
Abstract
Understanding the impact of magmatic plumbing systems on explosive volcanic activity is important for hazard management. This study describes phase equilibria experiments using a high-silica andesite (HSA; SiO2 = 62.5 wt%) from the 2006 eruption of Augustine Volcano, Alaska. Experiments were conducted under H2O saturated conditions, \(f_{{0_{2} }}\) = RRO (Re–ReO2 or ~ Ni–NiO + 2), at pressures of 50–200 MPa (PTotal = PH2O), and at temperatures of 800–1060 °C. Run durations varied from 23 to 539 h, inversely scaled with temperature. The natural Augustine HSA phase assemblage (plagioclase, two pyroxenes, Fe–Ti oxides, magnesio-hornblende) was reproduced at 860–880 °C and 120–200 MPa. Comparing experimental and natural glass and plagioclase compositions further refined those conditions to ~ 870 °C and 120–170 MPa. Crystallization of euhedral quartz was accompanied by biotite and small amounts of cummingtonite at T ≤ 850 °C. The relatively high temperature appearance of these typically low-T phases indicates that higher \(f_{{0_{2} }}\) could enhance their stability. The storage conditions estimated from our experiments compare well with previous magma plumbing system models for Augustine from geophysical and petrological data. The refined experimental pressure range suggests a storage depth of 4.6–6.6 km, assuming a crustal density of 2650 kg/m3. The strong petrological and geochemical similarities between the products of the 2006, 1986, and 1976 eruptions suggest that the Augustine magmatic system had generally consistent crystallization conditions for the HSA lithology during that > 30-year time interval. The experimental results broad implications for understanding higher \(f_{{0_{2} }}\) magmas at andesitic arc volcanoes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 2006 eruption of Augustine Volcano, a Pleistocene–Holocene stratovolcano located in Cook Inlet, Alaska (Fig. 1), continued a trend for renewed unrest on a 10- to 30-year cycle. With documented eruptions in 1812, 1883, 1935, 1964, 1976, 1986, and 2006, Augustine is the most active volcano in Cook Inlet. Ash fall from recent eruptions of Augustine has impacted local communities, while older historical and prehistorical eruptions have been linked to regional tsunamis, the most recent of which was in 1883 (Waythomas and Waitt 1998). In addition, ash from Augustine eruptions impacts on the busy airspace above southern Alaska (Neal et al. 2010). Given these facts, understanding the volcanic system at Augustine is important for hazard management, and consequently, the volcano is one of the most comprehensively monitored and studied in the region (Power et al. 2010).

Map of Cook Inlet volcanoes. Inset map shows Alaska with the Cook Inlet region highlighted
The three most recent eruptions of Augustine (2006, 1986, and 1976) were similar in both products (crystal-rich andesite) and temporal progression from initial explosive activity to effusion over a scale of weeks to months; all were all triggered by the injection of mafic magma into a cooler, shallow-stored andesite (Johnston 1978; Larsen et al. 2010; Roman et al. 2006; Steiner et al. 2012). The wide range of magma compositions produced and their incomplete hybridization indicates that Augustine has a complex crustal storage region comprising interconnected dikes and sills (De Angelis et al. 2013; Roman et al. 2006).
Pre-eruptive magma crystallization conditions (e.g., P–T–XH2O–\(f_{{0_{2} }}\)) play a significant role in defining the physico-chemical properties of magmas and, by extension, the style of associated volcanic activity. Experimentally recreating natural phase assemblages and phase compositions through the varying of those key conditions is an important tool for modeling magmatic and eruption processes at arc volcanoes. However, such an approach requires the assumption that samples were erupted in a state of equilibrium representing a unique set of P–T–XH2O–\(f_{{0_{2} }}\) conditions (Blundy and Cashman 2008). Augustine eruptions are marked by pervasive pre- and syn-eruptive magma mixing and mingling (Larsen et al. 2010; Roman et al. 2006), and many erupted products exhibit numerous disequilibrium features rendering them unsuitable targets for a phase equilibria study. The high-silica andesite (HSA) lithology erupted in 2006 appears to represent a relatively unmodified end member, although it too shows some signs of compositional and textural heterogeneity and complexity (Benage et al. 2018). Compared with other andesite lithologies from the 2006 eruption, the HSA exhibits little whole-rock compositional variation (62.2–63.3 wt% SiO2), fewer disequilibrium textures (Larsen et al. 2010), a homogenous low-Al amphibole (magnesio-hornblende) population (De Angelis et al. 2013), and the lowest and most uniform apparent temperatures among the eruptive products (880 ± 13 °C; Coombs and Vazquez 2014; Larsen et al. 2010; Tilman 2008). The HSA is inferred to represent the Augustine lithology that is the least impacted by mingling and hybridization. Conversely, it could also represent a crystal mush, resident in the shallow crust since at least the last episode of activity, that was remobilized during replenishment prior to the eruption (Larsen et al. 2010).
Here, we present an experimental study that examines the pressure–temperature (PH2O–T) stability of magmatic phases within the HSA. Given the complexity of all Augustine andesite lithologies, we use the experimental results largely to refine and corroborate prior geophysical and petrological models of pre-eruptive storage at Augustine, rather than as a stand-alone product. At XH2O = 1 and = \(f_{{0_{2} }}\) RRO (Re–ReO2 or ~ Ni–NiO + 2), we found that the natural HSA is best reproduced at 860–870 °C and ~ 120–170 MPa (~ 4.6–6.6 km depth). The phase diagram also shows interesting shifts to higher temperatures of the phase stability for low temperature quartz, biotite, and cummingtonite, likely because of the higher \(f_{{0_{2} }}\) used. Therefore, our results are also broadly applicable to examining the crystallization conditions of other high-silica arc andesites at relatively oxidized conditions and can be applied to other volcanic systems that contain those phases.
Methodology
Starting materials
Experiments were conducted using the HSA pumice sample 06AUMC004c (SiO2 = 62.52 wt%; Table 1), emplaced during the Rocky Point pyroclastic flow on 27 January 2006 (Vallance et al. 2010). Starting material powders were prepared in three different ways to allow comparison of the kinetic effects imposed on experiments by their starting texture: (1) crushed natural powders (CH); (2) sintered (SNS); and (3) sintered and seeded (SS). For each, the HSA starting material was lightly crushed using an agate mortar and pestle. The sintered starting material was then produced by twice fusing 15 g of crushed HSA powder in a platinum crucible, in air at 1400 °C, for 30 min apiece, re-grinding the fused glass to a fine powder after the heating steps. Electron microprobe analyses of the twice-fused sintered glass verified that the composition remained close to that of the original HSA sample and that there was no appreciable Fe loss to the platinum crucible. For seeded experiments, this sintered powder was combined with 20 wt% plagioclase and 5 wt% amphibole (magnesio-hornblende) grains. The plagioclase seeds were extracted from 06AUMC004c using heavy liquid separation. The relative scarcity of amphibole in Augustine samples renders the extraction of seed grains inefficient; therefore, seeds were taken from samples erupted from Soufrière Hills Volcano, Montserrat in 2008. The compositions of the amphibole (magnesio-hornblende) in 06AUMC004c and of those in the Montserrat samples are very similar (Table 1). Amphibole were separated from their host using heavy liquids and then picked by hand.
Experimental methods
Powdered starting material (0.1 g), ~ 10 wt% de-ionized H2O, and the \(f_{{0_{2} }}\) buffer capsules were loaded into 4 mm Au tubes. Melt inclusions from the 2006 HSA contain only trace CO2 (Webster et al. 2010); therefore, experiments were conducted under H2O saturated conditions. Experiments covered a PH2O–T range spanning 50–200 MPa, and 800–1060 °C (Table 2). Augustine magmas are relatively oxidized (Larsen et al. 2010) and experiments were buffered at \(f_{{0_{2} }}\) = RRO (Re + ReO2 or approximately NNO + 2 log units). The 3-mm Ag70 Pd30 \(f_{{0_{2} }}\) buffer capsules were loaded with Re and ReO2 powder in a 1:1 molar ratio. To verify the effectiveness of the buffer assemblage, we ran 7- to 14-day test experiments. The compositions of seeded Fe–Ti oxide pairs from these experiments showed re-equilibration, thereby verifying accurate experimental temperatures and \(f_{{0_{2} }}\) (based on the algorithm of Ghiorso and Evans 2008).
Experiments below 880 °C were conducted in externally heated, Tuttle-style, Waspaloy cold-seal pressure vessels placed in horizontal furnaces and quenched with compressed air, followed by immersion in water. Temperatures were monitored using K-type thermocouples, with accuracy verified to within 5 °C by direct measurement of the melting point of gold. The thermal gradient along experimental capsules was measured and did not exceed a total of 10 °C; however, because experimental charges are loaded into the base of capsules and positioned closest to the thermocouple, experimental temperatures were likely within 5 °C of reported temperatures. Experiments at temperatures above 880 °C were conducted using titanium–zirconium–molybdenum (TZM) alloy pressure vessels and DelTech furnaces. In addition to the internal buffer capsules, TZM runs were buffered against loss of hydrogen by adding ~ 2.5 bars CH4 to the Ar pressurizing gas. TZM runs were rapidly quenched following Sisson and Grove (1993). Experimental durations ranged from 23 to 539 h (Table 2), with longer run times used in lower temperature runs in order to achieve a sufficient approach to equilibrium.
Reversal experiments were performed employing CH and SNS starting materials in inner capsules within the same run at 1000 °C and 1060 °C and at 150 and 140 MPa (Table 2). Two series of controlled melting and cooling experiments were also performed to approximate reversals and to verify the positions of the phase stability curves (HSA4-1, HSA4-2 and HSA4-2st2; Table 2). The crystallization experiment was conducted by first equilibrating SNS starting material at 1100 °C and 125 MPa for 48 h. The resulting HSA4-1 material was then re-loaded into a sealed capsule with sufficient water and refreshed buffer materials, thermally equilibrated to 1100 °C, and then cooled to 1000 °C using a 0.03 °C per minute cooling ramp to induce crystallization. The melting experiment started with CH material loaded into the capsules and held at 100 MPa and 840 °C for 120 h. The sample was then heated to 870 °C and held for a further 120 h before quenching.
Analytical methods
Phase identification and acquisition of major oxide compositional data for experimental glasses and mineral phases were conducted using a Cameca SX50 electron microprobe and a JEOL JXA-8530F field emission electron microprobe, both at the University of Alaska Fairbanks Advanced Instrumentation Laboratory. The Cameca SX50 is equipped with one EDAX energy-dispersive (EDS) spectrometer and four wavelength-dispersive (WDS) spectrometers. The JEOL JXA-8530F is equipped with a Thermo System 7 silicon drift detector (SDD) EDS system and five WDS spectrometers.
Mineral phase analyses were conducted with a 1–5 μm, 15 kV, 10 nA beam. Amphibole compositions were re-calculated to their mineral formula and classified according to Leake et al. (1997), based on the occupancy of the A (Na + K), B (Ca, Na), C (Mg, Fe2+, Fe3+, Ti, Mn, Alvi), and T (Si, Aliv) sites. Recalculation to the mineral formula was based on 23 oxygens, with Fe2+/Fe3+ estimation assuming 13 cations for magnesio-hornblende and 15 cations for cummingtonite. Error propagation during mineral formal recalculation was performed using MINERAL (MINeral ERror AnaLysis), a MATLAB® based program that calculates the error on formula unit cations though the propagation of analytical uncertainties during mineral formula recalculations (De Angelis and Neill 2012).
Areas of matrix glass in the experimental charges were often marginal in size compared with the defocused (~ 10 μm diameter) beams routinely used for analyzing hydrous glasses; therefore, we used a semi-focused (2–4 μm) low current (3 nA) beam with a voltage of 15 kV to help reduce Na migration. In addition, a linear time-dependent correction to Na counts using the Time Dependent Intensity (TDI) function in the Probe for EPMA (PFE) software was applied; however, consistently low Na concentrations in measured glasses suggest that this correction was insufficient. Other researchers have reported similar deficiencies with this correction (Sisson 2017, pers. com.). Working standards for the experimental glass analysis included both dry and hydrous glass samples (KN-18, VNM50-15, 508, and 510), as described by Devine et al. (1995) and originally utilized for calibrating electron probe micro-analysis (EPMA) hydrous glasses data against FTIR data.
A Qemscan® WellSite instrument (Quantitative Evaluation of Minerals by SCANning electron microscopy) at the University of Liverpool was used to quantify mineral proportions. Qemscan®, manufactured by FEI, is an Automated SEM–EDS system that uses a Tungsten filament to produce a 15 kV electron beam. Two Bruker energy dispersive X-ray spectrometers record discrete secondary X-rays emitted by the sample. X-Ray spectra are then processed to identify and quantify the elements present, and to output a chemical composition. This chemical composition is matched to known chemical compositions for minerals, glasses, and other amorphous phases.
Controlled using the iMeasure® software, data were collected from polished and carbon coated experimental charges at 2-µm resolution. The electron beam dwelled on each point until sufficient X-rays were collected to allow for successful phase matching (i.e., 1,000 X-rays in this study). Using the iDiscover® software, the resulting data were stitched to form continuous color images of each sample. The numbers of pixels for each phase were then summed, and normalized to give mineral abundances as an area-percentage. Unclassified pixels (e.g., vesicles, epoxy) and ‘other’ pixels (i.e., unidentified phases and/or compositions produced when the beam hit two grains at the same time) were removed, and the remaining data were re-normalized to provide a final area-percentage. In this study, the ‘other’ category was < 1 area %, and generally ≪ 1 area %, for all experimental charges. Minerals with abundances of < 0.1 area % were considered to represent noise and were reclassified as ‘other’. The QEMSCAN beam has an interaction volume of ~ (~ 10 µm3); as such, the abundances of phases with small crystal size (e.g., groundmass phases) may be misrepresented. In general, we found that the minerals identified by Qemscan® were consistent with those identified during traditional, manual SEM analysis. However, there was sometimes a mismatch for minerals present in low amounts. In particular, manual SEM analysis failed to identify clinopyroxene in many of the experiments where Qemscan® identified it in low quantities (< 5 area %). In contrast, minor amounts of cummingtonite were identified during manual SEM analysis, but this mineral was only identified by Qemscan® in two experiments (yielding abundances 0.5 area % for both).
Results
Augustine HSA summary from prior studies
The 2006 HSA samples are generally uniform in whole-rock composition (62.2–63.3 wt% SiO2) and matrix glass composition (74–77 wt% SiO2; Larsen et al. 2010); although, a single low-K HSA sample was also identified. For most oxides, the matrix glass composition of this low-K HSA was consistent with other HSA samples; however, it contained elevated SiO2 (76.7–79.7 wt%) and depleted K2O (1.1–1.6 wt% compared with 1.5–2.6 wt% in the majority of HSA samples; Larsen et al. 2010). The HSA contains 45–60 modal % microlite-poor glass, plagioclase (32 modal.%), augite (6 modal.%), orthopyroxene (4 modal.%), Fe-Ti oxides (2 modal.%), and minor amphibole (magnesio-hornblende); some samples also contain minor olivine, and apatite (< 0.5 modal.%; Larsen et al. 2010). Larsen et al. (2010) used the QUILF algorithm to calculate \(f_{{0_{2} }}\) conditions of NNO + 2 and a temperature range of 841–898 °C (Fig. 2), which they then corrected by 30 °C to account for a QUILF overestimation for high fugacity magmas (Rutherford and Devine 1996). A recalculation of the same and additional oxide data using the Ghiorso and Evans (2008) algorithm yields slightly less oxidized \(f_{{0_{2} }}\) conditions (NNO + 1.5 or 0.5 log units below RRO) and slightly higher temperatures (880 °C ± 13 °C; Coombs and Vazquez 2014; Fig. 2).

Estimated high-silica andesite storage temperature and \(f_{{0_{2} }}\) conditions calculated from the compositions of euhedral touching Fe–Ti oxide pairs in natural samples (Tilman 2008; Larsen et al. 2010; Coombs and Vazquez 2014). White circles denote calculations following the algorithm of Ghiorso and Evans (2008). Grey circles denote recalculations, on a subset of the same data, following the QUILF algorithm (Andersen et al. 1993). QUILF derived temperatures have been corrected by 30 °C to account for overestimation by the algorithm when applied to highly oxidized magmas (Rutherford and Devine 1996)
Webster et al. (2010) used Fourier transform infrared (FTIR) to analyze plagioclase melt inclusions in 2006 HSA samples and found that all (n = 49) had CO2 contents below the limit of detection (< 0.003 wt%), while H2O contents ranged from 3.08 ± 1.3 to 4.1 ± 0.001 wt%. Based on the model of Papale et al. (2006) and assuming a temperature of 880 °C, the mean of these analyses corresponds to a saturation pressure of ~ 77 MPa (~ 2.9 km depth, assuming a crustal density of 2650 kg/m3 here and throughout this manuscript), while the upper end of the analyzed range yields a saturation pressure of 104 MPa (~ 4.0 km depth). Similar results are found when applying the model of Newman and Lowenstern (2002), which yields saturation pressures of ~ 85 MPa (~ 3.3 km depth) and 115 MPa (~ 4.4 km depth) for the mean and maximum values, respectively. In general, OH bearing amphibole is stable at pressures greater than ~ 100 MPa (~ 4 km) and in melts containing at least 4 wt% H2O (Rutherford and Hill 1993); although Moore and Carmichael (1998) found amphibole stable in andesitic melt with just 3.5 wt% H2O. As such, most of the melt inclusion water contents are below those typically needed to stabilize amphibole in a melt; therefore, the inclusions likely experienced leakage and/or grew during decompression crystallization. As a result, storage pressures represent minimum possible values and are likely underestimated. However, the deepest storage pressure estimates are consistent with the conclusions of De Angelis et al. (2013), who used the existence of amphibole in the 2006 HSA to suggest storage at pressures of greater than ~ 100 MPa (~ 4 km depth).
Experimental run products
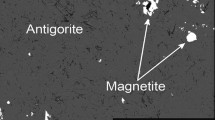
The crystallinity of experimental charges ranged with experimental conditions, from crystal free or crystal poor (< 20 area % crystals; Table 2) at high temperatures (Fig. 3a, c) to crystal and microlite rich (from ~ 40 to 80 area % crystals; Table 2) at low temperatures (Fig. 3b, d). Based on crystal-free experimental products (using both the CH and SNS starting materials; Table 2), at PH2O = 140 MPa the liquidus is located between 1000 and 1060 °C (Fig. 4); however, liquidus temperature increases with decreasing pressure, with plagioclase (32.2 area %), orthopyroxene (6.3 area %), and clinopyroxene (2 area %) observed at 1000 °C and PH2O = 125 MPa. Plagioclase and orthopyroxene were observed in all experimental runs at < 1060 °C; however, experimental resolution was insufficient to identify which was the liquidus phase. At PH2O = 150 MPa and 1000 °C, plagioclase crystals (13.9 area %) in the CH experiment were anhedral and embayed; however, stable euhedral grains were observed in the groundmass. Clinopyroxene crystallized at temperatures below 1000 °C and was most abundant (up to 4.5 area %) in experiments run between 840 and 860 °C at PH2O = 130–200 MPa; at P–T conditions above and below this, clinopyroxene abundance was generally less than 1% by area. Fe–Ti oxides (dominated by magnetite) crystallized in all experiments conducted at ≤ 1000 °C; however, the abundance of Fe–Ti oxides remained low in all experiments (< 1 area %).

Images of experimental groundmass. Backscatter electron (BSE) images of a Aug 73b (sintered-not-seeded [SNS] starting material held at 1000 °C/150 MPa for 69 h) and b Aug 37 (sintered-not-seeded [SNS] starting material held at 840 °C/130 MPa for 190 h). Qemscan® (Quantitative Evaluation of Minerals by SCANning electron microscopy) automated energy dispersive spectrometer (EDS) phase maps of c Aug 50 (sintered-not-seeded [SNS] starting material held at 960 °C/130 MPa for 30 h) and Aug 24 (sintered-and-seeded [SS] starting material held at 820 °C/120 MPa for 237 h). In Qemscan® images: grey, glass; light blue, plagioclase; light purple, orthopyroxene; green, clinopyroxene; orange, hornblende; pink, cummingtonite; brown–red, biotite; dark blue, quartz; light red, Fe–Ti oxides; black, unidentified phase; white, voids

Experimental phase diagram for the Augustine 2006 high-silica andesite (HSA). Solid curves denote confirmed boundaries, while dashed curves indicate inferred but unresolved phase boundaries. Right pointing triangles denote crushed HSA starting material, left pointing triangles denote sintered-and-unseeded (SNS) starting material, black circles denote sintered-and-seeded (SS) starting material, and grey squares denote experiments conducted using the experimental charge from SNS experiment HSA4-1. Arrows connect the start and end conditions of variable temperature experiments. Opx orthopyroxene, Pl plagioclase, Ox Fe–Ti oxides, Cpx clinopyroxene, Hb hornblende, Cum cummingtonite, Qtz quartz, Bt biotite. The lightly shaded horizontal grey box denotes the minimum pressure range for HSA storage based on natural melt inclusions (Webster et al. 2010) and the models of Newman and Lowenstern (2002) and Papale et al. (2006). The lightly shaded vertical grey box denotes the temperature range for HSA storage based on Fe–Ti oxides in natural HSA samples. The dark grey horizontal shaded area denotes the area in which experimental glass compositions most closely match that of the natural HSA starting material. The hashed grey area denotes the region in which experiment plagioclase exhibit mol.% An contents consistent with those of the natural HSA starting material
Magnesio-hornblende was clearly identified in experiments run at ≥ 120 MPa and ≤ 880 °C using both the SS and SNS starting materials, with most crystals euhedral and 5–30 μm in diameter; the overall abundance was less than 1 area % in all experiments. In addition, trace amphibole (magnesio-hornblende) was observed in a CH reversal experiment run at PH2O = 100 MPa, with a starting temperature of 840 °C and a final temperature of 870 °C.
Using manual SEM analysis, we observed both cummingtonite and euhedral quartz in experiments conducted at ≤ 850 °C using all starting materials; both phases were also sometimes observed as intergrowths with orthopyroxene. The crystallization of cummingtonite and quartz was concurrent with a reduction in augite. We verified the positions of the quartz- and cummingtonite-in curves with experiment HSA4-2st2. This experiment was first equilibrated within the cummingtonite–quartz fields for 120 h at 840 °C and 100 MPa, and then heated and re-equilibrated for 120 h at 870 °C, outside of the cummingtonite and quartz fields. In automated Qemscan® analyses, quartz abundances vary from 0.2 area % (850 °C/200 MPa) to 19 area % (840 °C/100 MPa). Using Qemscan®, cummingtonite was only confirmed in two experiments, both with abundances of 0.5 area % (820 °C/120 MPa and 840 °C/120 MPa, both using the SS starting material).
Using automated Qemscan® analysis, biotite was identified in two low P–T experiments [840 °C/120 MPa and 820 °C/120 MPa (Fig. 3d)], with abundances of 5.1 and 6.4 area %. In manual SEM analysis, a phase rich in K, Mg, Al, Fe, and Si was identified in other experiments run below ~ 850 °C run using the sintered starting material. This phase was unstable under the electron beam and quantitative analysis was not successful. As a result, we were unable to confirm with 100% certainty the nature of this phase; however, EDS spectra and textural characteristics suggest that it was biotite. Given the uncertainty, an exact phase appearance curve was not delineated.
Using manual SEM identification, accessory apatite was identified, even in experiments run at > 900 °C; however, its small size made verification challenging and we were not able to constrain the phase boundary; it was not identified in Qemscan® analysis.
Experimental mineral compositions
Plagioclase was the most commonly observed mineral in all experimental products (Table 3; Fig. 5). The An (anorthite) content of plagioclase ranged from An47–An81 and was strongly controlled by experimental temperature. In general, An was positively correlated with temperature (Fig. 5a) and negatively correlated with estimated undercooling (ΔT; Fig. 5c). At temperatures of < 900 °C, we also observed a positive correlation between An and PH2O (Fig. 5b). However, as most experiments at temperatures of > 900 °C were conducted at intermediate pressure (PH2O = 120–150 MPa), the resulting An values of > 70 plot above the general trend. At temperatures of < 900 °C, PH2O < 180, and undercooling of > ~ 180 °C, plagioclase compositions were An50–60 (i.e., the compositional range of natural HSA plagioclase).

Experimental and natural plagioclase compositions: a variation in experimental mol.% An (anorthite) as a function of temperature (°C); b variation in experimental mol.% An as a function of pressure (PH2O in MPa) and; c variation in experimental mol.% An as a function of undercooling (ΔT in °C). Data points represent the mean of all analyses from each experiment. One standard deviation (1σ) error was calculated following De Angelis and Neill (2012). Right pointing triangles denote crushed high silica andesite (HSA) starting material, left pointing triangles denote sintered-and-unseeded (SNS) starting material, black circles denote sintered-and-seeded (SS) starting material, and grey squares denote experiments conducted using the experimental charge from SNS experiment HSA4-1. The dashed black line and surrounding lightly shaded grey area represents the mean and 1σ of the natural HSA An content
Experimental pyroxene showed a range of compositions, some of which overlapped with the compositions of natural HSA phenocrysts (Table 4; Fig. 6). Both orthopyroxene (enstatite) and clinopyroxene (augite), showed weak positive correlations between Mg# (Mg/Mg + Fe) and both PH2O (Fig. 6b) and experimental temperature (Fig. 6c). In general experimental orthopyroxene overlapped with the natural orthopyroxene Mg#’s (~ 0.67) between ~ 135 and 200 MPa, and between 840 and 860 °C. Experimental clinopyroxene only overlapped with natural HSA clinopyroxene compositions (Mg# ~ 0.74) at pressures (PH2O) of 120 MPa and temperatures of 820 °C. At higher PH2O–T conditions, clinopyroxene were significantly Mg-enriched.

Experimental and natural pyroxene compositions: a Pyroxene classification quadrilateral. Black squares denote rim compositions of pyroxene in the natural high-silica andesite (HSA) starting material. Experimental data symbol shapes denote pressure (PH2O in MPa), while symbol color denotes temperature (white symbols = ≥ 1000 °C, dark grey symbols = 870–960 °C, and light grey symbols = ≤ 860 °C). b Variation in the Mg# (Mg/Mg + Fe) of experimental pyroxene with changing experimental pressure (PH2O in MPa). c Variation in the Mg# of experimental pyroxene with changing experimental temperature (°C). Data points represent the mean of all analyses from each experiment. One standard deviation (1σ) error was calculated following De Angelis and Neill (2012); however, the 1σ error is always smaller than the data symbols. In b, c: solid black lines and the surrounding lightly shaded grey areas denote the mean and standard deviation (1σ) of Mg# for clinopyroxene (cpx) in the natural HSA starting material; dashed black lines and the surrounding shaded grey areas denote the mean and 1σ of Mg# for orthopyroxene (opx) in the natural HSA starting material; symbol shape denotes pyroxene type (circles = cpx, squares = opx) and; symbol shading denotes starting material (white = sintered-and-unseeded/SNS, dark grey = sintered-and-seeded/SS, black = crushed HSA, and light grey = experiments conducted using the experimental charge from SNS experiment HSA4-1)
Experimental amphibole were classified as magnesio-hornblende or cummingtonite (Fig. 7; Table 5). Magnesio-hornblende compositions were consistent with those of both natural HSA amphibole and the seed grains. Regardless of experimental starting material (SS or SNS) or the PH2O–T conditions (PH2O = 120–200 MPa, T = 820–860 °C), differences in composition were minor; however, magnesio-hornblende in seeded experiments were slightly Mg-depleted with respect to both the natural HSA and seed grain compositions, while those that crystallized from the unseeded starting material were either slightly Mg- or Si-enriched (Fig. 7b). Cummingtonite Mg# ranged from 0.75 to 0.86. The number of data points was insufficient to comprehensively assess any correlation between Mg# and experimental conditions; however, lower Mg# (< 0.8) values tended to be found at higher PH2O (> 170 MPa).

Experimental and natural amphibole compositions: a classification diagram for magnesio-hornblende crystals, and b classification diagram for cummingtonite crystals. Data points represent the mean of all analyses from each experiment. One standard deviation (1σ) error was calculated following De Angelis and Neill (2012). The black square denotes amphibole from the natural high-silica andesite (HSA) starting material, while the black circle denotes the amphibole seed compositions (Soufrière Hills Volcano andesite)
Experimental glass compositions
Despite the small low-amperage beam utilized for glass analysis, locating areas of exposed glass in some experimental charges was unsuccessful. In particular, low PH2O–T experiments often contained a microlite-rich groundmass (Fig. 3b, d) and as a result, in a number of experimental charges, observed glass areas were too small to analyze. For experiments where glass was successfully analyzed, the range in compositions reflected the changing experimental conditions (and in particular changing T) and phase assemblages (Fig. 8).

Experimental glass compositions. a–f Harker plots showing the variation in experimental and natural glass wt% SiO2 with wt%: a Al2O3; b CaO; c FeO; d MgO; e K2O; and f Na2O. g Experimental melt H2O estimated using the model of Waters and Lange (2015). Data points represent the mean of all analyses from each experiment with error bars showing one standard deviation (1σ). Black squares denote the groundmass glass composition of the natural high silica andesite (HSA) starting material, black diamonds denote the whole rock composition of the natural HSA starting material, and black circles denote the groundmass glass composition of a natural low-K HSA sample (Larsen et al. 2010). Experimental data symbol shapes denote pressure (PH2O in MPa), while symbol color denotes temperature (white symbols = ≥ 1000 °C, light grey symbols = 880–960 °C, and dark grey symbols = ≤ 860 °C)
In general, experimental glass compositions formed a tie line between the natural HSA whole rock and glass end-member compositions. At 1060 °C and PH2O = 140 MPa, experimental charges (experiments Aug 76a and Aug 76b) were completely aphyric. With the exception of Na2O, major oxide glass compositions were similar to the whole rock composition of the HSA starting material (to within ~ 1.3 wt% or less); for N22O, the experimental charges were depleted (1.99 and 2.44 wt%, respectively) in comparison with the natural HSA whole rock (3.78 wt%). With decreasing experimental temperature, glasses became progressively more silicic, increasing from 63.18 wt% SiO2 (Aug 76b; 1060 °C/140 MPa; Fig. 8) to 77.82 wt% SiO2 (Aug 49; 850 °C/160 MPa). Concurrent decreases were seen in Al2O3 (from 18.03 wt% at 1100 °C/125 MPa to 12.67 wt% at 850 °C/160 MPa), CaO (from 6.78 wt% at 1060 °C/140 MPa to 2.04 wt% at 840 °C/130 MPa), FeO (from 5.05 wt% at 1060 °C/140 MPa to 1.08 wt% at 850 °C/160 MPa), and MgO (from 3.75 wt% at 1060 °C/140 MPa to 0.60 wt% at 860 °C/150 MPa).
Alkali elements (i.e., K and Na) were depleted in almost all experimental glasses (Fig. 8e, f). Despite the application of a TDI correction, low Na2O values (Fig. 8f) most likely reflect Na migration under the electron microprobe beam, and can also partially explain the consistently low experimental glass totals (~ 88–97 wt%; Table 6), which is a common feature of hydrous experimental glass analysis (e.g., Hammer et al. 2002; Rutherford and Devine 2003). In contrast, the K2O values in glasses from liquidus experiments (0.86–0.89 wt% at 1060 °C/140 MPa; Fig. 8e) showed only minor depletion in comparison to natural whole rock K2O contents (0.99 wt%); however, with decreasing experimental temperature and increasing SiO2, the experimental glasses failed to become significantly or consistently K2O enriched, despite the progressive crystallization with decreasing temperature and pressure. The veracity of these values was tested by reanalyzing some of the samples on a different microprobe (Cameca SX5 at the Cameca Demonstration Laboratory in Madison, Wisconsin), following re-polishing and reapplication of the carbon coat. The low-T (840–860 °C) experimental glasses had K2O values of just 1.00–1.47 wt%, compared with a natural HSA glass composition of 2.22 wt%. However, the K2O values of the low-T experimental matrix glasses showed excellent agreement with a single low-K HSA identified in the 2006 deposits (K2O = 1.1–1.6 wt%; Larsen et al. 2010).
For experiments where data pairs were available, the Waters and Lange (2015) hygrometer was used to estimate experimental melt water contents from mean plagioclase and glass compositions (Fig. 8g). We found no clear relation between experimental pressure and melt H2O; however, there was a clear negative correlation between experimental temperature and melt H2O, ranging from 5.0–5.5 wt% H2O at T = ≤ 880 °C to ~ 3.5 wt% H2O at T = 1000 °C.
With the exception of the alkali elements, low-T experimental glasses (840–860 °C) showed the greatest degree of consistency with natural HSA glass compositions. However, they were slightly enriched in SiO2 (77.68–78.03 wt% vs. 75.86 wt% in the natural HSA matrix glass), making them more consistent with the SiO2 values of the natural low-K HSA sample (76.7–79.7 wt%). A similar result was seen for FeO, although the difference in FeO between the normal and low-K natural HSA glasses was minor, while Al2O3 and CaO showed greater consistency with the standard natural HSA matrix glass compositions.
Discussion
Approach to equilibrium
Experiments on silicic systems can be challenging because of slow reaction timescales, especially in crystal-rich charges with evolved groundmass melts (e.g., Brugger et al. 2003; Lange et al. 2009). Furthermore, systems characterized by mixing and mingling are generally unsuitable for phase equilibria studies because by their very nature they are out of equilibrium. To address the issue of natural disequilibrium, we selected the most homogenous lithology erupted from Augustine during the 2006 activity. This HSA is crystal rich (36–55 vol.% phenocrysts), and this study produced experiments that evolved to melt SiO2 of > 75 wt%. We experienced a number of ‘failed’ experiments in which sluggish kinetics resulted in a failure to approach equilibrium. Given these challenges, a systematic assessment of the approach to equilibrium was critical.
First, we used three different types of starting materials (completely glassy, seeded, and crystal-rich natural powders) to assess the achievement of equilibration in experiments with both melt and crystal-rich starting points, similar to traditional reversal experiments. Crushed natural powders most closely replicated natural textural conditions; however, the solid-state re-equilibration of many silicate minerals, including plagioclase and amphibole, is an extremely slow process, taking many years to accomplish for complete equilibration (e.g., Scaillet and Evans 1999). On the scale of days to weeks, as is practical for experimental durations, only local equilibrium between crystal rims and coexisting glass may be achieved, and some phases may show no evidence for re-equilibration (Scaillet and Evans 1999). In contrast, crystallization experiments using crystal-free glass starting materials may achieve local equilibrium more quickly (e.g., Pichavant et al. 2007). However, the lack of crystals in the starting material to act as ‘seed crystals’ can dramatically affect experimental kinetics. High nucleation rates may inhibit the growth of experimental ‘phenocrysts’, and the resulting fine-grained texture leaves few large areas of glass exposed between the crystals, making analysis problematic.
Second, in order to achieve sufficient local equilibrium, at least between crystal rims and the adjacent melt, we employed very long run times (up to > 500 h) as suggested by a variety of experimental studies (e.g., Brugger et al. 2003; Martel and Schmidt 2003). Martel and Schmidt (2003) found that rhyolitic melts (SiO2 > 75 wt%) achieved equilibrium within 7 days (168 h) for pressures ≥ 100 MPa. Brugger et al. (2003) found that in their most silicic experiments (SiO2 ~ 75.7–77.6 wt%) run times of 3.5 days (84 h) or longer were required for the attainment of equilibrium. In our study of 21 experiments, 12 were for held for greater than 84 h, and seven were held for greater than 168 h (Table 2). Only nine experimental runs had durations of less than 84 h (Table 2), and those were all at temperatures > 900 °C, at which equilibration timescales should be much shorter.
Despite the fine-grained textures, particularly at low-T conditions with significant undercooling (ΔT = 100–300 °C), the experiments using the glassy starting material (SNS) were most successful. Some seeded (SS) and crushed HSA (CH) experiments exhibited unstable crystal textures (e.g., dissolution or breakdown) and showed significant scatter in the compositional data of glass and mineral phases. These features suggest a failure to equilibrate well, despite long experimental durations, and on this basis these experiments were not used to construct the Augustine phase diagram. We also conducted two double capsule experiments, in which CH and SNS starting materials were loaded in separate compartments within a single capsule (Aug 73, Aug 76). These experiments served as ‘reversals’ to help assess equilibration. For the double-capsule experiment at 1060 °C (PH2O = 140 MPa), both starting materials transformed into crystal-free experimental charges with mean major oxide glass values that were all within 0.52 wt% of each other (Fig. 8). The 1σ envelopes around these values converged for all oxides with the exception of SiO2, where uncertainties were ± 0.01 wt%. For the double-capsule experiment at 1000 °C (PH2O = 150 MPa), the resulting phase assemblages were the same (Table 2), while the glass compositions were similar, with all major oxides except SiO2 and Al2O3, showing compositional overlap between the experiments. The mean SiO2 and Al2O3 values differed by 1.5 and 0.67 wt%, respectively, while the 1σ envelopes around these values approached each other, but did not converge. The lower SiO2 and Al2O3 values were both seen for the CH experiment and it is likely that minor amounts of a non-stable phase (e.g., plagioclase) failed to completely dissolve; however, the remaining amount was insufficient to be observed during extensive microprobe sessions. On the basis of these results, we inferred that both double capsule experiments exhibited an acceptable approach to equilibrium.
For the experiments included in the final dataset, the 1σ envelopes around the mean major oxide glass compositions were relatively low and were consistent with those deemed acceptable by other experimental studies (e.g., Brugger et al. 2003; Hammer et al. 2002; Sisson et al. 2005). For example, the mean 1σ on Al2O3 for glass analyses in this study was 0.35, with a maximum of 0.86 (Aug 42). In contrast, Hammer et al. (2002), Brugger et al. (2003), and Sisson et al. (2005) report maximum standard deviations of 0.7, 0.7, and 0.9, respectively; thus, our experimental glasses are sufficiently homogeneous as compared to similar studies to exhibit an acceptable approach to equilibrium.
Despite our efforts to ensure that experiments reached equilibrium, some experiments with a rhyolitic groundmass, and in particular those in which quartz saturation was reached, may have had experimental kinetics so sluggish as to prohibit the achievement of equilibrium. For these experiments in particular, we endeavored to probe crystals with clear growth textures and/or euhedral shapes consistent with approach to equilibrium.
Low temperature phase stability
While the positions and shapes of stability limit curves for magnesio-hornblende, plagioclase, and pyroxene (Fig. 4) are consistent with the results from other experimental studies of similar volcanic systems (e.g., Browne and Gardner 2006; Rutherford and Devine 2003), we observed the unusually early (hot) appearance of normally low-T phases (i.e., quartz, cummingtonite, and biotite). In comparison with experimental studies on similar volcanic systems, including the Soufrière Hills Volcano andesite (Barclay et al. 1998; Rutherford and Devine 2003) and Unzen Volcano dacite (Holtz et al. 2005), our Augustine quartz appearance curve is ~ 20–40 °C hotter. However, similarities are seen between our results and the experimental study of the 1991 Mount Pinatubo dacite (Rutherford and Devine 1996), where the intersection between the quartz and magnesio-hornblende phase appearance curves was observed at ~ 120 MPa and 850–860 °C, despite the ~ 0.8 wt% higher SiO2 content of Mount Pinatubo glasses (Rutherford and Devine 1996). The Mount Pinatubo experiments were conducted at high \(f_{{0_{2} }}\) conditions (NNO + 3), even more oxidizing that Augustine.
Biotite was only confirmed in two of our experiments (820 °C and 840 °C at 120 MPa), although there is evidence to suggest that it may also be present in other experiments of < ~ 850 °C. Biotite is generally not observed above ~ 800 °C in experimental studies of calc-alkaline andesitic systems (e.g., Holtz et al. 2005; Rutherford and Devine 2003; Venezky and Rutherford 1999), although in studies of more mafic and crystal poor magmas, biotite has been seen to crystallize at temperatures greater than 900 °C (Barclay and Carmichael 2004). The only exception that we are aware of is in an experimental study of a similarly highly oxidized system (Mt. Pinatubo, Philippines; NNO + 2 to NNO + 3), where biotite is also seen to crystallize at relatively high temperatures (810–830 °C at 150–250 MPa; Rutherford and Devine 1996).
It is likely that the relatively higher temperature appearance of quartz and biotite reflects both the high crystallinity of the experiments and the high oxygen fugacity (RRO), which forced the melt to evolve to higher SiO2 at higher PH2O–T conditions relative to systems with \(f_{{0_{2} }}\) closer to NNO. Magmatic \(f_{{0_{2} }}\) influences both the PH2O–T range of mafic (Fe-bearing) mineral crystallization and the order in which phases appear. In the context of basaltic magmas, Sisson et al. (2005) found that \(f_{{0_{2} }}\) exerts a strong influence on near-solidus phases and on melting and crystallization behavior. They found that at constant temperature, increasing the \(f_{{0_{2} }}\) of basaltic magmas encourages the consumption of both melt and Fe2+ amphibole components and results in the crystallization of plagioclase and Fe–Ti oxides and the production of H2O (Sisson et al. 2005). This should result in a reduction of melt fraction, driving the liquid toward more evolved compositions that encourages the stabilization of late stage minerals such as quartz and biotite.
These conditions are also likely responsible for the extremely high-T crystallization of cummingtonite. However, while quartz and biotite are common in calc-alkaline magmatic systems, cummingtonite is much rarer, although it has been identified in some magmas similar to those studied here; for example, the Mount St. Helens (Geschwind and Rutherford 1992) and Mt. Pinatubo (Rutherford and Devine 1996; Scaillet and Evans 1999) dacites. Experimental work on suggests that cummingtonite in silicic magmas is only stable at temperatures of < 800 °C, pressures of ~ 250–300 MPa, \(f_{{0_{2} }}\) of > NNO + 1, and PH2O that is ≥ 75–80% of Ptotal (e.g., Geschwind and Rutherford 1992; Nicholls et al. 1992; Wood and Carmichael 1973). Scaillet and Evans (1999) identified cummingtonite in dacites of the 1991 eruption of Mount Pinatubo; however, they failed to crystallize it in experiments run at a range of \(f_{{0_{2} }}\) conditions (NNO to NNO + 2.7). Further experimental work on the Pinatubo dacite was able to synthesize cummingtonite through the addition of sulfur (Evans et al. 2007) and through the use of an unfused natural starting material (Rutherford and Devine 1996), inferred to have a higher natural sulfur content than the dry-glass starting materials used by Scaillet and Evans (1999).
At Augustine, cummingtonite has been identified in late Pleistocene rhyolites (Coombs and Vasquez 2014; Nadeau et al. 2015) and prehistoric pumicious tephras (Tappen et al. 2009), where it coincides with magnesio-hornblende; however, to our knowledge, despite extensive work by various researchers, it has not been identified in the products of the 2006 eruption. Despite this, the 2006 products are consistent with the conditions required for cummingtonite stability, including high \(f_{{0_{2} }}\) (NNO + 2) and high PH2O (in our experiments, PH2O–PTOTAL). Furthermore, a number of studies have suggested a link between low-K glass, as observed in our low P–T experiments and in one natural HSA sample from 2006, and the stabilization of cummingtonite (e.g., Coombs and Gardner 2004; Tomiya et al. 2010). In terms of sulfur, rhyodacite and rhyolitic melt inclusions from Augustine generally have lower contents than those seen at other calc-alkaline subduction zone volcanoes; however, the high \(f_{{0_{2} }}\) of the system means that the majority of this sulfur would be present in an oxidized state, leading to fluids rich in sulfur species (Webster et al. 2010); this is consistent with the significant SO2 measured in volcanic vapor during the 2006 eruption (McGee et al. 2010). Even so, experimental cummingtonite at such high temperatures (up to 850 °C) is unprecedented. Higher cummingtonite Mg# has been linked with higher temperatures (e.g., Evans and Ghiorso 1995; Geschwind and Rutherford 1992); at Mg#s of 0.75–0.86, the cummingtonite in our study are consistent with higher temperature occurrence. Tomiya et al. (2010) suggested that cummingtonite in some of their experiments was not a truly stable phase, but instead a function of a powdered dry glass starting material. It is possible that the same may be true for our experiments; however, its appearance in starting materials of all types is contrary to this theory.
Low alkalis in experimental glasses
Low alkali values are a feature of almost all experimental glasses in this study. With respect to Na, low values are also seen in working standards and likely stem from Na migration away from the electron beam, as noted by previous authors (e.g., Devine et al. 1995; Nielsen and Sigurdsson 1988). The TDI correction applied to Na count data was insufficient to account for this Na migration, which is consistent with the observations of other researchers (Sisson 2017, pers. com.).
In terms of potassium, the K2O in experimental glasses from experiments approaching the liquidus behaved as expected, with compositions approaching those of the whole rock (~ 1 wt%; Fig. 8e). However, as an element that is highly incompatible in most minerals, K2O in experimental glasses should increase with increasing SiO2 and with an increasing degree of crystallization as experimental temperatures and pressures decrease. However, in no experiment did K2O values reach the level seen in natural HSA glasses (~ 2–3 wt%).
Similar to Na, the migration of K away from the electron beam during electron microprobe analysis of hydrous glass has also been documented. However, the effects of K migration are significantly less pronounced than those for Na (Reed 2005). Furthermore, our electron microprobe data do not show similar K depletion in working standards, and time-dependent K count data on experimental glasses do not exhibit systematic decreases; therefore, while it may contribute to low K2O in analyzed glass, K migration in our samples cannot completely explain the results. Potassium is relatively insoluble in super-critical H2O fluid and is not known to alloy with gold capsules during hydrothermal experiments like Fe does (Ratajeski and Sisson 1999). Therefore, viable explanations for the depletion of K include its incorporation in modest quantities into more abundant phases (e.g., plagioclase) or the crystallization of K-rich mineral phases such as K-feldspars (e.g., sanidine) or biotite micas.
A small number of the experimental plagioclase show significant K enrichment, with some having orthoclase (Or) contents of > Or-5. While this could be the result of the electron beam partially overlapping with areas of K-enriched glass, both the relative ease of plagioclase analysis (resulting from the abundance of relatively large crystals) and the low-K nature of the experimental glasses in general, render this unlikely. Furthermore, similar levels of Or enrichment have also been reported from experiments on dacite starting materials from the 1912 Katmai eruption (Hammer et al. 2002) and the 1991 Pinatubo eruption (Prouteau and Scaillet 2003; Scaillet and Evans 1999), which were also conducted under high oxidation conditions (i.e., ≥ NNO + 2).
More importantly, significant biotite was identified in two of our samples (6.4 and 5.1 area %) using Qemscan® analysis; furthermore, using manual SEM analysis, a biotite-like K-rich phase was identified in other low P–T experiments using the sintered starting material. On this basis, the low-K nature of the most evolved experimental glasses most likely reflects and confirms the crystallization of biotite. Interestingly, our glass compositions were similar to those from a single low-K HSA sample identified in the natural Augustine 2006 deposits and mass balance calculations predict that the crystallization of ~ 2 to 3 wt% biotite in the natural HSA would produce the observed low-K composition.
Experimental constraints on HSA storage
Given the heterogeneous nature of the broad spectrum of andesites produced by the Augustine volcanic system, including the HSA end member, we considered our phase equilibrium constraints within the broader context of estimates from both geophysics and natural sample petrology and geochemistry; as such, we use them largely as a refinement of prior models. Using petrological and geophysical data, a number of storage models have previously been proposed for Augustine. For the HSA of the 1976 eruption (62.1–62.3 wt% SiO2), Harris (1994) used the compositions of Fe–Ti oxide touching pairs to calculate a temperature range of 867–885 °C and \(f_{{0_{2} }}\) conditions of Ni–NiO (NNO) + 2. Similarly, for HSA of the 1986 eruption (60.7–63.3 wt% SiO2), Roman et al. (2006) calculated temperature and conditions of 877–890 °C and NNO + 2 (based on Fe–Ti touching pairs and using the QUILF algorithm; Anderson et al. 1993), and a storage depth of at least 4–5 km below the summit (based on melt inclusion water contents). For the 2006 HSA, Fe–Ti oxide data indicate \(f_{{0_{2} }}\) conditions of NNO + 1.5 (or 0.5 log units below RRO) and temperatures of 880 °C ± 13 °C (Coombs and Vazquez 2014; Fig. 2). The presence of stable amphibole places the minimum storage depth at ~ 4 km (De Angelis et al. 2013). Furthermore, based on the H2O contents of natural plagioclase. storage depth has been estimated to be no less than 3.3–4.4 km (Webster et al. 2010). As discussed, this range includes some inclusion water contents that permit amphibole (> ~ 4 wt% H2O) and some that are too low. It is possible that some melt inclusions experienced leakage, or were entrapped at lower pressures during decompression crystallization. Experimental melt H2O was found to be sufficient for amphibole stabilization at temperatures of ≤ 960 °C (Fig. 8g).”
Geophysical models for magma storage at Augustine are consistent with petrological data. Based on pre-eruptive 1976 seismic activity and on post-eruptive 2006 seismic activity, Power and Lalla (2010) inferred a storage region at 3.5–5 km below sea level (bsl). Based on geodetic data from the 2006 eruption, Cervelli et al. (2010) proposed a cylindrical pipe storage model with a top at 2.5–4.5 km bsl and a base at 6.5–10.5 km bsl.
Overall, the experimental mineral stabilities and compositions observed in this study are consistent with past estimates and provide further refinement to the range of possible PH2O-T conditions for the Augustine HSA. In particular, HSA storage conditions are constrained between the amphibole (hornblende) in-curve (~ 200 MPa at 890 °C and ~ 110 MPa at > 860 °C) and the cummingtonite and quartz in-curves (~ 200 MPa at 840 °C and ~ 100 MPa at 860 °C; Fig. 4). Within this region, conditions are further constrained by the upper temperature and pressure limits at which experimental and natural plagioclase An compositions are consistent (~ 870 °C and 170 MPa) and by the lower pressure limit and upper temperature limit at which natural and experimental glass compositions are consistent (~ 120 MPa and ~ 860 °C, respectively). Clinopyroxene occurs in relatively low abundance, and compositions show no systematic changes with changing experimental conditions (Fig. 6); similarly, while orthopyroxene is significantly more abundant, there is only weak correlation between changing compositions and changing experimental PH2O–T conditions, which is consistent with the results of other experimental studies (e.g., Prouteau and Scaillet 2003; Rutherford 1985). However, in general, our experimental orthopyroxene overlapped with the natural orthopyroxene Mg#’s (~ 0.67) between ~ 135 and 200 MPa, and between 840 and 860 °C.
When all phase stability and compositional trends are considered, the resulting storage temperature range (~ 860 to 870 °C) provides a refinement on past estimates (838–908 °C). The refined pressure range (120–170 MPa) suggests a storage depth of 4.6–6.6 km, which fits particularly well with geodetic observations (Cervelli et al. 2010) that indicate a storage region with a top at 2.5–4.5 km bsl and a base at 6.5–10.5 km bsl, and meets the storage pressure constraints from petrological data (e.g., > ~ 4 km based on the presence of stable amphibole).
Wider implications for petrological modeling
The results of this study have potential implications for existing petrological and thermodynamic models of volcanic systems; for example, rhyolite-MELTS (Gualda et al. 2012) and the plagioclase-liquid hygrometer-thermometer (Lange et al. 2009; Waters and Lange 2015). In particular, our findings offer new insights and data for the calibration of such models because: (1) few experimental studies of arc-andesite systems have produced glasses with such high-SiO2 rhyolitic compositions; (2) the unprecedented shift to higher temperatures of the quartz, biotite, and cummingtonite stability curves; (3) that these findings appear to be linked to the elevated experimental \(f_{{0_{2} }}\), which has been reported for a small, but important sub-set of arc-andesite volcanoes [e.g., Augustine Volcano, Alaska (e.g., Larsen et al. 2010); Pinatubo, Philippines (e.g., Scaillet and Evans 1999; Rutherford and Devine 1996); Volcan de Colima, Mexico (e.g., Moore and Carmichael 1998; Reubi et al. 2013)]; and (4) given the challenges involved in this study, particular care was taken to ensure well constrained experimental conditions and an approach to equilibrium in experimental charges.
Summary and conclusions
In this study, we performed phase equilibria experiments using a high-silica andesite (HSA; SiO2 = 62.5 wt%) from the 2006 eruption of Augustine Volcano, Alaska. Experiments were conducted under H2O saturated conditions, \(f_{{0_{2} }}\) = RRO (~ Ni–NiO + 2), pressures of 50–200 MPa (PTotal = PH2O), and at temperatures of 800–1060 °C. The natural phase assemblage from the Augustine HSA was reproduced at 860–880 °C and 120–200 MPa. Comparison with natural glass and plagioclase compositions further refined those conditions to ~ 870 °C and 120 to 170 MPa. A significant finding of this work was the unprecedented shift to higher temperatures of the quartz, biotite, and cummingtonite stability curves. Crystallization of euhedral quartz was accompanied by small amounts of cummingtonite at T ≤ 850 °C; biotite was also identified in two low P–T experiments (840 °C/120 MPa and 820 °C/120 MPa). The relatively high temperature appearance of these typically low-T phases indicates that higher \(f_{{0_{2} }}\) could enhance their stability.
Given the challenges associated with experimental studies of silicic systems (e.g., slow reaction timescales, crystal-rich charges, evolved groundmass melts), particular care was taken to ensure that experimental conditions were accurate and well constrained (e.g., temperature accuracy was verified to within 5 °C by direct measurement of the melting point of gold), and to ensure that a true approach to equilibrium was achieved in experimental charges. We employed three different starting materials to assess the achievement of equilibration in experiments with both melt and crystal-rich starting points. To ensure local equilibrium between crystal rims and melt, we employed very long run times (up to > 500 h), in particular for those experiments run at the lowest P/T conditions. We monitored the 1σ envelopes around mean major oxide compositions to confirm low data spreads within experiments and the similarities/differences among data values for different experiments. Microprobe data were collected from crystals with clear growth textures and/or euhedral shapes. Experiments with an insufficient approach to equilibrium were not used in the final model.
The results of this study have allowed us to refine previous geophysical and petrological estimates for the location of HSA storage beneath the volcano prior to the 2006 unrest [120–170 MPa (or 4.6–6.6 km) at ~ 860–870 °C]. Prevailing models conclude that magmas beneath Augustine are housed in a complex network of interconnected dikes (Roman et al. 2006; Larsen et al. 2010; De Angelis et al. 2013), rather than one contiguous, stable and stratified magma reservoir. The pressures and temperatures of the magmas stored in these dikes would vary, depending on their size, age, relative depth, and level of interaction with hotter replenishing magmas; this variation is supported by the range of eruptive products observed for recent eruptions. However, the strong similarity of the 2006 HSA to the HSAs erupted during both 1976 and 1986 (e.g., bulk composition, matrix glass composition, mineralogy, crystal compositions; Johnston 1978; Roman et al. 2006; Larsen et al. 2010; Steiner et al. 2012) suggest that the pre-eruptive crystallization conditions of the HSA magma beneath Augustine remained more or less consistent during the > 30-year time interval encompassing the 1976 through 2006 eruptions. Future eruptions of Augustine, which have recently occurred on a 10- to 20-year interval, are thus likely to produce a similar suite of low- tohigh-silica andesite deposits.
From a broader perspective, the results of this study (e.g., the high-SiO2 rhyolitic experimental melts; relatively high-T stability of quartz, biotite, and cummingtonite; high experimental \(f_{{0_{2} }}\); and strong focus on approach to equilibrium) will be of interest to petrological and thermodynamic modelers with a focus on evolved magmas from arc volcanic systems.
References
Anderson DJ, Lindsley DH, Davidson PM (1993) QUILF: a pascal program to assess equilibria among Fe–Mg–Mn–Ti oxides, pyroxenes, olivine, and quartz. Comput Geosci 19:1333–1350. https://doi.org/10.1016/0098-3004(93)90033-2
Barclay J, Carmichael ISE (2004) A hornblende basalt from western Mexico: water-saturated phase relations constrain a pressure-temperature window of eruptibility. J Petrol 45(3):485–506. https://doi.org/10.1093/petrology/egg091
Barclay J, Rutherford MJ, Carroll MR, Murphy MD, Devine JD, Gardner J, Sparks RS (1998) Experimental phase equilibria constraints on pre-eruptive storage conditions of the Soufriere Hills magma. Geophys Res Lett 25(18):3437–3440. https://doi.org/10.1029/98GL00856
Benage M, Wright H, Coombs M (2018) The response of andesites to pulsatory eruptions. 2018 Fall AGU Meeting
Blundy J, Cashman K (2008) Reconstruction of magmatic system variables and processes. In: Putirka KD, Tepley FJ (eds) Minerals, inclusions and volcanic processes: reviews in mineralogy and geochemistry 69. Mineralogical Society of America, Chantilly, Virginia, pp 179–239
Browne BL, Gardner JE (2006) The influence of magma ascent path on the texture, mineralogy, and formation of hornblende reaction rims. Earth Planet Sci Lett 246:161–176. https://doi.org/10.1016/j.epsl.2006.05.006
Brugger CR, Johnston D, Cashman KV (2003) Phase relations in silicic systems at one-atmosphere pressure. Contrib Mineral Petrol 146:356–369. https://doi.org/10.1007/s00410-003-0503-9
Cervelli P, Fournier T, Freymueller J, Power J, Lisowski M, Pauk BA (2010) Geodetic constraints on magma movement and withdrawal during the 2006 eruption of Augustine Volcano. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 427–452. https://doi.org/10.3133/pp1769
Coombs ML, Gardner JE (2004) Reaction rim growth on olivine in silicic melts: implications for magma mixing. Am Min 89(5–6):748–758. https://doi.org/10.2138/am-2004-5-608
Coombs ML, Vazquez JA (2014) Cogenetic late Pleistocene rhyolite and cumulate diorites from Augustine Volcano revealed by SIMS 238U–230Th dating of zircon, and implications for silicic magma generation by extraction from mush. Geochem Geophys Geosyst 15(12):4846–4865. https://doi.org/10.1002/2014GC005589
De Angelis SMH, Neill OK (2012) MINERAL: a program for the propagation of analytical uncertainty through mineral formula recalculations. Comput Geosci 48:134–142. https://doi.org/10.1016/j.cageo.2012.05.023
De Angelis SH, Larsen J, Coombs M (2013) Pre-eruptive magmatic conditions at Augustine Volcano, Alaska, 2006: evidence from amphibole geochemistry and textures. J Petrol 54(9):1939–1961. https://doi.org/10.1093/petrology/egt037
Devine JD, Gardner JD, Brack HP, Layne GD, Rutherford MJ (1995) Comparison of microanalytical methods for estimating H2O contents of silicic volcanic glasses. Am Min 80:319–328
Evans BW (2007) Take dacite, add fire and brimstone, get peraluminous. Abstract, XXth Hutton Granite Conference, Stellenbosch. South Africa
Evans BW, Ghiorso MS (1995) Thermodynamics and petrology of cummingtonite. Am Min 80:649–663
Geschwind CH, Rutherford MJ (1992) Cummingtonite and the evolution of the Mount St Helens (Washington) magma system: an experimental study. Geology 20(11):1011–1014. https://doi.org/10.1130/0091-7613(1992)020%3c1011:CATEOT%3e2.3.CO;2
Ghiorso MS, Evans BW (2008) Thermodynamics of rhombohedral oxide solid solutions and a revision of the Fe–Ti two-oxide geothermometer and oxygen-barometer. Am J Si 308:957–1039. https://doi.org/10.2475/09.2008.01
Gualda GAR, Ghiorso MS, Lemons RV, Carley TL (2012) Rhyolite-MELTS: A modified calibration of MELTS optimized for silica-rich, fluid-bearing magmatic systems. J Petrol 53:875–890. https://doi.org/10.1093/petrology/egr080
Hammer JE, Rutherford MJ, Hildreth W (2002) Magma storage prior to the 1912 eruption at Novarupta, Alaska. Contrib Mineral Petrol 144:144–162. https://doi.org/10.1007/s00410-002-0393-2
Harris G (1994) The petrology and petrography of lava from the 1986 eruption of Augustine Volcano. Ph.D. thesis, University of Alaska Fairbanks
Holtz F, Sato H, Lewis J, Behrens H, Nakada S (2005) Experimental petrology of the 1991–1995 Unzen dacite, Japan. Part 1: phase relations, phase composition, and pre-eruptive conditions. J Petrol 46:319–337. https://doi.org/10.1093/petrology/egh077
Johnston D (1978). Volatiles, magma mixing, and the mechanism of eruptions at Augustine Volcano, Alaska. Ph.D. thesis, University of Washington, Seattle.
Lange RA, Frey HM, Hector J (2009) A thermodynamic model for the plagioclase-liquid hygrometer/thermometer. Am Min 94(4):494–506. https://doi.org/10.2138/am.2009.3011
Larsen J, Nye C, Coombs M, Tilman M, Izbekov P, Cameron C (2010) Petrology and geochemistry of the 2006 eruption of Augustine Volcano. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 335–382. https://doi.org/10.3133/pp1769
Leake B, Woolley A, Arps C, Birch W, Gilbert C, Grice J, Hawthorne F, Kisch H, Kato A, Krivovichev V, Laird J, Linthougt K, Mandarino J, Maresch W, Nickel E, Rock N, Schumacher J, Smith D, Stephenson N, Ungaretti L, Whittaker E, Youzhi G (1997) Nomenclature of amphiboles: report of the subcommittee on amphiboles of the international mineralogical associations commission on new minerals and mineral names. Can Min 3:219–246
Martel C, Schmidt BC (2003) Decompression experiments as an insight into ascent rates of silicic magmas. Contrib Mineral Petrol 144:397–415. https://doi.org/10.1007/s00410-002-0404-3
McGee KA, Doukas MP, McGimsey RG, Neal CA, Wessels RL (2010) Emission of SO2, CO2, and H2 S from Augustine Volcano, 2002–2008. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 609–627. https://doi.org/10.3133/pp1769
Moore G, Carmichael ISE (1998) The hydrous phase equilibria (to 3 kbar) of an andesite and basaltic andesite from western Mexico: constraints on water content and conditions of phenocryst growth. Contrib Mineral Petrol 130:304–319. https://doi.org/10.1007/s004100050367
Nadeau PA, Webster JD, Mandeville CW, Goldoff BA, Shimizu N, Monteleone B (2015) A glimpse into Augustine Volcano's pleistocene past: insight from the petrology of a massive rhyolite deposit. J Volcanol Geotherm Res 304:304–323. https://doi.org/10.1016/j.jvolgeores.2015.07.034
Neal, CA, Murray TL, Power JA, Adleman JN, Whitmore PM, Osiensky JM (2010) Hazard information management, interagency coordination, and impacts of the 2005–2006 eruption of Augustine Volcano. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 645–667. https://doi.org/10.3133/pp1769
Newman S, Lowenstern JB (2002) Volatile-calc: a silicate melt-H2O–CO2 solution model written in visual basic for excel. Comput Geosci 28:597–604. https://doi.org/10.1016/S0098-3004(01)00081-4
Nicholls IA, Oba T, Conrad WK (1992) The nature of primary rhyolitic magmas involved in crustal evolution: evidence from an experimental study of cummingtonite-bearing rhyolites, TaupoVolcanic Zone. New Zealand Geochim Cosmochim Acta 56(3):955–962. https://doi.org/10.1016/0016-7037(92)90039-L
Nielsen CH, Sigurdsson H (1988) Quantitative methods of electron microprobe analysis of sodium in natural and synthetic glasses. Am Min 66:547–552
Papale P, Moretti R, Barbato D (2006) The compositional dependence of the saturation surface of H2O + CO2 fluids in silicate melts. Chem Geol 229:78–95. https://doi.org/10.1016/j.chemgeo.2006.01.013
Pichavant M, Costa F, Burgisser A, Scaillet B, Martel C, Poussineau S (2007) Equilibration scales in silicic to intermediate magmas—implications for experimental studies. J Petrol 48(10):1955–1972. https://doi.org/10.1093/petrology/egm045
Power J, Lalla D (2010) Seismic observations of Augustine Volcano, 1970–2007. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 3–40. https://doi.org/10.3133/pp1769
Power JA, Coombs ML, Freymueller JT (eds) (2010) The 2006 eruption of Augustine volcano. Alaska, U.S. Geol. Sur. Professional Paper, p 1769. https://doi.org/10.3133/pp1769
Prouteau G, Scaillet B (2003) Experimental constraints on the origin of the 1991 Pinatubo dacite. J Petrol 44(12):2203–2241. https://doi.org/10.1093/petrology/egg075
Ratajeski K, Sisson T (1999) Loss of iron to gold capsules in rock-melting experiments. Am Min 84:1521–1527. https://doi.org/10.2138/am-1999-1005
Reed SJB (2005) Electron microprobe analysis and scanning electron microscopy in geology. Cambridge University Press, Cambridge
Reubi O, Blundy J, Varley NR (2013) Volatiles contents, degassing and crystallisation of intermediate magmas at Volcan de Colima, Mexico, inferred from melt inclusions. Contrib Mineral Petrol 165:1087–1106. https://doi.org/10.1007/s00410-013-0849-6
Roman DC, Cashman KV, Gardner CA, Wallace PJ, Donovan JJ (2006) Storage and interaction of compositionally heterogeneous magmas from the 1986 eruption of Augustine Volcano. Alaska Bull Volcanol 68(3):240–254. https://doi.org/10.1007/s00445-005-0003-z
Rutherford M (1985) The May 18, 1980 eruption of Mount St Helens 1 Melt composition and experimental phase equilibria. J Geophys Res 90(B4):2929–2947. https://doi.org/10.1029/JB090iB04p02929
Rutherford MJ, Devine JD (1996) Pre-eruption pressure-temperature conditions and volatiles in the 1991 dacitic magma of Mount Pinatubo. In: Newhall CG, Punongbayan RS (eds) Fire and Mud: eruptions and lahars of Mount Pinatubo, Philippines. University of Washington Press, Seattle, pp 751–766
Rutherford MJ, Devine JD (2003) Magmatic conditions and magma ascent as indicated by hornblende phase equilibria and reactions in the 1995–2002 Soufrière Hills Volcano magma. J Petrol 44(8):1433–1454. https://doi.org/10.1093/petrology/44.8.1433
Scaillet B, Evans B (1999) The 15 June 1991 eruption of Mount Pinatubo I Phase equilibria and pre-eruption P-T–fO2–fH2O conditions of the dacite magma. J Petrol 40(3):381–411. https://doi.org/10.1093/petroj/40.3.381
Sisson T, Grove T (1993) Experimental investigations of the role of H2O in calc-alkaline differentiation and subduction zone magmatism. Contrib Mineral Petrol 113:143–166. https://doi.org/10.1007/BF00283225
Sisson TW, Ratajeski K, Hankins WB, Glazner AF (2005) Voluminous granitic magmas from common basaltic sources. Contrib Mineral Petrol 148:635–661. https://doi.org/10.1007/s00410-004-0632-9
Steiner AR, Browne BL, Nye CJ (2012) Quenched mafic inclusions in ≤2200 years B.P. deposits at Augustine volcano. Alaska Int Geol Rev 54:1241–1270. https://doi.org/10.1080/00206814.2011.636641
Tappen CM, Webster JD, Mandeville CW, David R (2009) Petrology and geochemistry of ca. 2100–1000 aBP magmas of Augustine volcano, Alaska, based on analysis of prehistoric pumiceous tephra. J Volcanol Geotherm Res 183(1/2), 42–62. Doi: 10.1016/j.jvolgeores.2009.03.007.
Tilman MR (2008) An investigation of symplectite-rimmed olivine and magmatic processes during the 2006 eruption of Augustine Volcano. Alaska. M.S. thesis, University of Alaska Fairbanks
Tomiya A, Takahashi E, Furukawa N, Suzuki T (2010) Depth and Evolution of a Silicic Magma Chamber: Melting Experiments on a Low-K Rhyolite from Usu Volcano. Japan. J Petrol 51(6):1333–1354. https://doi.org/10.1093/petrology/egq021
Vallance JW, Bull KF, Coombs ML (2010) Pyroclastic flows, lahars, and mixed avalanches generated during the 2006 eruption of Augustine Volcano. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 219–268. https://doi.org/10.3133/pp1769
Venezky DY, Rutherford MJ (1999) Petrology and Fe–Ti oxide reequilibration of the 1991 Mount Unzen mixed magma. J Volcanol Geotherm Res 89(1–4):213–230. https://doi.org/10.1016/S0377-0273(98)00133-4
Waters LE, Lange RA (2015) An updated calibration of the plagioclase-liquid hygrometer–thermometer applicable to basalts through rhyolites. Am Min 100(10):2172–2184. https://doi.org/10.2138/am-2015-5232
Waythomas CF, Waitt RB (1998) Preliminary volcano-hazard assessment for Augustine Volcano, Alaska. U.S. Geol. Sur. Open-File Report 98–106, p 39
Webster JD, Mandeville CW, Goldoff B, Coombs M, Tappen C (2010) Augustine Volcano—the influence of volatile components in magmas erupted A.D. 2006 to 2,100 years before present. In: Power JA, Coombs ML, Freymueller JT (eds) The 2006 eruption of Augustine Volcano, Alaska. U.S. Geol. Sur. Professional Paper 1769, pp 383–423. https://doi.org/10.3133/pp1769
Wood BJ, Carmichael ISE (1973) P total, and P H2O the occurrence of cummingtonite in volcanic rocks. Contrib Mineral Petrol 40(2):149–158. https://doi.org/10.1007/BF00378172
Acknowledgements
The authors are indebted to Ken Severin and to the staff of the Alaska Volcano Observatory. A significant amount of analytical work for this study was conducted in the Advanced Instrumentation laboratory housed in the College of Natural Sciences and Mathematics at the University of Alaska Fairbanks (UAF). We would also like to thank the FEI Company of Hilsboro Oregon, and Richard H. Worden of the University of Liverpool, for significant support with the Qemscan® WellSite SEM hardware and iDiscover® software. This work was supported by a grant from the National Science Foundation to J. Larsen (NSF EAR 0911694). We thank Tom Sisson, Tim Grove, and an anonymous reviewer for constructive comments and suggestions that improved this manuscript. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Timothy L. Grove.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
De Angelis, S.H., Larsen, J.F., Coombs, M.L. et al. Phase equilibrium of a high-SiO2 andesite at \(f_{{0_{2} }}\) = RRO: implications for Augustine Volcano and other high-\(f_{{0_{2} }}\) arc andesites. Contrib Mineral Petrol 175, 24 (2020). https://doi.org/10.1007/s00410-020-1663-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-020-1663-6