
Abstract
Experiments are applied to constrain the composition of primary kimberlitic magmas which were in equilibrium with lithospheric peridotite and could resorb the entrained diamond to form typical dissolution features. The experiments are run on samples of a model carbonatite and a melt of the Udachnaya kimberlite at 6.3 GPa and 1400 °C, and at unbuffered or Re–ReO2-buffered oxygen fugacity (1–2 log units above Ni–NiO). Near-liquidus dry Fe3+-free carbonatitic melt (derived from carbonated harzburgite) is saturated with the Ol–Grt–Opx–Mgs assemblage and is almost inert to diamond. Carbonatitic melts that bear 4.6–6.8 wt% Fe2O3 or 1.5 wt% H2O are in equilibrium only with Mgs ± Ol near the liquidus. Dissolution of diamond by these melts produces surface textures uncommon (corrosion sculptures) or common (negative-oriented trigons, shield-shaped laminae and elongate hillocks) to kimberlitic diamonds. The near-liquidus melt of the Udachnaya kimberlite (Yakutia) with 10–12 wt% H2O is saturated with the Ol–Grt–Cpx assemblage and may result from melting of carbonated garnet-bearing wehrlite. Hydrous kimberlitic melt likewise resorbs diamonds forming typical negative-oriented trigons, shield-shaped laminae and elongate hillocks on their surfaces. Therefore, the melts that could originate in the thermal conditions of subcratonic lithosphere, entrain diamond and dissolve it to produce dissolution features on crystal surfaces, were compositionally close to kimberlite (16–19 wt% SiO2) and rich in H2O. Dry Fe3+-bearing carbonatites with fO2 controlled by the ferric/ferrous equilibrium slightly above the Ni–NiO buffer cannot be diamond carriers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Kimberlite magma attracts much interest being a host of diamonds. Its generation involved metasomatism, plume magmatism and fluids and required specific redox conditions in the mantle. Although a wealth of data has been collected, many aspects of kimberlite genesis remain poorly understood. A key issue among them is the primary composition the magma had at the time when it separated from the source lithology and began its ascent to the Earth’s surface. This separation was inferred to occur at temperatures from 1400 to 1600 °C (Mitchell 2008; Brey et al. 2009) and oxygen fugacities from the enstatite–magnesite–olivine–diamond/graphite (EMOD/G) to fayalite–magnetite–quartz (FMQ) and nickel–bunsenite (NNO) buffers (Mitchell 1973; Fedortchouk and Canil 2004; Fedortchouk et al. 2005; Höfer et al. 2009; Stagno et al. 2013).
The reconstructed primary magma compositions proceed from petrology and geochemistry data on hypabyssal kimberlites from South Africa (le Roex et al. 2003; Harris et al. 2004; Becker and le Roex 2006) and the Lac de Grace field (Kjarsgaard et al. 2009), as well as on quenched kimberlite samples of a hypabyssal dike that cuts the Jericho pipe, both in Canada (Kopylova et al. 2007). According to these publications, the proto-kimberlite magma had a carbonate–silicate composition with high contents of volatiles (H2O and CO2) and MgO (~25–31 wt%) but low SiO2 (~26–30 wt%), Al2O3 and alkalis. Kamenetsky et al. (2012) suggest that the primary composition recorded in ultrafresh kimberlite from the Udachnaya-East pipe in Yakutia, as well as other kimberlites of group I, correspond to an alkalic anhydrous carbonatite melt (SiO2 ~5 wt%) which became saturated with olivine and acquired kimberlitic features by dissolution of peridotitic mantle during its ascent. Note that carbonatite compositionally similar to this anhydrous magma (aillikite) was found recently in cratonic areas, though its genetic relation to kimberlite was questioned (Mitchell and Tappe 2010).
The composition of primary magma equilibrated with a lithospheric peridotite source is known to mainly depend on H2O content and temperature. Small-degree partial melting of anhydrous carbonated peridotite under thermal conditions of subcratonic lithosphere at T ≤ 1400 °C and 6–10 GPa produces essentially carbonatitic liquid (Dalton and Presnall 1998; Dasgupta and Hirschmann 2007; Brey et al. 2008; Dasgupta et al. 2013) remaining carbonate rich even at 1600–1700 °C (Brey et al. 2008). Addition of H2O into the system both reduces the solidus temperature of carbonated peridotite and increases the Si solubility of the melt, thus transforming the carbonatite into a carbonate–silicate liquid, which shares much similarity with typical kimberlite magmas (Brey et al. 2009; Foley et al. 2009). According to multiple saturation experiments, kimberlite-like melts may reach equilibrium with garnet peridotite at >1400 °C and under a broad range of pressures and CO2 and H2O (Eggler and Wendlandt 1979; Edgar et al. 1998; Edgar and Charbonneau 1993; Mitchell 2004; Ulmer and Sweeney 2002; Girnis et al. 1995, 2011; Sokol et al. 2013a, 2014).
Diamond may store information on the kimberlite magma at different stages of its evolution. The available knowledge includes (1) composition of carbonate and carbonate–silicate growth environments of fibrous diamond related to kimberlite melts (Navon 1999; Klein-BenDavid et al. 2007, 2009) and (2) experimentally constrained potential conditions of diamond crystallization in kimberlite melts (Arima et al. 1993; Palyanov et al. 2015). However, there are little experimental data on diamond dissolution in kimberlite-like melts at P–T conditions of the kimberlite source region that allow interpreting faithfully the dissolution features typical of natural crystals. Kozai and Arima (2005) studied diamond dissolution at 1.0 GPa, 1300–1420 °C and fO2 controlled by the iron-wüstite (IW), wüstite-magnetite (WM) and magnetite-hematite (MH) buffers in the aphanitic kimberlite samples from Wesselton Mine (South Africa). Fedortchouk et al. (2007) considered dissolution of diamond in synthetic kimberlite, alkaline basalt and the CaO–MgO–SiO2–H2O–CO2 system at 1 GPa and 1150–1500 °C and fO2 near the NNO buffer. The study by Khokhryakov and Palyanov (2007) focused on the morphology evolution in diamonds exposed to dissolution in the hydrous melt of the Udachnaya kimberlite at 5.7 GPa and 1400 °C. The obtained data show that the degree of diamond dissolution strongly depends on temperature, fO2, and the presence of free fluid in kimberlite magma.
Little is known about diamond dissolution in carbonated melts. They generally do not react with diamond at 1 GPa under reduced conditions (Kozai and Arima 2005), whereas dry carbonated melts at 5.7–7.0 GPa and 1400–1750 °C are rather active agents that can maintain both growth and dissolution of diamond (Pal’yanov et al. 2002; Khokhryakov and Palyanov 2010).
This study provides experimental constraints on the composition of primary magma (carbonatite and hydrous archetypical kimberlite melt) at the time when it separated from the source region. The verification criteria include: (1) possibility for generation of melts multiply saturated with peridotite under subcratonic thermal conditions recorded by the deepest xenoliths from the Udachnaya kimberlite and (2) reproducibility in the experiments of surface dissolution features common to natural diamonds.
Methods
Starting materials
We used samples of fresh group I kimberlite from the Udachnaya-East pipe in Yakutia (referred to as Udachnaya kimberlite), which has been documented by Kamenetsky et al. (2009, 2012). Before the experiments, the samples were cleaned from xenogenic material and preconditioned as reported by Sokol et al. (2013a).
The composition of a melt derived from K-rich carbonated harzburgite at 1400 °C and 6 GPa (hereafter B6) (Brey et al. 2011; run M-277) was used to model carbonatite magma. The carbonatite composition was modified in some experiments. Since carbonatite melts can efficiently extract water from nominally anhydrous minerals (NAMs) of peridotite (Sokol et al. 2013b), 1.5–5 wt% H2O were added to some charges. To model carbonatite melts equilibrated with ilmenite and/or magnetite, which may bear high contents of Fe3+ and have fO2 notably above the CCO buffer (Bataleva et al. 2012), 4.6–6.8 wt% Fe2O3 were added to some carbonatite samples. The summary of starting compositions is presented in Table 1.
The starting mixture corresponding to B6 carbonatite was made of chemically pure carbonates (MgCO3, CaCO3, Na2CO3, K2CO3), oxides (MgO, SiO2, Al2O3, Cr2O3, TiO2, FeO, Fe2O3, MnO, NiO) and natural olivine (9 wt% FeO) from a xenolith in the Udachnaya kimberlite. Then the kimberlite powder and B6 samples were dried and stored in a desiccator. The charge weight was in the range from 111 to 113 mg. Distilled water was added by a micro-syringe, immediately before the final capsule assembly. The Pt capsules were placed inside a vessel filled with liquid nitrogen and welded shut. The total error on addition of water was ±0.3 wt%, as estimated by weighing the sealed capsules.
In each experimental run, we used three octahedral crystals of natural diamond from the Udachnaya and Aikhal kimberlites (Yakutia). They were colorless flat-faced crystals (0.3–0.43 mg, 0.93–1.22 mg total weight or 0.8–1.1 wt% of charge), free from resorption signatures, inclusions or cracks visible in an optical microscope. The diamond crystals were cleaned in a mixture of concentrated sulfuric acid and aqueous solution of potassium permanganate (volume ratio 3:1) before each run and in hydrochloric acid after each run. The measured weight of crystals was accurate to ±0.01 mg.
Redox conditions
The performed explorative runs (Ud-1, Ud-2, Ud-3) showed that diamond dissolution in the melt obtained from the Udachnaya kimberlite in a Pt capsule (without fO2 buffering) was independent of water amount. It stopped after 3 h upon saturation with carbon at the account of hydrogen flow into the capsule from the high-pressure cell where fO2 is controlled by the furnace assembly (Boettcher et al. 1973). Hydrogen input into the charge was observed in experiments with CO2–C and CO2–H2O–C systems sealed in Pt capsules as notable H2O enrichment of the fluid (Sokol et al. 2004; Pal’yanov et al. 2005). Sokol et al. (2004) showed that the furnace assembly used in this study controls fO2 in the Pt capsule with C–O–H fluid slightly above “water maximum” (CW, corresponding to the maximum H2O content in C–O–H fluids).
Given that diamond dissolution depends on experimental P–T–fO2 conditions (Kozai and Arima 2005; Fedortchouk et al. 2007; Khokhryakov and Palyanov 2007, 2010), the fO2 control had to be adapted to the objectives of this study. Unlike the routine control of pressure and temperature, that of oxygen fugacity is a challenge. The widespread double-capsule method of fO2 control (Fig. 1a) works due to incorporation of water into the outer capsule with metal-oxide buffers, such as IW, WM, MH or NNO. The redox reaction of the buffer components with water gives rise to equilibrium fH2. Since platinum is permeable to hydrogen which can freely pass through the walls of the inner Pt capsule, the hydrogen fugacity is fixed in the sample capsule (Boettcher et al. 1973; Luth 1989); the oxygen fugacities in both the buffer and the sample are equal when they have equal fH2O.

Schemes used for fO2 buffering of samples in high-pressure experiments. a Classical double-capsule assembly; b new assembly with an insulating outer container and a Re-lined Pt capsule. 1 outer capsule; 2 inner capsule; 3 sample; 4 metal-oxide buffer; 5 NiO + CsCl or Fe2O3 + CsCl (2:1) insulating container; 6 Re liners; 7 CsCl
The classical double-capsule method is typically applied below 1400 °C because at higher temperatures the inner capsule alloys with the metal component of the metal-oxide buffer and thus loses chemical inertness. Note that Pt can absorb iron from Fe-bearing systems as a result of alloying with Fe0, and this process changes the oxidation state of the sample (Brey et al. 2011).
In some cases, Pt capsules are lined with Re foil in order to prevent Fe loss and sometimes to fix fO2 in the sample (Girnis et al. 2011). Re lining used with oxidized systems may give rise to ReO2 films on the surface indicating fO2 conditions of the Re–ReO2 buffer, or 1–2 log units above Ni–NiO (Pownceby and O’Neill 1994). This technique is, however, difficult to apply when fH2 in the capsule should be lower than that controlled by the furnace assembly. Without external buffering of fH2, uncontrolled hydrogen leakage into the capsule leads to reduction of ReO2 to Re0 (or prevents oxidation of Re liners) and the buffer fails. Thus, Re–ReO2 buffering of fO2 in a H2O-bearing sample requires preventing hydrogen leakage from outside to the capsule.
In this study, the double-capsule technique of fO2 control was modified: We used Pt capsules which were lined with Re foil and placed in special NiO + CsCl or Fe2O3 + CsCl (weight ratio 2:1) containers (Fig. 1b). Such containers reduced fH2 around the capsule to below the Re–ReO2-buffered fH2 in the water-bearing sample. The capsules with samples were insulated from the containers with a CsCl layer (after long drying at ~100 °C). In the case of a NiO + CsCl outer container, oxygen fugacity (\(f{\text{O}}_{2}^{S}\)) in the sample receiving hydrogen from the environment was (Whitney 1972)
where KNNO is the equilibrium constant of the Ni + 1/2O2 = NiO reaction and fH2OS and fH2OB are the H2O fugacities of the sample and the buffer, respectively.
As long as the H2O fugacity in the outer container with NiO (fH2OB) remains much lower than that in the sample (fH2OS), hydrogen leakage into the inner capsule becomes actually blocked. Note that the outer container with Fe2O3 impeded the hydrogen leakage to the charge still more strongly.
With this approach, the system was protected against hydrogen input from the furnace assembly for the whole run duration, provided that NiO or Fe2O3 remained in the outer container. Furthermore, low fH2 in the insulating container caused some hydrogen leakage from the sample and partial oxidation of the liner surface to ReO2 (Fig. 2). Thus, fO2 in the charge was buffered by Re–ReO2, while Re foil prevented the iron-bearing systems from Fe loss.

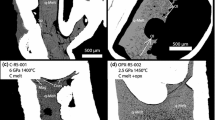
A typical quench texture of melt and products of Re oxidation in samples (run# B6-5, 6.8 wt% Fe2O3)
High-pressure apparatus
The experiments were carried out in a split-sphere multianvil high-pressure apparatus (Palyanov et al. 2010). The 8/6 multianvil sphere consisted of two anvils with square faces on top and bottom and four side anvils with rectangular faces placed in an octahedral cavity formed by truncating the vertices of eight steel anvils [for detail, see Fig. 1 in Palyanov et al. (2010)]. The sizes of the high-pressure cell were 21.1 × 21.1 × 25.4 mm; the graphite heater had an inner diameter of 12 mm and a height of 18.8 mm. Pressure was calibrated by recording the change in the resistance of Bi at 2.55 GPa and of PbSe at 4.0 and 6.8 GPa at room temperature and by bracketing the graphite–diamond equilibrium (Kennedy and Kennedy 1976) at high temperatures. Temperature was monitored in each experiment with a PtRh6/PtRh30 thermocouple calibrated at 6.3 and 7.5 GPa using the melting points of Ni and Pt. For details of the pressure and temperature calibration, see (Palyanov et al. 2010; Sokol et al. 2015). Pressure and temperature were measured to an accuracy of 0.1 GPa and 20 °C. The temperature gradient within the Pt capsule was within 10 °C/mm. The runs were terminated by power cutoff. The quench rate was about 200 °C/s.
Analytical technique
The recovered Pt capsules were cleaned from CsCl, dried, weighted and then sawed along their axes into halves producing a top-to-bottom section. One half was polished without the use of water. Samples were studied using optical and scanning electron (Tescan MYRA 3 LMU) microscopy. Electron microprobe analyses (EMPA) were performed at 20 kV accelerating voltage and 20 nA beam current on a Cameca Camebax and a Jeol JXA-8100 microanalyzers; the beam diameters for silicate and carbonate phases were 1–2 µm. The standards used were as follows: pyrope for Si, Al and Fe; diopside for Mg and Ca; albite for Na; orthoclase for K; ilmenite for Ti; and spinel for Ni. The uncertainty of the measurements was within two relative percent for all components.
In spite of fast cooling rates, the synthesized melts were not quenched to glass but rather formed aggregates of feather-like phases consisting of 50–500 µm dendritic silicate and carbonate crystals. The quenched melts were analyzed by a defocused beam with an area of 100 × 100 µm at 20 kV accelerating voltage and 40 nA beam current on a Cameca Camebax and a Jeol JXA-8100 microprobes.
The phase compositions of the NiO or Fe2O3 containers were analyzed by powder X-ray diffraction after the experiments.
Results
Phase equilibria
The duration of the main runs (10 h) was selected proceeding from explorative runs (3–10 h) and known rates of diamond dissolution in carbonate and carbonate–silicate melts rich in volatiles (Kozai and Arima 2005; Fedortchouk et al. 2007; Khokhryakov and Palyanov 2007, 2010). The dry melt and the residual silicate and carbonate phases reached equilibrium, as it follows from the results of experimental melting in the system “dry peridotite–CO2” under similar P–T conditions (Brey et al. 2008), when the compositions of both melt and peridotite minerals became time invariant already after the first few hours at 1500 °C. The observed homogeneity of liquid and crystalline phases indicates that the duration of 10 h at 1400 °C was enough for reaching equilibrium in anhydrous and hydrous samples (except for diamond). Note that the system became saturated with carbon and the diamond dissolution stopped after first 3 h of non-buffered explorative runs, in the presence of hydrogen leakage into the sample.
In the main series of experiments, the redox conditions in the samples were set using Re lining of Pt capsules. As shown by powder X-ray diffraction (Table 2), NiO and Fe2O3 were predominant phases in the outer containers after the experiments. The Re liner developed a film of oxide on the inner surface, detectable from energy-dispersive spectra. Furthermore, ReO2 grains were found in the quenched melt near the Re liner surface, which were especially abundant when Fe2O3 was added (Fig. 2). All this provides evidence of Re–ReO2 buffering of fO2 in the samples. In two runs with hydrous kimberlite, the Pt capsules had no Re lining, and Fe loss may have increased fO2 close to the MH buffer, according to data of Brey et al. (2011).
The weight proportions of phases in the main series experiments were estimated by least square mass-balance calculations for a restricted number of components in the system: SiO2, TiO2, Al2O3, MgO and CaO. The calculations were made using the starting compositions of charges and mineral phases and yielded a sum of squared residuals ≤1. The approximate proportions of phases were additionally controlled from BSE images. The results are summarized in Tables 2 and 3 and in Figs. 2, 3, 4 and 5.

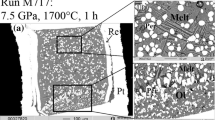
SEM images of the recovered samples. a run# B6-1 (dry); b run# B6-2 (1.5 wt% H2O); c run# B6-5 (6.8 wt% Fe2O3); d run# Ud-5 (12 wt% H2O). Ol, olivine; Opx, orthopyroxene; Cpx, clinopyroxene; Grt, garnet; Liq, quenched liquid

Experimentally determined phase relations of the Udachnaya kimberlite at 6.3 GPa, from (Sokol et al. 2013a) with new data obtained in this study

Liquid compositions and phase relations as a function of kimberlite and carbonatite compositions at 6.3 GPa (the analyses were not normalized to 100 wt% volatile-free compositions). a SiO2 content in the liquid and equilibrium phases as a function of CO2/(CO2 + H2O) molar ratios in the charge. Pentagons correspond to assemblages present in various regions of the diagram; solid sectors = phase present; void sectors = phase absent. B09 is after (Brey et al. 2009); S13 is after (Sokol et al. 2013a). Ol, olivine; Opx, orthopyroxene; Cpx, clinopyroxene; Grt, garnet; Mgs, magnesite; L, quenched liquid. b Compositions of obtained melts (open diamonds); compositions of kimberlites from (Sparks et al. 2009; Kamenetsky et al. 2009). W, Wesselton; WR, Wesselton (reconstructed); J, Jericho; B, Benfontain; IH, Igwesa Hills; U, Uintjiesberg; Ud, Udachnaya-East; reconstructed primary magmas are after Le Roex et al. 2003 (a), Harris et al. 2004 (b), Becker and Le Roex 2006 (c), Kopylova et al. 2007 (e, f) and Kjarsgaard et al. 2009 (g, h); (A) composition trend of primary melts in the systems CMAS-CO2 at 1380–1500 °C (Dalton and Presnall 1998); shaded fields are kimberlite compositions after (Mitchell 1986); c K2O in the melt versus CO2/(CO2 + H2O) molar ratio in the charge; d Al2O3 in the melt versus CO2/(CO2 + H2O) molar ratio in the charge
The starting charge compositions simulated potential primary magma compositions of carbonatite to kimberlite (Table 1). In experiments with dry and wet B6 carbonatite, the weight fraction of melt at 6.3 GPa and 1400 °C varied from 0.78 to 0.84. In the sample obtained by melting of B6 without adding H2O or Fe2O3, the liquid coexisted with the Ol–Grt–Opx–Mgs assemblage (Fig. 3a). Thus, the liquid of B6 near liquidus was multiply saturated with the same solid phases that were equilibrated with the liquid in the harzburgite–K2CO3–MgCO system at 6 GPa and 1400 °C (Brey et al. 2011, run M-277). In fact, the melt of dry carbonatite became saturated with respect to carbonated harzburgite under the experiment conditions.
As H2O and/or Fe2O3 were added to the carbonatite system, (1) magnesite remained the only stable solid phase, instead of the four-phase assemblage, at 1.5 wt% H2O (Fig. 3b) and carbonatite became completely molten at 5 wt% H2O; (2) multiple saturation of the melts with minor stable phases of olivine and magnesite was lost at 4.6–6.8 wt% Fe2O3 (Fig. 3c).
The phase assemblages of the Udachnaya kimberlite samples obtained in non-buffered explorative runs were similar to those reported by Sokol et al. (2013a) and were not analyzed in detail. Complete melting of kimberlite was detected at 13 wt% H2O. The main focus was on the assemblages and chemistry of phases in kimberlite melting experiments with controlled oxygen fugacity. The samples with 10–12 wt% H2O at 1400 °C had the melt weight fractions from 0.73 to 0.8 (Table 2). At 10 wt% H2O, the liquid became saturated with the Ol–Grt–Cpx assemblage. The melt was in equilibrium with the Ol–Grt–Cpx assemblage in one run with 12 wt% H2O but was saturated with Ol and Cpx only in another run (Figs. 3d, 4, 5a).
Compositions of liquid and solid phases
The compositions of quenched melts depend on the concentrations of main components in carbonate–silicate charges and on water content (Table 3; Fig. 5). The melt fraction varied from 0.73 to 1, and the melt occupied a large part of the capsule’s high-temperature region. As \(X_{{{\text{CO}}_{2} }}\) (CO2/(CO2 + H2O) mole ratio) in the starting composition decreased from 1 to ~0.2, the SiO2 content increased from ~5 to ~19 wt% (Fig. 5a). When H2O and Fe2O3 were added to B6 carbonatite, the content of SiO2 in the melt slightly increased and decreased, respectively. At 10–12 wt% H2O in kimberlite, the melt contained from 16 to 19 wt% SiO2. Note that the analyses of bulk melt compositions had quite large standard deviations (Table 3).
According to the experimental results we obtained at 6.3 GPa and 1400 °C, as well those reported by Brey et al. (2009) and Sokol et al. (2013a), the concentration of SiO2 in multiply saturated carbonate–silicate melts correlates with the composition of volatiles (Fig. 5a): SiO2 increases as \(X_{{{\text{CO}}_{2} }}\) in the system decreases. Thus, increased contents of silicate phases in the system potentially can recover multiphase saturation of H2O-bearing B6 carbonatite. At 1.5 and 5 wt% H2O in B6 (\(X_{{{\text{CO}}_{2} }}\) = 0.9 and 0.7), the melt should contain >9 and >11 wt% SiO2, respectively. In fact, this already a carbonate–silicate rather than carbonatitic melt.
The obtained melts only partly fell within the kimberlite field in the SiO2/MgO versus MgO/CaO diagram (Fig. 5b). The melts derived from hydrous kimberlite were the most proximal to the Benfontain, Igwesa Hills, Wesselton (reconstructed) kimberlite compositions (Sparks et al. 2009), as well as to the primary magma compositions reconstructed by Becker and le Roex (2006), Kopylova et al. (2007) and Kjarsgaard et al. (2009). Note that the melt compositions of this study had MgO/CaO markedly lower than in the Udachnaya kimberlite.
The melts derived from dry and wet carbonatite, with high MgO/CaO, generally fell beyond the primary kimberlite field (Fig. 5b), while MgO was the highest in those synthesized from hydrous carbonatite. On the other hand, dry B6 generated a melt that followed the trend Dalton and Presnall (1998) observed for the CMAS-CO2 system. Changes in the contents of Al2O3 and K2O in the synthesized melts were observed when \(X_{{{\text{CO}}_{2} }}\) changed in the starting charges (Fig. 5c, d), but were largely due to bulk composition variations in the latter.
Neither sample produced in this study showed fluid phase bubbles, i.e., there is no indication that a free fluid formed within the studied range of water concentrations. The added water corresponds to 5 and 14 wt% in the carbonatite and kimberlite melts, respectively, and both melts remained unsaturated with fluid.
Olivine occurred as subhedral, sometimes prismatic, colorless crystals up to 100 µm. The olivines obtained by melting of B6 carbonatite had lower Mg# (83–88) than those derived from hydrous kimberlite (93–95) (Table 3; Fig. 6a). The apparent melt-olivine Fe–Mg partition coefficient KD (Fe/Mg) was systematically higher for low-Fe olivines from wet kimberlites than for the high-Fe varieties from carbonatite (Fig. 6b).

Olivine compositions as a function of the starting charge and melt composition. a Mg# of olivine versus CO2/(CO2 + H2O) molar ratio in the charge; b melt-olivine Fe–Mg partition coefficient K D = (X Fe(melt)·X Mg(olivine))/(X Fe(olivine)·X Mg(melt)), e.g., X Fe(melt) = Fe/(Fe + Mg + Ca)
Garnet formed as compositionally uniform isometric unzoned crystals, up to 50 μm, in both water-bearing kimberlite and dry carbonatite charges. The garnets stable in hydrous kimberlite melt saturated with Cpx had Ca# (34–36) slightly higher than those derived from dry carbonatite (Stagno and Frost 2010). Given that MgO and CaO in the starting charges and the resulting melts were rather similar while the amount of silicates was small, the difference was apparently due to different phase assemblages of kimberlite and carbonatite samples: Ol–Grt–Cpx–L or Ol–Grt–Opx–Mgs–L, respectively. The contents of Cr2O3 in kimberlite garnet varied from 1.7 to 2.7 wt% (Table 3).
Orthopyroxene was present as a solid phase in melts derived from B6 carbonatite. Most of its crystals were 30–50 μm in size, had subhedral prismatic habits and contained 0.8 wt% Al2O3 and 1.3 wt% Ca. Magnesite was the main subliquidus phase of B6, with Ca enrichment varying slightly from 2.2 to 2.6 wt% as the starting charge compositions changed, while FeO increased in the series “wet B6–dry B6–B6 with Fe2O3.”
Note that mass-balance calculations for the runs of hydrous kimberlite melting showed notably greater Fe loss from the Pt capsules without Re lining than from the Re-lined ones (Tables 2, 3).
Features of diamond dissolution in hydrous kimberlite melts
Re lining of Pt capsules (Re–ReO2-buffered fO2) affected the dissolution rate and morphology of diamonds (Table 4). Namely, dissolution was 1.5 times faster without Re lining than with it, the content of water in kimberlite being the same (12 wt% H2O). The faster dissolution correlates with higher Fe loss from samples in the Pt capsules, as well as with higher fO2 according to Brey et al. (2011).
After all runs, diamond crystals had transitional morphologies with approximately equal percentages of resorbed surfaces and original {111} faces (Fig. 7). The crystals generally exhibited the same features of dissolution as in melting of hydrous carbonate–silicate systems: negative-oriented trigons, elongate hillocks and shield-shaped laminae, the latter having polycentric patterns on most {111} faces. However, there was some difference, especially in striation on rounded surfaces: The surfaces of tetrahexahedroids were covered with a fine striation in the case of fO2 buffered by Re–ReO2 but had round-stepped features and more elongate hillocks at higher fO2 (about MH).

SEM images of diamond crystals partly dissolved in hydrous kimberlite melt. a run# Ud-4 (10 wt% H2O, Pt capsule without Re lining); b run# Ud-5 (12 wt% H2O, Pt capsule without Re lining); c, d run# Ud-6 (12 wt% H2O, Pt capsule with Re lining)
Features of diamond dissolution in carbonatite melts
After melting of dry B6 carbonatite, the diamond crystals showed no weight loss, were smooth-faced and free from dissolution (resorption) features. Unlike those, diamonds examined after the runs with high-Fe2O3 carbonatite were more or less strongly resorbed (Fig. 8), having numerous irregularly shaped pits on the surfaces with their sizes proportional to the resorption degree. The weight losses in diamonds observed upon incorporation of 4.6 and 6.8 wt% Fe2O3 into carbonatite were 0.13 and 0.33 mg, respectively, or 12 and 33 % of the initial weight of the crystals. In the former case, they preserved their original octahedral habits but had rounded edges and corners (Fig. 8a, b), and matted surfaces produced by abundant <10 µm pits. As the amount of added Fe2O3 increased to 6.8 wt%, the diamonds lost their original habits (Fig. 8c, d) and acquired rough surfaces with some raised relict original surfaces.

SEM image of diamond crystals resorbed in carbonatite melt with added Fe2O3. a, b run# B6-4 (4.6 wt% Fe2O3, Pt capsule with Re lining); c, d run# B6-5 (6.8 wt% Fe2O3, Pt capsule with Re lining); b, d enlarged fragments of crystal surfaces
Melting of hydrous carbonatite with 1.5 and 5.1 wt% H2O led to resorption of diamonds and 0.21 and 0.29 mg weight losses, respectively, or 18 and 26 % of the initial weight. At 1.5 wt% H2O, the crystals preserved the original {111} faces (Fig. 9a) with sporadic flat-bottomed etch pits (negative-oriented trigons) and shield-shaped dissolution laminae. The diamond crystals from the run with 5.1 wt% H2O became more spherical (Fig. 9b), and their {111} faces became glossy rounded surfaces of tetrahexahedroids, with shield-shaped laminae and elongate hillocks.

SEM images of diamond crystals partly dissolved in water-bearing carbonatite melt. a run# B6-2 (1.5 wt% H2O, Pt capsule with Re lining); b run# B6-3 (5.1 wt% H2O, Pt capsule with Re lining)
Discussion
The composition of kimberlite magmas generated within a lithospheric source region and ascending to the surface largely depends on temperature and oxygen fugacity. The temperatures of subcratonic lithospheric mantle (SLM) beneath the Udachnaya site show bimodal distribution, according to xenolith data (Boyd et al. 1997; Goncharov et al. 2012) (Fig. 10). The upward “inflection” from the ~40 to ~45 mW/m2 geotherm of Pollack and Chapman (1977) observed in the lower one-third of the lithospheric mantle (Goncharov et al. 2012) was interpreted as preceding the eruption of kimberlite and resulting either from intrusion of hot material in the lower lithosphere (Goncharov et al. 2012) or from plume heat (Howarth et al. 2014). Given that thickness of the lithospheric mantle beneath Udachnaya ~220 km (Goncharov et al. 2012), the intersection of the 45 mW/m2 geotherm with the mantle adiabat corresponds to temperatures at the potential kimberlite source in a range of ~1320 to ~1400 °C (Fig. 10).

P–T estimates for peridotite xenoliths from the Udachnaya kimberlite and experimental conditions of this study. Model conductive geotherms are according to Pollack and Chapman (1977)
Data from the deepest mantle xenoliths cannot provide rigorous constraints on the redox conditions in the source region of kimberlite magma. The fO2 values for these xenoliths correspond to stability of silicate melts which may contain either H2O or ≪10 wt% CO3 2− (Stagno et al. 2013). Thus, only tentative estimates of possible fO2 range are possible (Fig. 11), with its lower bound slightly below EMOG/D but above CW (“water maximum”). The fO2 corresponding to CW is assumed to be the lowest limit as C–O–H fluids in these conditions have the highest H2O contents but very low CO2 of a few percent (Foley 2011). The upper fO2 bound is chosen with reference to the redox conditions of the formation of ilmenite megacrysts in kimberlite (Arculus et al. 1984). Note that this oxygen fugacity is not the limit for ilmenite which remains stable at still higher values (Fig. 11). The selected fO2 range for kimberlite magmas comprises the values valid for a polymictic breccia (Höfer et al. 2009) as well as for xenoliths metasomatized by the most strongly oxidizing melts/fluids (Creighton et al. 2009). Therefore, the applied 6.3 GPa and 1400 °C correspond to the available estimates of conditions within a lithospheric source region beneath Udachnaya, while the Re–ReO2 buffer constrains the highest possible fO2 in kimberlite magma.

P–fO2 conditions of runs and oxygen fugacity estimates for kimberlite magma (P–T paths: 6.3 GPa and T = 1400 °C near the source region and T = 1000 °C near the surface). Open star marks unbuffered explorative runs; bold star marks experiments at Re–ReO2-buffered fO2; bold square corresponds to fO2 of the kimberlite melt at around 1150 °C and 3.5 GPa (Höfer et al. 2009); bold vertical bar corresponds to conditions of olivine–chromite pair crystallization from kimberlite melt (1 GPa, ~1080 °C) according to (Fedortchouk and Canil 2004); gray square is fO2 of xenoliths oxidized by metasomatic fluid/melt (Creighton et al. 2009); fO2 of megacryst ilmenite from kimberlite is according to Arculus et al. (1984). Oxygen fugacities of most strongly oxidized xenoliths are according to Stagno et al. (2013). The mantle xenolith samples are from the Kaapvaal (1), Slave (2) and Siberian (3) cratons. The reaction Ilm + O2 = Mgt + Ru is constrained using the method of Bataleva et al. (2012). The Re–ReO2 buffer is constrained from joint data of Pownceby and O’Neill (1994) and Rohrbach and Schmidt (2011). Data on NNO and EMOG/D buffers and on water maximum (CW) are borrowed from Foley (2011) and Stagno et al. (2013)
Multiple saturation of melts
The experiments demonstrated multiple saturation of the Udachnaya kimberlite melt with Ol–Grt–Cpx at 10–12 wt% H2O, 6.3 GPa, 1400 °C, and fO2 slightly above the NNO buffer (Fig. 11). Thus, the stability limit of the kimberlite melt saturated with this assemblage as reported by Sokol et al. (2013a) were extended toward lower temperatures and higher water contents and fO2 (Fig. 4). Proceeding from complete melting of kimberlite at 1400 °C with 13 and 14 wt% H2O, we infer multiple saturation of the melt near the liquidus in the experiments with 10–12 wt% H2O (Fig. 4). This shows that the Udachnaya hydrous kimberlite (10–12 wt% H2O) to have originated in the Siberian SLM by melting of carbonated garnet-bearing wehrlite under the thermal conditions close to the 45 mW/m2 geotherm.
It is interesting to compare the results of multiple saturation experiments for group I and group II (micaceous) kimberlites. The melt of group II kimberlite can reach near-liquidus multiple saturation with Ol–Grt–Opx at 6.3 GPa, only at ~1500–1600 °C, and no more than 5 wt% H2O (Sokol et al. 2014). The absence of multiple saturation in the case of more hydrous (>5 wt% H2O) compositions with \(X_{{{\text{CO}}_{2} }}\) < 0.37 indicates the lack of equilibrium between group II kimberlite magma and garnet harzburgite. Thus, unlike the group I Udachnaya kimberlite, generation of group II kimberlite is impossible at <1500 °C.
The results for dry B6 carbonatite multiply saturated with the Ol–Grt–Opx–Mgs assemblage at 6.3 GPa, 1400 °C and fO2 slightly above the NNO buffer are also consistent with its origin by melting of carbonated harzburgite stable in the Siberian SLM at ~200 km depth about the 45 mW/m2 geotherm (Brey et al. 2011). The presence of H2O (even as little as 1.5 wt%) and Fe2O3 (4.6–6.8 wt%) in carbonatite lead to the loss of multiple saturation: The melt becomes saturated with magnesite only in the former case and only olivine and magnesite remain in equilibrium with it in the latter case. However, multiple saturation of both hydrous kimberlite and anhydrous carbonatite with respect to the peridotite assemblage implies that their mixtures will be saturated as well. Therefore, dissolution of additional silicate phases in B6 carbonatite with added H2O is expected to provide multiple saturation and SiO2 increase in the melt. Silica contents in multiply saturated hydrous carbonate–silicate melts may range from ~9 to ~19 wt%, according to interpolation of the data of Fig. 5a. This gives rise to a wide spectrum of primary mantle melts, depending on H2O activity, at the thermal conditions of SLM beneath Udachnaya.
Thus, when separating from the source lithology, the melts equilibrated with peridotite in the subcratonic lithospheric mantle at 6.3 GPa and 1400 °C may have had compositions from dry K-rich carbonatite to hydrous kimberlite with 10–12 wt% H2O and SiO2 slightly below 20 wt%. Generation of multiply saturated kimberlite magma with more than 20 wt% SiO2 was possible also if the peridotite source region was heated to >1400 °C (Eggler and Wendlandt 1979; Edgar et al. 1998; Edgar and Charbonneau 1993; Ulmer and Sweeney, 2002; Girnis et al. 1995, 2011; Sokol et al. 2013a, 2014).
Diamond dissolution
Dry carbonate and hydrous kimberlite melts may be growth or dissolution environments for diamond at the P–T–fO2 conditions of subcratonic mantle (Pal’yanov et al. 1999, 2002; Palyanov and Sokol 2009; Khokhryakov and Palyanov 2010; Palyanov et al. 2015). Diamond or graphite carbon can dissolve in carbonate or hydrous silicate melts either by oxidation to CO2 or CO3 2−, or in the elemental form, depending on oxygen fugacity (Sokol and Pal’yanov 2008; Palyanov and Sokol 2009). At fO2 about CW, dissolved C0 in hydrous H2O–C systems may predominate and reach 8 wt% at 7.5 GPa and 1600 °C, judging by the amount of quench carbon (Sokol and Pal’yanov 2008).
According to the available estimates, the temperature of erupting kimberlite magma changes from ~1400 °C in the source region (Mitchell 2008) to ~1080 °C at 1 GPa (Fedortchouk and Canil 2004) and ≤ 1000 °C at 1 bar (Nechaev and Khokhryakov 2013). The rapidly ascending originally dry magma reacts neither with xenoliths nor with wall rock. The position of the CO2 isopleth for carbonate–silicate melts (Dasgupta et al. 2013) at the P–T conditions in ascending kimberlite magma indicates that interaction with mantle peridotite can only slightly change the mostly carbonate composition of dry magma, which remains with >25 wt% CO2 at 2–6 GPa. Furthermore, the entrapped xenoliths cannot provide significant water input to dry magma because the amount of fluid phase is negligibly small, while water extraction from nominally anhydrous minerals is difficult (Demouchy et al. 2006). Therefore, initially dry magma should remain dry and mostly carbonate. The value of fO2 in dry magma remains close to the EMOD/G buffer at sufficient amounts of diamond and/or graphite, or otherwise depends on the Fe2+/Fe3+ ratio (Stagno et al. 2013).
The absence of weight loss in diamonds upon melting of B6 indicates that dry Fe3+-free carbonatite melts are weak solvents of diamond at the potential conditions of magma separation from the source region (~6.3 GPa and ~1400 °C) even at fO2 slightly above the NNO buffer. Proceeding from the data for low pressures (Rudenko et al. 1979; Kozai and Arima 2005), we infer that Fe3+-free ascending carbonate melts are inert to the diamonds they host and thus can carry diamonds almost without dissolving them. However, most of kimberlitic diamonds show resorption signatures as shield-shaped laminae, hillocks, etc. (Orlov 1977; Robinson et al. 1989); few non-resorbed crystals may have been transported inside xenoliths and got into the magma near the surface where the latter disintegrated.
Generation of carbonate magma in which ferric/ferrous equilibria control fO2 is associated with Fe3+ release upon decomposition of skiagite garnet and ensuing carbon oxidation (Rohrbach and Schmidt 2011; Stagno et al. 2013). Since ∆fO2 between the EMOD/G and ferric/ferrous equilibria increases as pressure decreases (Stagno et al. 2013), Fe3+-bearing magma becomes progressively a more stronger oxidizer for diamond, and the effect may be even greater at higher Fe3+ contents. In the case of an external control of fO2 in the melt, the Fe3+/Fe2+ ratio can adjust to the equilibrium value of wall rock. However, the xenoliths entrained from greatest depths hold relatively low fO2 in the oxidized carbonate–silicate magma (Goncharov et al. 2012; Stagno et al. 2013) and the two never reach fO2 equilibrium.
Diamond dissolution in dry magma with fO2 controlled by ferric/ferrous equilibria was simulated by incorporation of Fe2O3 into carbonatite B6. Note that in our experiments, the ferric/ferrous ratio in the melt was fixed by the Re–ReO2 buffer, i.e., fO2 in the melts with Fe2O3 was the same as in hydrous melts and generally close to the upper fO2 limit in kimberlite magma (Fig. 11). Diamond resporption in the melting experiments with the Fe3+-bearing carbonatite occurred only as formation of corrosion sculptures. Such sculptures are widespread in natural diamonds and look like pitted hemispherical cavities and coarse or fine frosting. However, they are rather late-stage etch features superposed on earlier dissolution features, and thus, postdate kimberlite emplacement associated with oxidation or graphitization (Robinson et al. 1989; Davis et al. 1999). Therefore, either the dry carbonatite melts separating from the source region carried diamonds while being depleted in Fe3+ and having fO2 controlled by equilibrium between C0, silicate phases, and carbonate–silicate liquid, or they never entrained diamond.
The redox conditions of hydrous melts were buffered by Re–ReO2 due to the reaction Re + 2H2O = ReO2 + 2H2. Judging by possible hydrogen leakage, the concentration of H2O in the melt decreased slightly during the experiments. The absence of bubbles in the quenched samples indicates that all added water dissolved in the melt, which thus lacked equilibrium fluid phases. In mafic and ultramafic magmas, water dissolves either as hydroxyl (OH) or as molecular H2O. The OH−/H2O ratio decreases as the melts become more mafic (Persikov 1998). The maximum amount of chemically bound water (OH−) dissolved in ultramafic melts can reach 2 wt% (Persikov 1998) or a few percent higher. Thus, the resorption features of diamonds in hydrous carbonatite and kimberlite melts may be due to the effect of dissolved molecular H2O. This inference is consistent with the fact that diamond dissolution in pure H2O under similar P–T conditions likewise produces negative-oriented trigons and shield-shaped laminae (Kanda et al. 1977), with the only difference that the trigons are larger and more abundant and they form at higher dissolution rates.
Diamond dissolution in both hydrous carbonatite and kimberlite melts produces spherical tetrahexahedroid habits. The surface features in such diamonds depend on the magnitude of the crystal lattice strains (Khokhryakov and Palyanov 2015) and on the composition of volatiles (Fedortchouk et al. 2007; Khokhryakov and Palyanov 2007, 2010). According to Yamaoka et al. (1980), oxygen fugacity influences the orientations of trigons on the {111} faces of diamonds. The texture of tetrahexahedroid surfaces can be used as a proxy of fO2: It is fine striation in the case of Re–ReO2-buffered fO2 in hydrous kimberlite melts or a round-stepped structure at higher fO2 about the MH buffer. Both features are common to natural spherical diamonds and may be indicators of fO2 in the host kimberlite magma. The revealed correlation between fO2 and the surface features in tetrahexahedroids hosted by kimberlite melts needs further investigation at lower pressures.
In our experiments at 6.3 GPa, 1400 °C and Re–ReO2-buffered fO2, diamond dissolved at rates of 0.020–0.024 and 0.021–0.029 mg/mm2 h in kimberlite (10–12 wt% H2O) and carbonatite (1.5 and 5.1 wt% H2O) melts, respectively. Although fO2 in the experiments was buffered at the upper limit for kimberlite magma (Fig. 11), the obtained rates are close to the slowest ones in the range 0.024–0.070 mg/mm2 h reported for compositionally different hydrous carbonate and silicate systems studied at 1–7.5 GPa and 1350–1400 °C (Fedortchouk et al. 2007; Khokhryakov and Palyanov 2007, 2010). At a dissolution rate within this range, a 10-mm diamond may convert to a tetrahexahedroid in ≥70 h. Judging by the predominant habits of natural diamonds and the estimated dissolution rates, the inferred duration of kimberlite magma ascent from a few hours to several days depending on temperature (Spera 1984; Meyer 1985; Pearson et al. 1997; Canil and Fedortchouk 1999; Kelley and Wartho 2000) appears quite reasonable. However, diamond can completely dissolve if hydrous magma stays long enough in intermediate reservoirs (Franz et al. 1996).
The carbonatite and kimberlite melts of our experiments were undersaturated with fluid and reacted with diamond over the entire range of H2O concentrations. However, Fedortchouk et al. (2007) showed that kimberlite melt at 1.0 GPa dissolved diamond only in the presence of a fluid phase. Thus, further studies are also required to constrain the role of free fluid in diamond dissolution. The presence of fluid bubbles in the primary magma was important for its separation from the source lithology and ascent along conduits by crack propagation (Brey et al. 2009). The compositions of the carbon-saturated fluid may have been from H2O–CO2 (CH4 < 5 %) at the lowest fO2 between the CW and EMOG/D (Foley 2011) to pure CO2 when fO2 was at CCO (Foley 2011 Stagno et al. 2013). At higher fO2 in the magma with unstable C0, the fluid composition apparently depended on the initial concentrations of CO2 and H2O and by their solubility changes with decreasing pressure. The presence of CO2-rich fluid in kimberlite magma can have resulted either from decarbonation of the source mantle peridotite (Brey et al. 2009) or from the stability loss of carbonates in the melt at P < 2 GPa.
Conclusions
Evidence of multiple saturation of carbonatite and nearly archetypical kimberlite melts with peridotite at 6.3 GPa, 1400 °C and fO2 slightly above the NNO buffer, which simulate the conditions of magma separation from the peridotite source lithology, as well as the observed dissolution (resorption) features in the hosted diamonds, has implications for the primary magma compositions. Dry Fe3+-free carbonatite melt separating from the source may become multiply saturated with peridotite and can carry diamond to the surface without reacting with it (an ideal transport environment). The studied Fe3+-rich dry carbonatite melt, in which fO2 is buffered by Re–ReO2, did not reach equilibrium with peridotite at the conditions applied in the experiments. Furthermore, the sculptures produced by this melt on diamond surfaces are uncommon to kimberlite diamonds.
In the thermal conditions of the subcratonic lithosphere base, a hydrous kimberlite melt with 10–12 wt% H2O may form by melting of carbonated garnet wehrlite. On the other hand, hydrous carbonatite containing ≤8 wt% SiO2 turns out to be non-equilibrated with peridotite. Multiply saturated H2O-bearing carbonatite melt apparently should contain more than 9–11 wt% SiO2. The dissolution features on diamonds from hydrous carbonatite and kimberlite melts, at Re–ReO2-buffered or higher fO2, are typical of natural diamonds. Thus, only hydrous carbonate–silicate melts can be both saturated with peridotite and produce the specific dissolution patterns in diamond. The obtained results, along with the evidence of resorbed surfaces in many natural kimberlitic diamonds (negative-oriented trigons, shield-shaped laminae and elongate hillocks), indicate that a large part of diamonds were transported by hydrous carbonate–silicate melts rather than by dry carbonatite melt with fO2 controlled by the ferric/ferrous equilibrium.
References
Arculus RJ, Dawson JB, Mitchell RH, Gust DA, Holmes RD (1984) Oxidation states of the upper mantle recorded by megacryst ilmenite in kimberlite and type A and B spinel lherzolites. Contrib Mineral Petrol 85:85–94
Arima M, Nakayama K, Akaishi M, Yamaoka S, Kanda H (1993) Crystallization of diamond from a silicate melt of kimberlite composition in high-pressure and high-temperature experiment. Geology 21:968–970
Bataleva YuV, Palyanov YN, Sokol AG, Borzdov YuM, Palyanova GA (2012) Conditions for the origin of oxidized carbonate–silicate melts: implications for mantle metasomatism and diamond formation. Lithos 128–131:113–125
Becker M, le Roex AP (2006) Geochemistry of South African on-and off-craton, Group I and Group II kimberlites: petrogenesis and source region evolution. J Petrol 47(4):673–703
Boettcher AL, Mysen BO, Allen JC (1973) Techniques for the control of water fugacity and oxygen fugacity for experimentation in solid-media high-pressure apparatus. J Geophys Res 78(26):5898–5901
Boyd FR, Pokhilenko NP, Pearson DG, Mertzman SA, Sobolev NV, Finger LW (1997) Composition of the Siberian cratonic mantle: evidence from Udachnaya peridotite xenoliths. Contrib Mineral Petrol 128:228–246
Brey GP, Bulatov VK, Girnis AV, Lahaye Y (2008) Experimental melting of carbonated peridotite at 6–10 GPa. J Petrol 49(4):797–821
Brey GP, Bulatov VK, Girnis AV (2009) Influence of water and fluorine on melting of carbonated peridotite at 6 and 10 GPa. Lithos 112:249–259
Brey GP, Bulatov VK, Girnis AV (2011) Melting of K-rich carbonated peridotite at 6–10 GPa and the stability of K-phases in the upper mantle. Chem Geol 281:333–342
Canil D, Fedortchouk Y (1999) Garnet dissolution and the emplacement of kimberlites. Earth Plan Sci Lett 167:227–237
Creighton S, Stachel T, Matveev S, Hofer H, McCammon C, Luth RW (2009) Oxidation of the Kaapvaal lithospheric mantle driven by metasomatism. Contrib Mineral Petrol 157:491–504
Dalton JA, Presnall DC (1998) The continuum of primary carbonatitic–kimberlitic melt compositions in equilibrium with lherzolite: data from the system CaO–MgO–Al2O3–SiO2–CO2 at 6 GPa. J Petrol 39(11-12):1953–1964
Dasgupta R, Hirschmann MM (2007) Effect of variable carbonate concentration on the solidus of mantle peridotite. Am Mineral 92:370–379
Dasgupta R, Mallik A, Tsuno K et al (2013) Carbon-dioxide-rich silicate melt in the Earth’s upper mantle. Nature 493:211–222
Davis RM, O’Relly SY, Griffin WL (1999) Diamonds from Wellington, NSW: insights into the origin of eastern Australian diamonds. Mineral Mag 63:447–471
Demouchy S, Jacobsen SD, Gaillard F, Stern CR (2006) Rapid magma ascent recorded by water diffusion profiles in mantle olivine. Geology 34:429–432
Edgar AD, Charbonneau HE (1993) Melting experiments on a SiO2-poor, CaO-rich aphanitic kimberlite from 5–10 GPa and their bearing on sources of kimberlite magmas. Am Mineral 78:132–142
Edgar AD, Arima M, Baldwin DK et al (1998) High-pressure high-temperature melting experiments on a SiO2-poor aphanitic kimberlite from the Wesselton mine, Kimberley, South Africa. Am Mineral 73:524–533
Eggler DH, Wendlandt RF (1979) Experimental studies on the relationships between kimberlite magma and partial melting of peridotite. In: Boyd FR, Meyer HOA (eds) Kimberlites, diatremes and diamonds: their geology, petrology, and geochemistry. American Geophysical Union, Washington, pp 331–378
Fedortchouk Y, Canil D (2004) Intensive variables in kimberlite magmas, Lac de Gras, Canada and implications for diamond survival. J Petrol 45:1725–1745
Fedortchouk Y, Canil D, Carlson JA (2005) Dissolution form in Las de Gras diamond and their relationship to the temperature and redox state of kimberline magma. Contrib Mineral Petrol 150:54–69
Fedortchouk Y, Canil D, Semenets E (2007) Mechanisms of diamond oxidation and their bearing on the fluid composition in kimberlite magmas. Am Mineral 92:1200–1212
Foley SF (2011) A reappraisal of redox melting in the Earth’s mantle as a function of tectonic setting and time. J Petrol 52:1363–1391
Foley SF, Yaxley GM, Rosenthal A, Buhre S, Kiseeva ES, Rapp RP, Jacob DE (2009) The composition of near-solidus melts of peridotite in the presence of CO2 and H2O between 40 and 60 kbar. Lithos 112:274–283
Franz L, Brey GP, Okrusch M (1996) Reequilibration of ultramafic xenoliths from Namibia by metasomatic processes at the mantle boundary. J Geol 104:599–615
Girnis AV, Brey GP, Ryabchikov ID (1995) Origin of Group IA kimberlites: fluid saturated melting experiments at 45–55 kbar. Earth Planet Sci Lett 134:283–296
Girnis AV, Bulatov VK, Brey GP (2011) Formation of primary kimberlite melts—constraints from experiments at 6–12 GPa and variable CO2/H2O. Lithos 127:401–413
Goncharov AG, Ionov DA, Doucet LS, Pokhilenko LN (2012) Thermal state, oxygen fugacity and C-O-H fluid speciation in cratonic lithospheric mantle: new data on peridotite xenoliths from the Udachnaya kimberlite, Siberia. Earth Planet Sci Lett 357:99–110
Harris M, le Roex A, Class C (2004) Geochemistry of the Uintjiesberg kimberlite, South Africa: petrogenesis of an off-craton, group I, kimberlite. Lithos 74:149–165
Höfer HE, Lazarov M, Brey GP, Woodland AB (2009) Oxygen fugacity of the metasomatizing melt in a polymict peridotite from Kimberley. Lithos 112:1150–1154
Howarth GH, Barry PH, Pernet-Fisher JF, Baziotis IP, Pokhilenko NP, Pokhilenko LN, Bodnar RJ, Taylor LA, Agashev AM (2014) Superplume metasomatism: evidence from Siberian mantle xenoliths. Lithos 184–187:209–224
Kamenetsky VS, Kamenetsky MB, Weiss Y, Navon O et al (2009) How unique is the Udachnaya-east kimberlite: comparison with kimberlites from the Slave Craton (Canada) and SW Greenland. Lithos 112:334–346
Kamenetsky VS, Kamenetsky MB, Golovin AV, Sharygin VV, Maas R (2012) Ultrafresh salty kimberlite of the Udachnaya_East pipe (Yakutiya, Russia): a petrological oddity or fortuitous discovery? Lithos 152:173–186
Kanda H, Yamaoka S, Setaka N, Komatsu H (1977) Etching of diamond octahedrons by high pressure water. J Cryst Growth 38:1–7
Kelley SP, Wartho JA (2000) Rapid kimberlite ascent and the significance of Ar–Ar ages in xenolith phlogopites. Science 289:609–611
Kennedy CS, Kennedy G (1976) The equilibrium boundary between graphite and diamond. J Geophys Res 81:2467–2470
Khokhryakov AF, Palyanov YN (2007) The evolution of diamond morphology in the process of dissolution: experimental data. Am Mineral 92:909–917
Khokhryakov AF, Palyanov YN (2010) Influence of the fluid composition on diamond dissolution forms in carbonate melts. Am Mineral 95:1508–1514
Khokhryakov AF, Palyanov YN (2015) Effect of crystal defects on diamond morphology during dissolution in the mantle. Am Mineral 100:1528–1532
Kjarsgaard BA, Pearson DG, Tappe S, Nowell GM, Dowall DP (2009) Geochemistry of hypabyssal kimberlites from Lac de Gras, Canada: comparisons to a global database and applications to the parent magma problem. Lithos 112:236–248
Klein-BenDavid O, Izraeli ES, Hauri E, Navon O (2007) Fluid inclusions in diamonds from the Diavik mine, Canada and the evolution of diamond-forming fluids. Geochim Cosmochim Acta 71(3):723–744
Klein-BenDavid O, Logvinova AM, Schrauder M et al (2009) High-Mg carbonatitic microinclusions in some Yakutian diamonds a new type of diamond-forming fluid. Lithos 112S:648–659
Kopylova MG, Matveev S, Raudsepp M (2007) Searching for parental kimberlite melt. Geochim Cosmochim Acta 71:3616–3629
Kozai Y, Arima M (2005) Experimental study on diamond dissolution in kimberlitic and lamproitic melts at 1300–1420 °C and 1 GPa with controlled oxygen partial pressure. Am Mineral 90:1759–1766
le Roex AP, Bell DR, Davis P (2003) Petrogenesis of group I kimberlites from Kimberley, South Africa: evidence from bulk-rock geochemistry. J Petrol 44:2261–2286
Luth RW (1989) Natural versus experimental control of oxidation state: effects on the composition and speciation of C–O–H fluids. Am Mineral 74:50–57
Meyer HO (1985) Genesis of diamond: a mantle saga. Am Mineral 70:344–355
Mitchell RH (1973) Composition of olivine, silica activity and oxygen fugacity in kimberlite. Lithos 6:65–81
Mitchell RH (1986) Kimberlites: mineralogy, geochemistry and petrology. Plenum Press, New York
Mitchell RH (2004) Experimental studies at 5–12 GPa of the Ondermatjie hypabyssal kimberlite. Lithos 76:551–564
Mitchell RH (2008) Petrology of hypabyssal kimberlites: relevance to primary magma compositions. J Volcanol Geotherm Res 174:1–8
Mitchell R, Tappe S (2010) Discussion of “Kimberlites and aillikites as probes of the continental lithospheric mantle”, by D. Francis and M. Patterson (Lithos v. 109, p. 72–80). Lithos 115:288–292
Navon O (1999) Diamond formation in the Earth’s mantle. In: Gurney JJ, Gurney JL, Pascoe MD, Richadson SH (eds) VII International Kimberlite conference 2. Cape Town: Red roof design, pp 584–604
Nechaev DV, Khokhryakov AF (2013) Formation of epigenetic graphite inclusions in diamond crystals: experimental data. Russ Geol Geophys 54:399–405
Orlov IL (1977) The mineralogy of the diamond. Wiley, New York
Pal’yanov YN, Sokol AG, Borzdov YM, Khokhryakov AF, Sobolev NV (1999) Diamond formation from mantle carbonate fluids. Nature 400:417–418
Pal’yanov YN, Sokol AG, Borzdov YM, Khokhryakov AF (2002) Fluid-bearing alkaline–carbonate melts as the medium for the formation of diamonds in the Earth’s mantle: an experimental study. Lithos 60:145–159
Pal’yanov YN, Sokol AG, Tomilenko AA, Sobolev NV (2005) Conditions of diamond formation through carbonate–silicate interaction. Eur J Mineral 17:207–214
Palyanov YN, Sokol AG (2009) The effect of composition of mantle fluid/melts on diamond formation processes. Lithos 112S:690–700
Palyanov YN, Borzdov YuM, Khokhryakov AF, Kupriyanov IN, Sokol AG (2010) Effect of nitrogen impurity on diamond crystal growth processes. Cryst Growth Des 10:3169–3175
Palyanov YN, Sokol AG, Khokhryakov AF, Kruk AN (2015) Conditions of diamond crystallization in kimberlite melt: experimental data. Russ Geol Geophys 56:196–210
Pearson DG, Kelley SP, Pokhilenko NP, Boyd FR (1997) Laser 40Ar/39Ar dating of phlogopites from Southern African and Siberian kimberlites and their xenoliths: constraints on eruption ages, melt degassing and mantle volatile compositions. Geol Geofiz 38:106–117
Persikov ES (1998) Viscosities of model and magmatic melts at the TP-parameters of the Earth’s crust and upper mantle. Geol Geofiz 39:1780–1792
Pollack HN, Chapman DS (1977) On the regional variation of heat flow, geotherms, and lithospheric thickness. Tectonophysics 38:279–296
Pownceby MI, O’Neill HSC (1994) Thermodynamic data from redox reactions at high temperatures. IV. Calibration of the Re–ReO2 oxygen buffer from EMF and NiO + Ni-Pd redox sensor measurements. Contrib Mineral Petrol 118:130–137
Robinson DN, Scott JA, Niekerk AV, Anderson VG (1989) The sequence of events reflected in the diamonds of some southern African kimberlites. In Ross J (ed) Kimberlite and related rocks, vol. 2. Geological Society of Australia. Special Publication, Blackwell, Carlton, 14, pp 1042–1053
Rohrbach A, Schmidt MW (2011) Redox freezing and melting in the Earth’s deep mantle resulting from carbon-iron redox coupling. Nature 472:209–212
Rudenko AP, Kulakova II, Sturman VL (1979) Oxidation of natural diamond. Novye dannye o mineralogii SSSR. Academiya Nauk SSSR, Fersman Mineralogical Museum, Moscow, pp 105–125 (in Russian)
Sokol AG, Pal’yanov YN (2008) Diamond formation in the system MgO–SiO2–H2O–C at 7.5 GPa and 1600°C. Contrib Miner Petrol 155:33–43
Sokol AG, Pal’yanov YN, Pal’yanova GA, Tomilenko AA (2004) Diamond crystallization in fluid and carbonate-fluid systems under mantle P–T conditions: 1 Fluid composition. Geochem Int 42:830–838
Sokol AG, Kupriyanov IN, Palyanov YN, Kruk AN, Sobolev NV (2013a) Melting experiments on the Udachnaya kimberlite at 6.3–7.5 GPa: implications for the role of H2O in magma generation and formation of hydrous olivine. Geochim Cosmochim Acta 101:133–155
Sokol AG, Kupriyanov IN, Palyanov YN (2013b) Partitioning of H2O between olivine and carbonate–silicate melts at 6.3 GPa and 1400 °C: implications for kimberlite formation. Earth Planet Sci Lett 383:58–67
Sokol AG, Kruk AN, Palyanov YN (2014) The role of water in generation of group II kimberlite magmas: constraints from multiple saturation experiments. Am Mineral 99:2292–2302
Sokol AG, Borzdov YuM, Palyanov YN, Khokhryakov AF (2015) High-temperature calibration of a multi-anvil high-pressure apparatus. High Press Res 35:139–147
Sparks RSJ, Brooker RA, Field M, Kavanagh J, Schumacher JC, Walter MJ, White J (2009) The nature of erupting kimberlite melts. Lithos 112:429–438
Spera FJ (1984) Carbon dioxide in petrogenesis III: role of volatiles in the ascent of alkaline magma with special reference to xenolith-bearing mafic lavas. Contrib Mineral Petrol 88:217–232
Stagno V, Frost DJ (2010) Carbon speciation in the asthenosphere: experimental measurements of the redox conditions at which carbonate-bearing melts coexist with graphite or diamond in peridotite assemblages. Earth Planet Sci Lett 300:72–84
Stagno V, Ojwang DO, McCammon CA, Frost DJ (2013) The oxidation state of the mantle and the extraction of carbon from Earth’s interior. Nature 493(7430):84–88
Ulmer P, Sweeney RJ (2002) Generation and differentiation of group II kimberlites: constraints from a high-pressure experimental study to 10 GPa. Geochim Cosmochim Acta 66:2139–2153
Whitney JA (1972) The effect of reduced H2O fugacity on the buffering of oxygen fugacity in hydrothermal experiments. Am Mineral 57:1902–1908
Yamaoka S, Kanda H, Setaka N (1980) Etching of diamond octahedrons at high temperatures and pressures with controlled oxygen partial pressure. J Mater Sci 15:332–336
Acknowledgments
We wish to thank Yury Borzdov, Galina Palyanova and Alexey Kruk for their assistance throughout the study. The manuscript profited much from editorial handling and comments by Max Schmidt, as well as from thoughtful reviews by Andrei Girnis and the anonymous reviewer. The research was performed by Grant of the Russian Science Foundation (Project 14-27-00054).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Max W. Schmidt.
Rights and permissions
About this article

Cite this article
Sokol, A.G., Khokhryakov, A.F. & Palyanov, Y.N. Composition of primary kimberlite magma: constraints from melting and diamond dissolution experiments. Contrib Mineral Petrol 170, 26 (2015). https://doi.org/10.1007/s00410-015-1182-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-015-1182-z