
Abstract
Although the monomer size, nucleotide sequence, abundance and species distribution of tandemly organized DNA families are well characterized, little is known about the internal structure of tandem arrays, including total arrays size and the pattern of monomers distribution. Using our rye specific probes, pSc200 and pSc250, we addressed these issues for telomere associated rye heterochromatin where these families are very abundant. Fluorescence in situ hybridization (FISH) on meiotic chromosomes revealed a specific mosaic arrangement of domains for each chromosome arm where either pSc200 or pSc250 predominates without any obvious tendency in order and size of domains. DNA of rye-wheat monosomic additions studied by pulse field gel electrophoresis produced a unique overall blot hybridization display for each of the rye chromosomes. The FISH signals on DNA fibres showed multiple monomer arrangement patterns of both repetitive families as well as of the Arabidopsis-type telomere repeat. The majority of the arrays consisted of the monomers of both families in different patterns separated by spacers. The primary structure of some spacer sequences revealed scrambled regions of similarity to various known repetitive elements. This level of complexity in the long-range organization of tandem arrays has not been previously reported for any plant species. The various patterns of internal structure of the tandem arrays are likely to have resulted from evolutionary interplay, array homogenization and the generation of heterogeneity mediated by double-strand breaks and associated repair mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The discovery of a high proportion of repeated DNA sequences in eukaryotic genomes (Britten and Kohn 1968) led to one of the first classification schemes, which divided repeats into two major classes according to their organization: (1) dispersed repetitive sequences (those interspersed with genes or other repeat families in different permutations); and (2) tandem arrays consisting of similar monomers monotonously following one another (Flavell 1980). Tandem repeats are now usually subdivided according to the size of monomer such as microsatellite, minisatellite, and satellite DNA (Charlesworth et al. 1994). The last is routinely considered as comprising “typical” or “classic” tandem repeats having a monomer size ranging from 100 to 400 bp. In more recent reports megasatellite DNA consisting of long monomers of several kilobases in length with complex internal structure has been described (Elisaphenko et al. 1998; Gondo et al. 1998; Kojima et al. 2002; O’Hare et al. 2002). Some eukaryotic genes, for example, those encoding the precursor of 18S, 5.8S and 25S ribosomal RNA (rRNA genes or rDNA), are arranged in tandem arrays and are clustered at one or a few sites, most of which are associated with the nucleolar organizing regions (Lapitan 1992). They have been characterized extensively in many plant species (Ellis et al. 1988; Lapitan 1992; Copenhaver and Pikaard 1996). The present discussion will concentrate on classic tandem repeats.
Studies of tandem repeats have generally focused on determining the size of the monomers, their nucleotide sequence, abundance in the genome and species distribution (for review, see Willard 1989; Ugarkovic and Plohl 2002). These general characteristics have been well documented for many species and the α-satellite human DNA is the best-studied family, presented on all human chromosomes and organized in clusters that range in size from 0.5 to 5 Mb (Willard 1989; Lee et al. 1997). As a rule, tandem repeats are localized at centromeres and telomeres and the abundance of tandemly repetitive DNA families corresponds to the size of the heterochromatic regions of chromosomes. In spite of their location near functionally important chromosomal regions, tandemly repeated sequences are commonly regarded as having little or no functional significance and have been proclaimed to be “selfish” or “junk” DNA (Doolittle and Sapienza 1980; Orgel and Crick 1980). Tandemly repeated DNAs show extreme diversity in sequence, copy numbers or both, even among closely related species, and are considered as a fast evolving part of the eukaryotic genome (Waye and Willard 1989; Vershinin et al. 1996; Alexandrov et al. 2002).
The precise sequence of long arrays of such non-coding DNA is notoriously difficult to determine, and despite large-scale genome sequencing projects these regions are generally uncharacterized. Therefore, during the last decade they represented a less explored territory of research than genes or mobile genetic elements. However, it has recently been shown that tandem repeats appear to be major constituents of centromeres in Drosophila melanogaster (Sun et al. 1997), maize (Ananiev et al. 1998a), Arabidopsis thaliana (Heslop-Harrison et al. 1999; Kumekawa et al. 2000), and rice (Nonomura and Kurata 2001; Cheng et al. 2002) and the functional centromere of the human X chromosome consists entirely of a homogeneous 2–4 Mb DXZ1 array of tandemly organized monomers of an X-specific α-satellite DNA subfamily (Schueler et al. 2001). Rapidly changing centromeric tandem DNA may be a driving force of adaptive evolution of centromeric histones, leading to the speciation process (Henikoff et al. 2001).
These findings prompt further attention to tandem repeats, which account for a significant part of the total genome in some species and even the majority of genomic DNA in the kangaroo rat Dipodomys ordii (Hatch and Mazrimas 1974). This class of repeats makes up at least 10% of human DNA (Willard 1989) and the proportion is higher in plant species with large genomes. Large heterochromatic blocks are a major structural characteristic of the rye genome and are present near the telomeres of most chromosome arms. They distinguish rye chromosomes from those of evolutionarily close genera, including wheat and barley. Subtelomeric, or telomere-associated (TA), rye heterochromatin is enriched for a few multi-copy tandemly organized DNA families (Bedbrook et al. 1980; Appels et al. 1981; Jones and Flavell 1982; McIntyre et al. 1990; Vershinin et al. 1995). Most of these families of telomere-associated repeats (TARs) are essentially genus (Secale) specific (Bedbrook et al. 1980). Two of them, pSc200 and pSc250, have monomer lengths of 379 and 571 bp, respectively. We previously demonstrated by double target fluorescence in situ hybridization (FISH) that both sequences occur together and give mostly overlapping strong signals at the ends of most arms of metaphase chromosomes (Vershinin et al. 1995). The overlapping signals did not allow us to dissect the localization of these families relative to each other and internal organization of their prominent hybridization bands.
In spite of the abundance of well-characterized monomers of tandem arrays, little is known about their long-range organization and mutual arrangement within arrays (Zhong et al. 1998). Because of the inherent difficulties of direct sequencing of these genomic regions, important questions remain unresolved. Monosomic additions allow the assignment of specific tandem array organizations to individual chromosomes (de Jong et al. 2000). In particular, it would be interesting to know how the long repetitive arrays of monomers are organized with respect to each other and what are the DNA sequences immediately flanking tandem arrays. Here we addressed these issues for telomere-associated rye heterochromatin using wheat-rye monosomic additions and pulse-field gel electrophoresis (PFGE) and FISH to extended DNA fibres. Fluorescence in situ hybridization to meiotic chromosomes from early prophase provided supplementary information on the large-scale organization of the repetitive arrays in TA heterochromatic regions of individual chromosomes.
Materials and methods
Plant material
For DNA extraction, chromosome slides and fibre DNA preparations, we used seedlings of the seven disomic chromosome wheat-rye addition lines (1R-7R) where pairs of rye (Secale cereale) chromosomes of the variety Imperial were added to wheat (Triticum aestivum) variety Chinese Spring, from Sears’s collection described in Driscoll and Sears (1971). DNA was extracted from etiolated seedlings following Ausubel et al. (1987).
DNA plug preparation, PFGE and blot-hybridization
The isolation of intact protoplasts from wheat-rye addition lines and subsequent mixing with 2% low-melting-point agarose, washing the blocks, and treating high molecular weight DNA with restriction enzymes were performed essentially as described by Cheung and Gale (1990). Gels were run with a CHEF-DRII pulse field electrophoresis system (Bio-Rad) in 1% agarose (Fast-Lane; FMC BioProducts) at 4.5 V/cm and 75 s pulse time, optimizing separation of fragments between 50 and 800 kb. DNA was transferred to a Hybond-N+ membrane. Blot hybridization was conducted at 65°C by the method of Church and Gilbert (1984) with washing once at 65°C in 0.1 M Na2HPO4, 0.1% SDS for 30 min, and once in 0.04 M Na2HPO4, 0.5% SDS for 30 min.
DNA probes and labelling
The telomeric repeat (TR) is contained in the telomeric probe pLT11, which represents the 400 bp insert of pAtT4 from Arabidopsis (Richards and Ausubel 1988) subcloned in pUC19. pLT11 was provided by L. Turner and T.H.N. Ellis (John Innes Centre, Norwich, UK). Two non-homologous repetitive sequences, pSc200 and pSc250, were cloned from S. cereale (Vershinin et al. 1995).
For in situ hybridization, the clones were labelled with digoxigenin-11-dUTP (Roche) and biotin-11-dUTP (Roche) by the polymerase chain reaction (PCR). For blot hybridization, the probes were labelled with 32P-dCTP (Amersham) by random priming.
Cytological preparations
Spikelets from meiotic inflorescences were fixed individually in 3:1 ethanol:acetic acid for at least 3 h and stored at −20°C. Fixed anthers were prepared for FISH following procedures essentially as described in Schwarzacher and Heslop-Harrison (2000). Before FISH, slides were pre-treated with RNase A (10 μg/ml) in 2×SSC for 1 h at 37°C, refixed for 10 min at room temperature in 1% formaldehyde, washed further in 2×SSC, dehydrated in an ethanol series and air-dried. Because there is some variation in stage even within the same anther, we have not rigidly assigned a stage to each cell analysed and have defined them generally as cells in early prophase.
Preparations of extended DNA fibres were made from isolated nuclei according to the protocol of Fransz et al. (1996). Individual 1R chromosomes were prepared by flow cytometric sorting of suspensions of intact chromosomes (Doležel et al. 1999). We are grateful to Dr. J. Doležel (Institute of Experimental Botany, Olomouc, Czech Republic) for providing us with sorted rye chromosomes.
In situ hybridization
Methods for chromosome slide preparation and in situ hybridization were adapted from protocols developed by Schwarzacher and Heslop-Harrison (2000). Briefly, root tips of seedlings were fixed, partially digested with enzymes and cells were spread on slides. Probes were labelled, denatured, applied onto the slides, and probes and slides denatured together at 70°C for 5 min. The concentration of formamide in the hybridization mixture, containing 2×SSC, was 40 or 50%. After overnight hybridization at 37°C, slides were washed, with the most stringent wash at 42°C in 20% formamide, 0.1×SSC. The hybridization signals were detected using anti-digoxigenin conjugated to fluorescein isothiocyanate (Roche). After detection, the slides were washed and analysed with appropriate filters on a Leica epifluorescence microscope.
Polymerase chain reaction, cloning and sequencing
Polymerase chain reaction was used for the analysis of the junction of the pSc200 and pSc250 monomers. Two pairs of primers were designed to different regions of these probes as follows: from pSc200: primer 1, 5′-GCACCAAGGACCTGAAC-3′; primer 2, 5′-CCACCCATGTATGGATAC-3′; primer 3, 5′-CTGTGTGTGCACGTATG-3′; primer 4, 5′-CACGTGCGTGGAAAATTC-3′; from pSc250: primer 1, 5′-GTTCGAAAATAATGGGCC-3′; primer 2, 5′-GTAGAAGAGATGGTATGC-3′; primer 3, 5′-CTTCTAGCGTCTACCACCC-3′; primer 4, 5′-CCAACCACTAAATCATTCG-3′. Polymerase chain reaction was performed with total genomic DNA under the following conditions: 5 min at 94°C; 35 cycles of 30 s at 94°C, 30 s at 56°C and 2 min at 72°C followed by 10 min at 72°C. The resultant products were TA-cloned into pCRII-TOPO vector (Invitrogen).
Sequencing was performed on an ABI377 automated sequencer. The GCG computer software was applied for analysis of primary structure. The occurrence of four different types of short repeats in the DNA spacer localized in pSc200/pSc250 junctions was assessed by measuring the complexity profile (Gusev et al. 1999), which was compared with that of neighbouring regions.
Results
Patterns of large-scale organization of tandem repeats on individual rye chromosomes
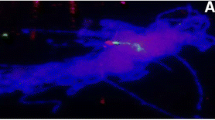
Since pSc200 and pSc250 are not detectable in the wheat genome by blot and in situ hybridization (Vershinin et al. 1996), rye chromosomes can be analysed individually using molecular and cytogenetic methods in wheat-rye addition lines. Meiotic chromosomes at the early prophase stage are less condensed than metaphase chromosomes and provide higher FISH resolution. We performed FISH on meiotic chromosomes of these lines and located pSc200 and pSc250 sequences on individual arms of rye chromosomes 3R, 5R and 7R as shown in Fig. 1. The concentrated spots of hybridization characteristic of metaphase chromosomes were found to be split over disjointed domains, as illustrated for example, by pSc200 on 5R and 7R (Fig. 1a,b,d) or by pSc250 on 3R (Fig. 1e). Some of these domains are separated by non-fluorescent (spacer) regions like several pSc200 domains on the 5R short arm (Fig. 1a). However, more frequently, the pSc200 and pSc250 domains juxtapose forming overlapping regions of yellow fluorescence. We demonstrate different types of domain disposition, namely the pSc250 domain surrounded by the pSc200 domains (Fig. 1a,b,d) and the reverse (Fig. 1e). Hence FISH on meiotic rye chromosomes revealed a specific mosaic arrangement of domains for each chromosome arm.

Fluorescence in situ hybridization on meiotic early prophase chromosomes of wheat-rye addition lines with rye specific tandemly organized DNA families, pSc200 (green) and pSc250 (red). Arrows denote the localization of these sequence domains on: a 5R short arm; b 5R long arm; c 7R short arm; d 7R long arm; 7R has nearly equal arm lengths; this classification is in agreement with Sybenga (1983); e 3R short arm. Bar represents 10 μm
The chromosome specific large-scale organization of the tandem repeat arrays was confirmed by PFGE, which allows separation of large DNA fragments up to several megabases in length and identification of the size of individual arrays. The DNAs of Chinese Spring-Imperial addition lines were cut with the restriction enzymes, BglII, Sau3A and RsaI, which do not cleave within the pSc250 or pSc200 monomers. For all chromosomes blot hybridization with these sequences after BglII digestion yielded strong smears ranging from 40 to 200 kb for pSc250 and longer for pSc200 and many discrete bands on the smear background (Fig. 2). This concentration of signal likely indicates that pSc250 and pSc200 monomers are interspersed within the same arrays and such arrays represent a widespread type of organization. Longer hybridization fragments are unique for each chromosome and consist largely of only pSc250 or only pSc200 monomers. The hybridization patterns obtained after restriction cleavage with 4-base cutters, Sau3A and RsaI, showed a significantly reduced size of the fragments, likely because of cryptic restriction sites within monomers and/or intervening alien DNA sequences in the monomers array. Thus, each wheat-rye addition line studied produced a chromosome specific overall hybridization pattern indicating a unique large-scale organization of the tandem repeat arrays on each rye chromosome.

Pattern of hybridization fragments after pulse field gel electrophoresis of DNAs from wheat-rye addition lines following hybridization with a pSc200 and b pSc250. 1R-6R designate rye chromosomes; 7R is absent. High molecular weight DNAs were digested with B—BglII, Rs—RsaI, S—Sau3A
Heterogeneous internal organization of tandem arrays
We studied the length of individual tandem DNA arrays and the composition and distribution of monomers within arrays by performing dual-label FISH to extended DNA fibres using TR (TTTAGGG) n , and two TARs, pSc200 and pSc250, as probes. Various arrangements of the monomers with respect to each other were visualized for both types of repeats and are summarized in Table 1. For conversion of microscopic length (μm) into molecular size (kb), a stretching degree of 3.27 kb/μm was applied (Fransz et al. 1996), which our measurements (Schwarzacher and Heslop-Harrison 2000) and others using this method show is typical.
The monotonous TR-containing fluorescing tracks, which were measured on the fibres, were shorter than those of the TAR and did not exceed 50 kb (Fig. 3a). This correlates with the rye telomere lengths measured by Bal31 treatment (Vershinin and Heslop-Harrison 1998). The homogeneous continuous dotted tracks consisting of identical monomers of pSc200 or pSc250 have significantly longer spans, from 90 to 600 kb (Fig. 3d). These sizes are consistent with those of the hybridization fragments obtained after PFGE (Fig. 2). The common characteristic of most FISH patterns is the interruption of fluorescing tracks by shorter non-fluorescing spacers, most probably representing other intervening uncharacterised DNA sequences. The spacers have different lengths that, as a rule, do not exceed 35 kb. Furthermore, we found fibres with scattered dots of TR and TAR (Fig. 3c,e), which suggests that these repeats are not always organized as tandemly reiterated monomers and their separate copies can be also dispersed among other types of DNA sequences.

Fluorescence in situ hybridization to extended DNA fibres obtained from wheat-rye monosomic additions. a, b, c Different patterns of telomere repeat (TR) organization: a monotonous tracks; b, c tracks with different spacer sizes (arrows in b); d different patterns of pSc200 tandem arrays. e Example of separate dispersed copies of pSc250; f track of pSc200 monomers (red dots) with separate intruding copies of pSc250 (green dots); g example of junction between pSc200 (red) and pSc250 (green) tracks. 1 buffer zone where monomers of both families are interspersed; 2 homogenous track of pSc250 monomers. h Track of TR (green) with separate dispersed copies of pSc250 monomers (red); i track of pSc250 monomers (red) with separate dispersed copies of TR (green). Bar represents 10 μm
In order to visualize the mutual organization of TR and TAR we next probed pSc200 and pSc250 together and one of them with TR. After double FISH with pSc200 and pSc250, stretches of interspersed double-coloured dots were frequently observed. The heterogeneous arrays show a number of different patterns of interspersion, such as the regular alternation of short stretches of pSc200 and pSc250 monomers, or stretches of monomers from one family being interrupted by single copies of the other repeat (Fig. 3f). The length of heterogeneous arrays, where the pSc200 and pSc250 monomers coexist in mixed order, varied from 60 to 450 kb, with most between 60 and 150 kb, in agreement with the most intensive hybridization signals after PFGE. Long homogeneous stretches of pSc200 and pSc250 were never juxtaposed. They partly overlapped, forming “buffer” zones of different sizes where the monomers of both sequences were interspersed and flanked by uniform blocks of monomers. An example of such buffer zone is given in Fig. 3g.
In spite of the observed localization of pSc200 and pSc250 at the very end of metaphase rye chromosomes, double-labelled FISH on extended DNA fibres did not corroborate the immediate juxtaposition of these sequence arrays with TR stretches. Two rare tracks were visualized: one track of TR that spanned 10 kb and was associated with separate pSc250 copies (Fig. 3h) and a stretch of the pSc250 signal spanned 58 kb and co-localized with the dots produced by TR (Fig. 3i). This indicates that some copies of TR can be scattered among subtelomeric repeat arrays.
Analysis of the pSc200/pSc250 junctions
The features of the pSc200 and pSc250 monomer junction (pSc200/pSc250 junctions) were studied by PCR with primers designed for different regions of monomers following sequencing of PCR products. Then, we cloned the PCR products and repeated the sequencing. Preliminary sequencing of the PCR products before cloning was essential since some products containing several identical monomers appear to be partly deleted or rearranged after transformation. For the final analysis we selected only those clones in which the inserts retained their initial structure.
Two patterns of pSc200/pSc250 junctions were identified: monomers of different families joined directly or joined (or separated) by inserted DNA sequences other than pSc200 and pSc250 (spacers). We did not reveal hotspots in the localization of breakpoints and, correspondingly, junction sites within the pSc200 and pSc250 monomers. All transitions between pSc200 and pSc250 monomers occurred at direct or symmetrical repeats in pSc200 and near one of the 22 inverted repeats of pSc250, which also overlapped with other types of short repeats. The general characteristic of these junctions is short regions of homology, which are present on both sides of the junction and, hence, belong to different monomers. For example, in one of the PCR products, fragment 12 (GenBank Accession number AY522380), the 3′ end truncation of the pSc200 monomer has the sequence 5′-GCTCATCGAAATTT-3′ following the immediate transition to the 5′ end of the pSc250 monomer starting from 5′-TTTCCGAGGCC-3′. Under junction formation one of the TTT triplets was deleted giving the junction sequence GCTCATCGAAATTTCGGAGGCC, the left part of which contains seven nucleotides out of 11 with the exact complement in reverse orientation of the right part, enabling pairing with stem formation. In fragment 13 (AY522381) the junction site is encompassed by imperfect direct repeats, ATACATGG, as the 3′ end of the pSc200 monomer and ATCTGG as the 5′ end of the pSc250 monomer.
The length of analysed fragments ranged from 500 to 1950 bp. Several PCR products containing inserted sequences (spacers) between pSc200 and pSc250 monomers were cloned, sequenced and analysed. Some of them did not reveal homology to known DNA sequences from databases and it is difficult to determine their molecular nature. Other spacers consisted of short DNA pieces of varied length from 35 to 250 bp with varying degrees of sequence identity, 80–90%, to various repetitive elements previously described, such as barley retrotransposons BAGY-1 (Panstruga et al. 1998) and cereba (Presting et al. 1998), or wheat telomere-associated sequence (Mao et al. 1997). Their general pattern is well illustrated by fragment 9 (AY522379) where the sequence between the pSc200 and pSc250 monomers is composed of a 611 bp region of homology to LTR BAGY-1 interrupted in the central part by 217 bp with 86% identity to repetitive DNA from Triticum monococcum (Dvorak et al. 1988).
Several specific features of the primary structure of fragment 2 (AY522378) are noteworthy (Fig. 4a). The spacer is 162 bp in length and represents a hairpin structure with a 30 bp stem of 80% identity. The 5′ end of the stem is derived from the truncated end of pSc200 and the 3′ end has a mosaic structure in which the first half represents the spacer sequence whereas the second half is derived from pSc250. The loop of this hairpin spacer structure is enriched for different types of short repeats even more than the flanking pSc200 and pSc250 sequences. With a minimum size of 7 bp and not less than 85% identity for repeats of up to 15 bp, and not less than 70% for longer repeats, we have found altogether 17 direct, complementary, inverted and symmetrical repeats. Most of these repeats are concentrated within the first 100 nucleotides and eight pairs of the longest direct repeats of 21–25 bp with identity 70% and higher are shown in Fig. 4a. The complexity profile (Gusev et al. 1999) calculated by scanning of the spacer sequence exhibits approximately the same level of complexity as neighbouring pSc200 and pSc250 with respect to inverted and symmetric repeats, whereas complexity with respect to direct repeats is close to the absolute minimum, depicting enrichment of this type of repeat.

Sequence organization and localization of spacer from fragment 2. a A scheme shows the loop structure of the spacer with the stem formed by inverted repeats consisting mostly of regions of the pSc200 and pSc250 monomers. The figures in italics mark the position of nucleotides. The black segment of the loop indicates the spacer region enriched in short direct repeats. Arcs correspond to the eight pairs of these repeats located between nucleotides: (1) 258–280, 279–302; (2) 266–287, 287–309; (3) 294–317, 322–346; (4) 295–316, 324–346; (5) 298–318, 327–348; (6) 300–324, 329–354; (7) 304–327, 333–357; (8) 306–329, 335–359. b Fluorescence in situ hybridization of this spacer on rye chromosome 1R. The chromosome was pre-hybridized with pSc200 (green) and pTa71 (red) shown by arrowhead, partly washed and hybridized with spacer probe (red) shown by arrows. Bar represents 10 μm
Since short regions of sequence similarity are associated with recombination events (for review, see Cromie et al. 2001), this spacer may be a powerful source of tandem array rearrangements and, therefore, we were interested in the evaluation of the approximate copy number of this sequence and its location on rye chromosomes. We performed FISH to rye chromosome 1R using a central part of the spacer sequence as probe. First chromosome 1R was hybridized to pSc200 and pTa71 (ribosomal probe). Then the slides were partially washed and re-probed with spacer probe, which showed two major locations (Fig. 4b). Several dots are visualized at the very end of the long arm and distributed among pSc200 signals. Two clear signals of spacer are also detected at the pericentromeric location.
Discussion
Based on the ladder pattern of blot hybridization, restriction mapping and strong compact FISH signals on metaphase chromosomes, the large-scale organization of tandemly organized repetitive DNA families has been traditionally regarded as long stretches of monotonous, nearly identical monomers. Until recently there have been few studies of the pattern and distribution of elements within tandem arrays by PFGE and FISH on DNA fibres (Zhong et al. 1998) and little is known about the structure of junctions between tandem arrays and other DNAs. There is a large gap between the physical information generated by blot hybridization and cytogenetic studies and the details of repetitive DNA sequences.
We have attempted to bridge this gap by investigation of the large-scale molecular organization of the telomere-associated heterochromatin of rye chromosomes, applying three complementary approaches. The bulk of DNA in the TA heterochromatin is composed of a few families of tandemly repeating units and we have used two of them, pSc200 and psc250, which represent ∼2.5% (pSc200) and ∼1% (pSc250) of the rye genome (Vershinin et al. 1995), as probes. Chromosomes in early meiotic prophase are longer and superior to metaphase chromosomes in FISH resolution (Cheng et al. 2001). Like the regions defined by FISH on metaphase chromosomes, these families are not dispersed, and have rather compact localization but are split into several domains (Fig. 1). Some of these domains are separated by non-fluorescent regions and some of them overlap each other, indicating a more complex pattern of distribution along the chromosomes than initially suggested (Vershinin et al. 1995). This pattern appears to be specific for each chromosome arm as regards domain order, size and spacer distribution.
Using PFGE (Fig. 2), we showed that arrays of pSc200 and pSc250 repeats are distributed across numerous fragment size classes, indicating possible interspersion of monomers within arrays. On the other hand, the presence of many distinct bands of high intensity demonstrates that the larger fragments are relatively homogenous and chromosome specific in accordance with the model of amplification of DNA families through unequal crossing over proposed by Smith (1976). In the light of this, it seems reasonable to propose that the process of homogenization of the given repetitive DNA family under mechanisms of concerted evolution (Dover 1982, 1986) was applied not only to the whole genome but also to single chromosomes during karyotype formation. Homogenization processes appear to be stronger within chromosomes than between chromosomes. Such homogenization would work better within high copy number repeat arrays as demonstrated by our PFGE data (Fig. 2).
Our second approach exploited FISH on DNA fibres, which provided new insights concerning the internal structure of tandem arrays. Fibre-FISH allowed visualization and therefore estimation of the tandem array size (Table 1) and revealed the complexity of their internal organization. Interspersion of different monomers within repetitive arrays was confirmed by this method (Fig. 3). Interspersed arrays of two satellite sequences were described in the Tribolium madens genome where they represent a major component of heterochromatin (Zinic et al. 2000). It is worth emphasizing that long homogeneous tracks of pSc200 and pSc250 were never precisely juxtaposed. They overlapped partly in “buffer” zones where the monomers of both sequences were interspersed (Fig. 3g). Heterogeneity in higher-order organization peculiar to the ends of repetitive arrays is consistent with the same tendency in divergence of monomer primary structure. This gradient observed near the edge of human α-satellite DNA arrays (Schueler et al. 2001) and the three most terminal repeats in the PSR2 array showed the higher sequence divergence relative to internal repeats (McAllister and Werren 1999). The gradient of increasing homogeneity in the large-scale organization of tandem arrays away from the buffer zone again suggests that the arrays have undergone successive rounds of homogenization events by a mechanism of unequal crossing over (Smith 1976).
Recombination between the arrays of different repetitive families resulting in various patterns of interspersion of different monomers as shown in Fig. 3 is not the only mechanism that would disturb homogenization of tandem arrays and their monotonous structure. Most of the fibre-FISH patterns consisted of continuous dotted tracks where signals were separated by non-fluorescent spacers of different size with various alterations. The spacers are characteristic features of both TARs and, to a lesser extent, TRs, and several examples of such patterns are given in Fig. 3. Earlier Zhong et al. (1998) demonstrated the occurrence of spacers in TR and TAR arrays in heterochromatin of tomato chromosomes. In general, different classes of transposable elements (TEs), including LTR-containing retrotransposons, LINEs and SINEs, represent another class of repetitive DNA that is disproportionately abundant in heterochromatin (Charlesworth et al. 1994). One possible reason for accumulation of TEs within noncoding regions such as tandem DNA is that there will be fewer deleterious consequences to fitness of TE insertions and hence this will not be opposed by selection. It has been shown that the retrotransposon Athila is frequently associated with a 180 bp repeat in pericentromeric regions of all Arabidopsis chromosomes (Pelissier et al. 1996). More studies on maize and rice confirmed the general pattern of centromere structure of higher plants where tandem repeats and retrotransposons are major components (Cheng et al. 2002; Zhong et al. 2002). In addition, basic 180 bp repeats isolated from maize knob heterochromatin form tandem arrays of different lengths that are interrupted by different types of other repeated sequences, mostly retrotransposons (Ananiev et al. 1998b).
In an attempt to dissect the molecular nature of spacer sequences and junctions between the tandem arrays, we applied PCR following cloning and sequencing of the PCR products. This approach revealed that, similar to pericentromeric and knob heterochromatin, TA rye heterochromatin contains a variety of repetitive sequences, a number of which show similarity to earlier identified cereal retrotransposons. The primary structure of some PCR products with scrambled regions of similarity to various known repetitive elements indicates that the formation of pSc200/pSc250 junctions through the spacers involves the concerted action of several DNA break-repair mechanisms. The basic recombination pathway repairing DNA double-strand breaks (DSBs) in plant cell is illegitimate recombination (IR) (reviewed in Gorbunova and Levy 1999; Vergust and Hooykaas 1999), which proceeds at frequencies that are several orders of magnitude higher than homologous recombination (HR) (Puchta and Hohn 1996; Vergust and Hooykaas 1999). Certain features in genomic sequences such as inverted repeats and AT-rich regions are thought to boost IR (Muller et al. 1999). Illegitimate recombination may be mediated by homology-dependent single-strand annealing and synthesis-dependent strand annealing, involving cis or trans multiple template switches (Gorbunova and Levy 1999), and by a double-stranded end-joining mechanism mediated by a complex of proteins including the DNA-binding heterodimer Ku 70/80 (Haber 2000). The latter mechanism is referred to as non-homologous end joining. All mechanisms may result in IR junctions consisting of complex filler (spacer) DNA between the non-contiguous fragments.
The analysis of the primary structure of pSc200/pSc250 junctions showed that most break points occurred at different sites but in close proximity to various types of short repeats. Also, the monomers contain blocks of adenines, in particular, pSc200 has five blocks with 5–6 consecutive adenines. The disposition of tandem arrays near telomeres implies their accessibility to the large amounts of Ku 70/80 protein because the latter is localized on telomeres, which play an important role in their maintenance (Hsu et al. 1999). Hence, in rye TA heterochromatin all the essential attributes for tandem arrays to be fragmented and undergo IR are present. The presence of microhomologies in the majority of our junctions or in their vicinity indicates that either any, or a combination, of these mechanisms may be involved in spacer formation. The scrambled structure of most spacers suggests that genomic fragments are integrated into DSBs as filler DNA reminiscent of the extensively scrambled regions of some transgene loci (Svitashev et al. 2002) or rearranged retrotransposons (Vershinin and Ellis 1999).
Although DSB repair is primarily mediated by IR, HR can make an important contribution as well, particularly if genomic DSBs are induced in close proximity to direct repeats (Siebert and Puchta 2002). The structure of the spacer in fragment 2 represents a hairpin loop with the stem formed mostly by adjoined regions of pSc200 and pSc250 monomers (Fig. 4a). This palindromic structure is expected to be prone to IR (Muller et al. 1999), frequently accompanied by gross deletions and translocations (Chuzhanova et al. 2003). The spacer structure is thus enriched in direct repeats and has the potential to be involved in both homologous and illegitimate recombination and rearrangement. It was shown that both repair pathways are partially complementary with each other in maintaining chromosomal DNA during the cell cycle as well as in repairing DSBs induced by γ-radiation (Takata et al. 1998). The numerous gross rearrangements in rye TA heterochromatin including deletions, translocations and other karyotypic structural changes were well documented by C-banding and FISH on metaphase chromosomes (Gustafson et al. 1983; Lapitan et al. 1984, 1986). For example, 12 of the 13 breakpoints in the chromosomes involved in translocations and deletions were in heterochromatic regions (Lapitan et al. 1984). In the light of this, the chromosomal localization of spacer in fragment 2 is noteworthy. On rye chromosome 1R several dots are detected at the very end of the long arm and distributed among pSc200 signals while two dots are localized at the pericentromeric region (Fig. 4b). Since pSc200 and pSc250 were detected only in TA rye heterochromatin, the spacer localization elsewhere in the genome means that the spacer sequences are not necessarily specific for tandem arrays. This conclusion is confirmed by the results of PCR with wheat DNA and FISH on metaphase wheat chromosomes, which revealed the presence of the spacer sequence in the wheat genome as well (data are not shown). The distribution of the spacer signal on rye chromosome 1R (Fig. 4b) is in good agreement with the localization of the breakpoints for deletions in TA heterochromatin (Gustafson et al. 1983) and for translocations of the 1R long arm with different wheat chromosomes, with apparent 1R centromeric breakpoints in most translocations (Lapitan et al. 1986). However, this is only coincidence in localization, and the direct involvement of spacer sequence in the production of DSBs accompanied by translocations needs to be checked by methods with higher resolution.
In summary, this level of complexity in the long-range organization of tandem arrays has not been reported previously. The various patterns of internal structure of arrays probably resulted from evolutionary interplay of the opposing forces of array homogenization and DSB-related repair mechanisms generating increased structural heterogeneity.
References
Alexandrov I, Kazakov A, Tumeneva I, Shepelev V, Yurov Y (2002) Alpha-satellite DNA of primates: old and new families. Chromosoma 110:253–266
Ananiev EV, Phillips RL, Rines HW (1998a) Chromosome-specific molecular organization of maize (Zea mays L.) centromeric regions. Proc Natl Acad Sci USA 95:13073–13078
Ananiev EV, Phillips RL, Rines HW (1998b) Complex structure of knob DNA on maize chromosome 9: retrotransposon invasion into heterochromatin. Genetics 149:2025–2037
Appels R, Dennis ES, Smith DR, Peacock WJ (1981) Two repeated DNA sequences from the heterochromatic regions of rye Secale cereale chromosomes. Chromosoma 84:265–277
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1987) Current protocols in molecular biology. Greene/Wiley Interscience, New York
Bedbrook JR, Jones J, O’Dell M, Tompson R, Flavell R (1980) A molecular description of telomeric heterochromatin in Secale species. Cell 19:545–560
Britten RJ, Kohn DE (1968) Repeated sequences in DNA. Science 161:529–541
Charlesworth B, Sniegowski P, Stephan W (1994) The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371:215–220
Cheng Z, Buell CR, Wing RA, Gu M, Jiang J (2001) Toward a cytological characterization of the rice genome. Genome Res 11:2133–2141
Cheng Z, Dong F, Langdon T, Ouyang S, Buell CR, Gu M, Blattner FR, Jiang J (2002) Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 14:1691–1704
Cheung WY, Gale MD (1990) The isolation of high molecular weight DNA from wheat, barley and rye for analysis by pulse-field gel electrophoresis. Plant Mol Biol 14:881–888
Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 81:1991–1995
Chuzhanova N, Abeysinghe SS, Krawczak M, Cooper DN (2003) Translocation and gross deletion breakpoints in human inherited disease and cancer II: potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat 22:245–251
Copenhaver GP, Pikaard CS (1996) Two-dimensional RFLP analyses reveal megabase-sized clusters of rRNA gene variants in Arabidopsis thaliana, suggesting local spreading of variants as the mode for gene homogenization during concerted evolution. Plant J 9:273–282
Cromie GA, Connelly JC, Leach DRF (2001) Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell 8:1163–1174
Doležel J, Macas J, Lucretti S (1999) Flow analysis and sorting of plant chromosomes. In: Robinson JP, Darzynkiewicz Z, Dean PN, Dressler LG, Orfao A, Rabinovitch PS, Stewart CC, Tanke HJ, Wheeless LL (eds) Current protocols in cytometry. Wiley, New York, pp 5.3.1.–5.3.33
Doolittle WF, Sapienza C (1980) Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603
Dover GA (1982) Molecular drive: a cohesive mode of species evolution. Nature 299:111–117
Dover GA (1986) Molecular drive in multigene families: how biological novelties arise, spread and are assimilated. Trends Genet 2:159–165
Driscoll CS, Sears ER (1971) Individual addition of the chromosomes of Imperial rye to wheat. Agron Abstr 1971:6
Dvorak JK, McGuire PE, Cassidy B (1988) Apparent sources of the A genome of wheat inferred from polymorphism in abundance and restriction fragment length of repeated nucleotide sequences. Genome 30:680–689
Elisaphenko EA, Nesterova TB, Duthie SM, Ruldugina OV, Rogozin IB, Brockdorff N, Zakian SM (1998) Repetitive DNA sequences in the common vole: cloning, characterization and chromosome localization of two novel complex repeats MS3 and MS4 from the genome of the East European vole Microtus rossiaemeridionalis. Chromosome Res 6:351–360
Ellis THN, Lee D, Thomas CM, Simpson PR, Cleary WG, Newman M-A, Burcham KWG (1988) 5S rRNA genes in Pisum: sequence, long range and chromosomal organization. Mol Genet Genomics 214:333–342
Flavell R (1980) The molecular characterization and organization of plant chromosomal DNA sequences. Annu Rev Plant Physiol 31:569–596
Fransz PF, Alonso-Blanco C, Liharska TB, Peeters AJM, Zabel P, de Jong JH (1996) High resolution physical mapping in Arabidopsis thaliana and tomato by fluorescence in situ hybridization to extended DNA fibre. Plant J 9:421–430
Gondo Y, Okada T, Matsuyama N, Saitoh Y, Yanagisawa Y, Ikeda J-E (1998) Human megasatellite DNA RS447: copy-number polymorphisms and interspecies conservation. Genomics 54:39–49
Gorbunova V, Levy AA (1999) How plants make ends meet: DNA double-strand break repair. Trends Plant Sci 4:263–269
Gusev VD, Nemytikova LA, Chuzhanova NA (1999) On the complexity measures of genetic sequences. Bioinformatics 15:994–999
Gustafson JP, Lukaszewski AJ, Bennett MD (1983) Somatic deletion and redistribution of telomeric heterochromatin in the genus Secale and in Triticale. Chromosoma 88:293–298
Haber JE (2000) Partners and pathways repairing a double-strand break. Trends Genet 16:259–264
Hatch FT, Mazrimas JA (1974) Fractionation and characterization of satellite DNAs of the kangaroo rat (Dipodomys ordii). Nucleic Acids Res 1:559–575
Henikoff S, Ahmad K, Malik HS (2001) The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293:1098–1102
Heslop-Harrison JS, Murata M, Ogura Y, Schwarzacher T, Motoyoshi F (1999) Polymorphism and genomic organization of repetitive DNA from centromeric regions of Arabidopsis chromosomes. Plant Cell 11:31–42
Hsu H-L, Gilley D, Blackburn EH, Chen D (1999) Ku is associated with the telomere in mammals. Proc Natl Acad Sci USA 96:12454–12458
Jones JDG, Flavell R (1982) The mapping of highly repeated DNA families and their relationship to C-bands in chromosomes of Secale cereale. Chromosoma 86:595–612
de Jong JH, Zhong X-B, Fransz PF, Wennekes-van Eden J, Jacobsen E, Zabel P (2000) High resolution FISH reveals the molecular and chromosomal organization of repetitive sequences of individual tomato chromosomes. In: Olmo E, Redi CA (eds) Chromosomes Today, vol 13. Birkhauser, Switzerland, pp 267–275
Kojima KK, Kubo Y, Fujiwara H (2002) Complex and tandem repeat structure of subtelomeric regions in the Taiwan cricet, Teleogryllus taiwanemma. J Mol Evol 54:474–485
Kumekawa N, Hosouchi T, Tsuruoka H, Kotani H (2000) The size and sequence organization of the centromeric region of Arabidopsis thaliana chromosome 5. DNA Res 8:285–290
Lapitan NLV (1992) Organization and evolution of higher plant nuclear genomes. Genome 35:171–181
Lapitan NLV, Sears RG, Gill BS (1984) Translocations and other karyotypic structural changes in wheat × rye hybrids regenerated from tissue culture. Theor Appl Genet 68:547–554
Lapitan NLV, Sears RG, Rayburn AL, Gill BS (1986) Wheat-rye translocations. J Hered 77:415–419
Lee C, Wevrick R, Fisher RB, Ferguson-Smith MA, Lin CC (1997) Human centromeric DNAs. Hum Genet 100:291–304
Mao L, Devos KM, Zhu l, Gale M (1997) Cloning and genetic mapping of wheat telomere-associated sequences. Mol Genet Genomics 254:584–591
McAllister BF, Werren JH (1999) Evolution of tandemly repeated sequences: what happens at the end of an array? J Mol Biol 48:469–481
McIntyre CL, Pereira S, Moran LB, Appels R (1990) New Secale cereale (rye) DNA derivatives for the detection of rye chromosome segments in wheat. Genome 33:317–323
Muller AE, Kamisugi Y, Gruneberg R, Niedenhof I, Horold RJ, Meyer P (1999) Palindromic sequences and A+T elements promote illegitimate recombination in Nicotiana tabacum. J Mol Biol 291:29–46
Nonomura K-I, Kurata N (2001) The centromere composition of multiple repetitive sequences on rice chromosome 5. Chromosoma 110:284–291
Orgel LE, Crick FHC (1980) Selfish DNA: the ultimate parasite. Nature 284:604–607
O’Hare K, Chadwick BP, Constantinou A, Davis AJ, Mitchelson A, Tudor M (2002) A 5.9-kb tandem repeat at the euchromatin–heterochromatin boundary of the X chromosome of Drosophila melanogaster. Mol Genet Genomics 267:647–655
Panstruga R, Buschges R, Piffanelli P, Schulze-Lefert P (1998) A contiguous 60 kb genomic stretch from barley reveals molecular evidence for gene islands in a monocot genome. Nucleic Acids Res 26:1056–1062
Pelissier T, Tutois S, Tourmente S, Deragon JM, Picard G (1996) DNA regions flanking the major Arabidopsis thaliana satellite are principally enriched in Athila retroelement sequences. Genetica 97:141–151
Presting GG, Malysheva L, Fuchs J, Schubert I (1998) A Ty3/gypsy retrotransposon-like sequence localizes to the centromeric regions of cereal chromosomes. Plant J 16:721–728
Puchta H, Hohn B (1996) From centimorgans to basepairs: homologous recombination in plants. Trends Plant Sci 1:340–348
Richards EJ, Ausubel FM (1988) Isolation telomeric repeats within the Arabidopsis thaliana genome. Cell 53:127–136
Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF (2001) Genomic and genetic definition of a functional human centromere. Science 294:109–115
Schwarzacher T, Heslop-Harrison JS (2000) Practical in situ hybridization. BIOS, Oxford
Siebert R, Puchta H (2002) Efficient repair of genomic double-strand breaks by homologous recombination between directly repeated sequences in the plant genome. Plant Cell 14:1121–1131
Smith GP (1976) Evolution of repeated DNA sequences by unequal crossover. Science 191:528–535
Sun X, Wahlstrom J, Karpen G (1997) Molecular structure of a functional Drosophila centromere. Cell 91:1007–1019
Svitashev SK, Pawlowski WP, Makarevitch I, Plank AW, Somers DA (2002) Complex transgene locus structures implicate multiple mechanisms for plant transgene rearrangement. Plant J 32:433–445
Sybenga J (1983) Rye chromosome nomenclature and homoeology relationships. Z Pflanzenzuchtg 90:297–304
Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17:5497–5508
Ugarkovic D, Plohl M (2002) Variation in satellite DNA profiles—causes and effects. EMBO J 21:5955–5959
Vergust AC, Hooykaas PJJ (1999) Recombination in the plant genome and its application in biotechnology. Crit Rev Plant Sci 18:1–31
Vershinin AV, Ellis THN (1999) Heterogeneity of the internal structure of PDR1, a family of Ty1/copia-like retrotransposons in pea. Mol Genet Genomics 262:703–713
Vershinin AV, Heslop-Harrison JS (1998) Comparative analysis of the nucleosomal structure of rye, wheat and their relatives. Plant Mol Biol 36:149–161
Vershinin AV, Schwarzacher T, Heslop-Harrison JS (1995) The large-scale organization of repetitive DNA families at the telomeres of rye chromosomes. Plant Cell 7:1823–1833
Vershinin AV, Alkhimova EG, Heslop-Harrison JS (1996) Molecular diversification of tandemly organized DNA sequences and heterochromatic chromosome regions in some Triticeae species. Chromosome Res 4:517–525
Waye JS, Willard HF (1989) Concerted evolution of alpha satellite DNA: evidence for species specificity and a general lack of sequence conservation among alphoid sequences of higher primates. Chromosoma 98:273–279
Willard HF (1989) The genomics of long tandem arrays of satellite DNA in the human genome. Genome 31:737–744
Zhong X-B, Fransz PF, Wennekes-van Eden J, Ramanna MS, van Kammen A, Zabel P, de Jong JH (1998) FISH studies reveal the molecular and chromosomal organization of individual telomere domains in tomato. Plant J 13:507–517
Zhong CX, Marshall JB, Topp C, Mroczek R, Kato A, Nagaki K, Birchler JA, Jiang J, Dawe RK (2002) Centromeric retroelements and satellites interact with maize kinetochore protein CENH3. Plant Cell 14:2825–2836
Zinic SD, Ugarkovic D, Cornudella L, Plohl M (2000) A novel interspersed type of organization of satellite DNAs in Tribolium madens heterochromatin. Chromosome Res 8:201–212
Acknowledgements
We are grateful to Dr. J. Doležel (Institute of Experimental Botany, Olomouc, Czech Republic) for providing us with sorted rye chromosomes, Dr. N. Chuzhanova (Cardiff University, Cardiff, UK) for measuring the complexity profile, and Prof. T.H.N. Ellis (John Innes Centre, Norwich, UK) for many valuable comments on the manuscript. The Institute of Cytology and Genetics is supported by the Russian Academy of Science; the Institute of Molecular Biology and Genetics is supported by the Ukraine National Academy of Sciences. This work was also supported by an INTAS grant (03-51-5908) and grants from the Russian Foundation for Basic Research (00-04-48992, 04-04-48813) and the Royal Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Shaw
Rights and permissions
About this article
Cite this article
Alkhimova, O.G., Mazurok, N.A., Potapova, T.A. et al. Diverse patterns of the tandem repeats organization in rye chromosomes. Chromosoma 113, 42–52 (2004). https://doi.org/10.1007/s00412-004-0294-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-004-0294-4