 1990, 1997). The majority-rule consensus sequences of the repeat units were determined by aligning the individual units extracted from the BLAST hits in CLUSTAL W (Thompson et al. 1994).

The consensus sequences of the Medicago satellite DNAs
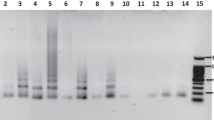
Within individual sequence reads these three repeats are not interrupted by other sequences. To analyze whether MtR1, MtR2 and MtR3 indeed form long tandem arrays, we performed Southern blot analysis of restriction enzyme-digested total genomic DNA. The restriction enzymes that were used have only a single restriction site in the repeat unit. The molecular weights of the laddered hybridizing fragments correspond to monomers and multimers of the appropriate unit size (Fig. 2a), showing that these repeats indeed form long tandem arrays. The observation of multimeric fragments may be explained by sequence heterogeneity between repeats, leading to loss of restriction enzyme sites, or due to chromatin modification (e.g., methylation) resulting in restriction enzyme resistance at certain sites.

Genomic organization of MtR1, MtR2 and MtR3 repeats in Medicago truncatula. a Southern blot analysis of satellite DNAs in the Jemalong A17 genome. Typical long tandem repeat ladder patterns are visible after digestion of genomic DNA and hybridization with the corresponding MtR repeat. b MtR3 is present in the R108-1 genome and is also organized in long arrays. Genomic DNA of both accessions was digested with DraI
Sequence comparison of the three MtR repeat units failed to detect similarity, suggesting that these repeats either arose independently or have diverged significantly since radiation from a common ancestor. To date, only two centromeric satellite repeats have been identified in legumes: the 488 bp tandem repeat pVuKB1 of Vigna unguiculata (Galasso et al. 1995) and a 120 bp repeat from Glycine max (L.) Merr. (Morgante et al. 1997). Pairwise alignments (ClustalW, version 1.82; http://www.ebi.ac.uk/clustalw) between the Medicago repeats and those of V. unguiculata or soybean failed to detect regions of sequence similarity. Furthermore, the MtR satellite DNAs lack detectable similarity to satellite repeats from other organisms.
Quantification of the Medicago satellite repeats
The frequency at which the repeats occur in the 20 Mb of random genome shotgun sequence was used to estimate the percentage of the Medicago genome that is composed of the three satellite repeats. The most abundant repeat is MtR2, constituting 4.5% of the genome, whereas that of MtR1 constitutes 1.1% and that of MtR3, 0.95%. We also determined the fraction of the Medicago genome corresponding to each MtR by means of quantitative dot-blot hybridization. Analysis of hybridization signals revealed that MtR1, MtR2 and MtR3 make up 1.4, 4.4 and 1.1%, respectively, of the Medicago genome. These values closely correspond to those obtained from the sequence data.
Mapping by FISH shows that MtR3 is located at the centromeres
We used FISH to locate the three MtR satellite repeats on Medicago pachytene chromosomes. Late pachytene microsporocytes stained with DAPI display brightly fluorescing (dark blocks in the inverted images) heterochromatic blocks in the pericentromeric regions (Fig. 3a,c,e) and a constriction of slightly weaker stained chromatin within the putative functional centromere (indicated by arrowheads in Fig. 3a). MtR1 was previously shown to be located within pericentromeric heterochromatin on the long arms of chromosome 1, 2 and 8, and on the short arms of chromosome 4 and both arms of chromosome 7 (Fig. 3d). MtR2 is located in all 16 pericentromeric heterochromatic blocks (Fig. 3b) and is typically proximal to that of MtR1, with the exception of chromosome 2 where the positions of the MtR1 and MtR2 regions are reversed (Fig. 3d). MtR3 is located in the centromeric regions (visible as primary constrictions) of all chromosomes (Fig. 3f,g). In addition to its centromeric location, the MtR3 region clearly extends into the pericentromeric regions of chromosomes 2, 3 and 5 (Fig. 5). Further evidence for the location of MtR3 at the functional centromere was obtained from FISH images of meiotic metaphase/anaphase I complements in which the fluorescence signals were located at the centromere domains of the homologous chromosomes pulled to opposite poles, suggesting that MtR3 resides at the kinetochores where spindle fibers are attached (Fig. 3h,i).

Localization of MtR satellites on Jemalong A17 chromosomes by fluorescent in situ hybridization (FISH) and counterstaining with 4′,6-diamidino-2-phenylindole (DAPI). a,c,e Pachytene complements. Centromeres (indicated by arrowheads in a) are located between heterochromatic blocks and visible as primary constrictions. b,d,f FISH of pachytene complements with MtR satellite probes. b MtR2 (red) is located in pericentromeric heterochromatin of all chromosomes. d Localization of MtR1 (green) and MtR2 (red) with respect to centromere. f FISH signals derived from MtR2 (green) and MtR3 (red); note that the MtR3 signal is located between two MtR2 signals on each bivalent. g A merged image of the bivalent chromosomes and the FISH signals. The MtR3 signals coincide with primary constrictions of all chromosomes and also clearly extend to pericentromeric heterochromatin in chromosomes 2, 3 and 5. h,i A pollen mother cell at metaphase I. i Merged image of chromosomes and MtR3 FISH signals. The signals are located on the stretched chromosomal regions. Images of DAPI-stained chromosomes are pseudocolored blue or inverted and in black and white to represent green FISH signals more clearly. Bar represents 5 μm
Occurrence of MtR repeats in Medicago R108-1
Although Medicago accession R108-1 has a markedly smaller genome than Jemalong A17, its total chromosome length at pachytene is hardly different: 406 μm for A17 (Kulikova et al. 2001) and 400 μm for R108-1 (data of ten fully analyzed pachytene cells). However, the relative length of the heterochromatic regions is much smaller in R108-1, namely 14.6% in A17 and 8.4% in R108-1. While R108 has only two loci of 5S rDNA (Fig. 4a), Jemalong A17 has three 5S rDNA loci (Gerbach et al. 1999; Kulikova et al. 2001). To determine whether the three MtR repeats occur in the R108-1 genome we used Southern blot analysis. We demonstrated that MtR1 and MtR2 repeats are both absent from the genome of R108-1. However, Southern blot hybridization with MtR3 reveals the characteristic ladder pattern, indicating that this repeat is present in the R108-1 genome as long tandem arrays (Fig. 2b). Furthermore, FISH on meiotic metaphase I chromosomes showed that MtR3 is located at the centromere positions of all chromosomes (Fig. 4b,c). These data are consistent with an essential function of MtR3 in Medicago centromeres.

Fluorescent in situ hybridization signals of repetitive DNA sequences on meiotic chromosomes of R108-1. a Localization of 5S rDNA (red signals) on pachytene bivalents. b A pollen mother cell at late metaphase I. c MtR3 (green signal) is located at the most poleward positions of metaphase I chromosomes. Bar represents 5 μm
Quantification of MtR3 regions in different chromosomes
To identify all chromosomes we used the 5S rDNA and MtR1 repeats as diagnostic FISH markers (Kulikova et al. 2001). Seven pachytene complements in which all eight bivalents could be unambiguously identified were selected for quantification. The intensities of each MtR3 signal are expressed as a percentage of the sum of the intensities of all MtR3 regions (Table 1). Given the estimate that MtR3 sequences encompass about 1% of the Medicago genome (560 Mb), they should account for 5.6 Mb. The largest MtR3 region occurs on chromosome 5, accounting for 22.3% of the total signal, or an estimated ∼1.2 Mb of contiguous sequence. The MtR3 region of chromosome 5 is detected not only in the primary constriction, but also clearly defines a heterochromatic block on the short arm (Figs. 3e, 5). The centromere of chromosome 3 is predicted to contain ∼0.9 Mb of contiguous MtR3 sequence. Chromosome 2 has ∼0.8 Mb of MtR3 sequence. For chromosomes 2 and 3 the MtR3 signals were localized on both primary constrictions and pericentromeric heterochromatin blocks. The smallest MtR3 blocks were detected in the centromeres of chromosomes 4 and 7, corresponding to an estimated ∼450 kb (∼8%) each. The centromeres of the other chromosomes (1, 6 and 8) have MtR3 regions in the range of 500–600 kb.

Schematic representation of all chromosomes of Medicago Jemalong A17 chromosomes stretched to equal length and showing the positions of the repetitive sequences MtR1, MtR2, MtR3, and 45S and 5S rDNAs in the (peri)centromeric regions
Discussion
In this study we used a combination of sequence analysis and FISH to identify satellite repeats located at the centromeres of Medicago chromosomes. The analysis of 20 Mb of whole genome shotgun sequence resulted in the identification of three so-called satellite repeats. As in other eukaryotes these satellite repeats form long arrays with a typical tail-to-head orientation. One of the repeats, MtR3, co-localizes with all primary constrictions. We speculate that MtR3 forms part of the functional centromere domains because it is located at the most poleward position of metaphase chromosomes during meiosis I. The conservation of MtR3 at the centromere of all eight chromosomes in Medicago genotype R108-1 is consistent with this conclusion.
MtR3 occupies regions of different size on individual Medicago chromosomes, varying from ∼450 bp to more than 1.0 Mb. Study of a Drosophila minichromosome has shown that a 420 kb region of centromeric repetitive DNA is the minimal length required for full centromere activity, since shorter regions lead to a progressive reduction of transmission (Sun et al. 1997). A similar minichromosome-based study in maize also concluded that 500 kb of centromeric satellite repeats is the minimum size necessary for fully functional B centromeres (Kaszas and Birchler 1998). Recently the first entire centromeric region of higher eukaryotic organisms has been sequenced in rice chromosome 8 (Nagaki et al. 2004; Wu et al. 2004). Chromatin immunoprecipitation with anti-CenH3 antibodies revealed that CenH3 is associated with the CentO satellite and the CRR family of retrotransposons and the cenH3-containing region spans ∼750 kb (Nagaki et al. 2004). Thus, the region occupied by MtR3 in Medicago is likely to be of sufficient size to function as a centromere.
Previously we calculated that the majority of the genome (80%) is located in the heterochromatic regions (Kulikova et al. 2001). This calculation was based on the length of pachytene chromosomes (406 μm), euchromatic regions (350 μm) and the condensation degree within one euchromatic region (300 kb/μm). Thus the total euchromatic fraction of Medicago DNA was estimated at 105 Mb (300 kb/μm × 350 μm). Using knowledge of genome size for two Medicago accessions and differences in heterochromatin length we can re-evaluate the size of the heterochromatic fraction of the Jemalong A17 genome. If we assume that the condensation degree of euchromatin is similar in both accessions, then 6% difference in heterochromatin length (14.6% of total length of pachytene bivalents in Jemalong A17 and 8.4% in R108-1) corresponds to the difference of 130 Mb between the Jemalong A17 and R108-1 genomes. Thus, heterochromatin accounts for ∼320 Mb of the Jemalong A17 genome, or 60% of the total genome size. This is consistent with the idea that the major part of the Medicago genome is composed of repeats within heterochromatic regions.
We identified three satellite repeats that together comprise 6.5% of genome. The other known tandem repeats in the Medicago genome are 5S rDNA and 45S rDNA. There are three loci of 5S rDNA and one locus of 45S rDNA on Medicago chromosomes (Gerbach et al. 1999; Kulikova et al. 2001). The frequencies of 5S rDNA and 45S rDNA in the 20 Mb random shotgun dataset are 0.25% and 1.2%, respectively. The positions of all highly repetitive DNA sequences are indicated in Fig. 5. The tandem repeats identified to date make up only ∼8% of the Medicago genome, suggesting that several other heterochromatic repeats, likely including transposons and retrotransposons, remain to be characterized.
References
Agarwal K, Gupta PK (1983) Cytological studies in the genus Medicago Linn. Cytologia 48:781–793
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Nucleic Acids Res 25:3389–3402
Blondon F, Marie D, Brown S, Kondorosi A (1994) Genome size and base composition in Medicago sativa and M. truncatula species. Genome 37:264–275
Bodenteich A, Chissoe S, Wang YF, Roe BA (1993) Shotgun cloning as the strategy of choice to generate templates for high-throughput dideoxynucleotide sequencing. In: Venter JC (ed) Automated DNA sequencing and analysis techniques. Academic, London, pp 42–50
Campell BR, Soung Y, Posch TE, Cullis CA, Town CD (1992). Sequence and organization of 5S ribosomal RNA-encoding genes of Arabidopsis thaliana. Gene 112:225–228
Cheng Z, Donga F, Langdon T, Ouyang S, Buell CR, Gu M, Blattner FR, Jiang J (2002) Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 14:1691–1704
Chissoe SL, Bodenteich A, Wang YF, Wang YP, Burian D, Clifton SW, Crabtree J, Freeman A, Iyer K, Jian L, Ma Y, McLaury HJ, Pan HQ, Sharon O, Toth S, Wong Z, Zhang G, Heisterkamp N, Groffen J, Roe BA (1995) Sequence and analysis of the human ABL gene, BCR gene and regions involved in the Philadelphia chromosomal translocation. Genomics 27:67–82
Choi HK, Mun JH, Kim DJ, Zhu H, Baek JM, Mudge J, Roe B, Ellis THN, Doyle J, Kiss GB, Young ND, Cook DR (2004) Estimating genome conservation between crop and model legume species. Proc Natl Acad Sci USA (in press)
Csink AK, Henikoff S (1998) Something from nothing: the evolution and utility of satellite repeats. Trends Genet 14:200–204
Ewing B, Green P (1998) Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl M, Green P (1998) Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Galasso I, Schmidt T, Pignone D, Heslop-Harrison JS (1995) The molecular cytogenetics of Vigna unguiculata (L.) Walp: the physical organization and characterization of 18S–5.8S–25S rRNA genes, 5S rRNA genes, telomere-like sequences, and a family of centromeric repetitive DNA sequences. Theor Appl Genet 928:935
Gerbach M, Kevel Z, Siljak-Yakovlev S, Kondorosi E, Kondorosi A, Trinh TH (1999) FISH chromosome mapping allowing karyotype analysis in Medicago truncatula Jemalong J5 and R-108-1. Mol Plant Microbe Interact 12:947–950
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Hall SE, Kettler G, Preuss D (2003) Centromere satellites from Arabidopsis populations: maintenance of conserved and variable domains. Genome Res 13:195–205
Hall AE, Keith KC, Hall SE, Copenhaver GP, Preuss D (2004) The rapidly evolving field of plant centromeres. Curr Opin Plant Biol 7:108–114
Henikoff S, Ahmad K, Malik HS (2001) The centromere complex: Stable inheritance with rapidly evolving DNA. Science 293:1098–1102
Heslop-Harrison JS (2000) Comparative genome organization in plants: from sequence and markers to chromatin and chromosomes. Plant Cell 12:617–635
Heslop-Harrison JS, Brandes A, Schwarzacher T (2003) Tandemly repeated DNA sequences and centromeric chromosomal region of Arabidopsis species. Chromosome Res 11:241–253
Hoffmann B, Trinh TH, Leung J, Kondorosi A, Kondorosi E (1997) A new Medicago truncatula line with superior in vitro regeneration, transformation, and symbiotic properties isolated through cell culture selection. Mol Plant Microbe Interact 10:307–315
Houben A, Schubert I (2003) DNA and proteins of plant centromeres. Curr Opin Plant Biol 6:554–560
Jiang J, Birchler J, Parrott W, Dawe R (2003) A molecular view of plant centromeres. Trends Plant Sci 8:570–575
Kaszas E, Birchler JA (1998) Meiotic transmission rates correlate with physical features of rearranged centromeres in maize. Genetics 150:1683–1692
Kulikova O, Gualtieri G, Geurts R, Kim D-J, Cook D, Huguet T, de Jong H, Fransz P, Bisseling T (2001) Integration of the FISH pachytene and genetic maps of Medicago truncatula. Plant J 27:49–58
Lamb JC, Theuri J, Birchler JA (2004) What’s in a centromere? Genome Biol 5:239
Maluszynska J, Heslop-Harrison JS (1991) Localization of tandemly repeated DNA sequences in Arabidopsis thaliana. Plant J 1:159–166
Martinez-Zapater JM, Estelle MA, Somerville CR (1986) A highly repeated DNA sequence in Arabidopsis thaliana. Mol Gen Genet 204:417–423
Morgante M, Jurman I, Zhu T, Keim P, Rafalski JA (1997) The STR120 satellite DNA of soybean: organization, evolution and chromosomal specificity. Chromosome Res 5:363–373
Nagaki K, Cheng Z, Ouyang S, Talbert PB, Kim M, Jones KM, Henikoff S, Buell CR, Jiang J (2004) Sequencing of a rice centromere uncovers active genes. Nat Genet 36:138–145
Ohmido N, Kijima K, Akiyama Y, de Jong JH, Fukui K (2000) Quantification of total genomic DNA and selected repetitive sequences reveals concurrent changes in different DNA families in indica and japonica rice. Mol Gen Genet 263:388–394
Pan HQ, Wang YP, Chissoe SL, Bodenteich A, Wang Z, Iyer K, Clifton SW, Crabtree JS, Roe BA (1994) The complete nucleotide sequence of the SacBII domain of the P1 pAD10-SacBII cloning vector and three cosmid cloning vectors: pTCF, svPHEP and LAWRIST16. GATA 11:181–186
Parsons JD (1995) Miropeats: graphical DNA sequence comparisons. Comput Appl Biosci 11:615–619
Rogers SO, Bendish AJ (1988) Extraction of DNA from plant tissue. In: Gelvin SB, Schilperoort RA (eds) Plant molecular manual. Kluwer, Dordrecht, pp 1–10
Ross K, Fransz PF, Jones GH (1996) A light microscopic atlas of meiosis in Arabidopsis thaliana. Chromosome Res 4:507–516
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, vols 1–3. Cold Spring Harbor Laboratory, NY
Sun X, Wahlstrom J, Karpen GH (1997) Molecular structure of a functional Drosophila centromere. Cell 92:1007–1019
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Wevrick R, Willard HF (1989) Long-range organization of tandem arrays of a satellite DNA at the centromeres of human chromosomes: high frequency array-length polymorphism and meiotic stability. Proc Natl Acad Sci USA 86:9394–9398
Wu J, Yamagata H, Hayashi-Tsugane M, Hijishita S, Fujisawa M, Shibata M, Ito Y, Nakamura M, Sakaguchi M, Yoshihara R, Kobayashi H, Ito K, Karasawa W, Yamamoto M, Saji S, Katagiri S, Kanamori H, Namiki N, Katayose Y, Matsumoto T, Sasaki T (2004) Composition and structure of the centromeric region of rice chromosome 8. Plant Cell 16:967–976
Acknowledgements
This work was financially supported by grants from the FP6 EU project Grain Legumes (FOOD-CT-2004-506223) (to R.G. and T.B.) and CW (NWO) (to T.B.), from the US National Science Foundation (DBI-0110206, to D.R.C. and D.J.K.), and from the Samuel Roberts Noble Foundation (to B.A.R.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Shaw
Rights and permissions
About this article
Cite this article
Kulikova, O., Geurts, R., Lamine, M. et al. Satellite repeats in the functional centromere and pericentromeric heterochromatin of Medicago truncatula. Chromosoma 113, 276–283 (2004). https://doi.org/10.1007/s00412-004-0315-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-004-0315-3